
Heterogeneous DNA Methylation Patterns in the GSTP1 Promoter Lead to Discordant Results between Assay Technologies and Impede Its Implementation as Epigenetic Biomarkers in Breast Cancer

Abstract
:1. Introduction

2. Experimental Section
2.1. Patient Material
2.2. Bisulfite Conversion
2.3. Methylation Specific PCR, MSP
2.4. Quantitative Methylation-Specific PCR, MethyLight
2.5. Pyrosequencing
2.6. MALDI Mass-Spectrometry
2.7. Cloning and Sequencing
2.8. RNA Expression
2.9. Statistical Analysis
3. Results and Discussion
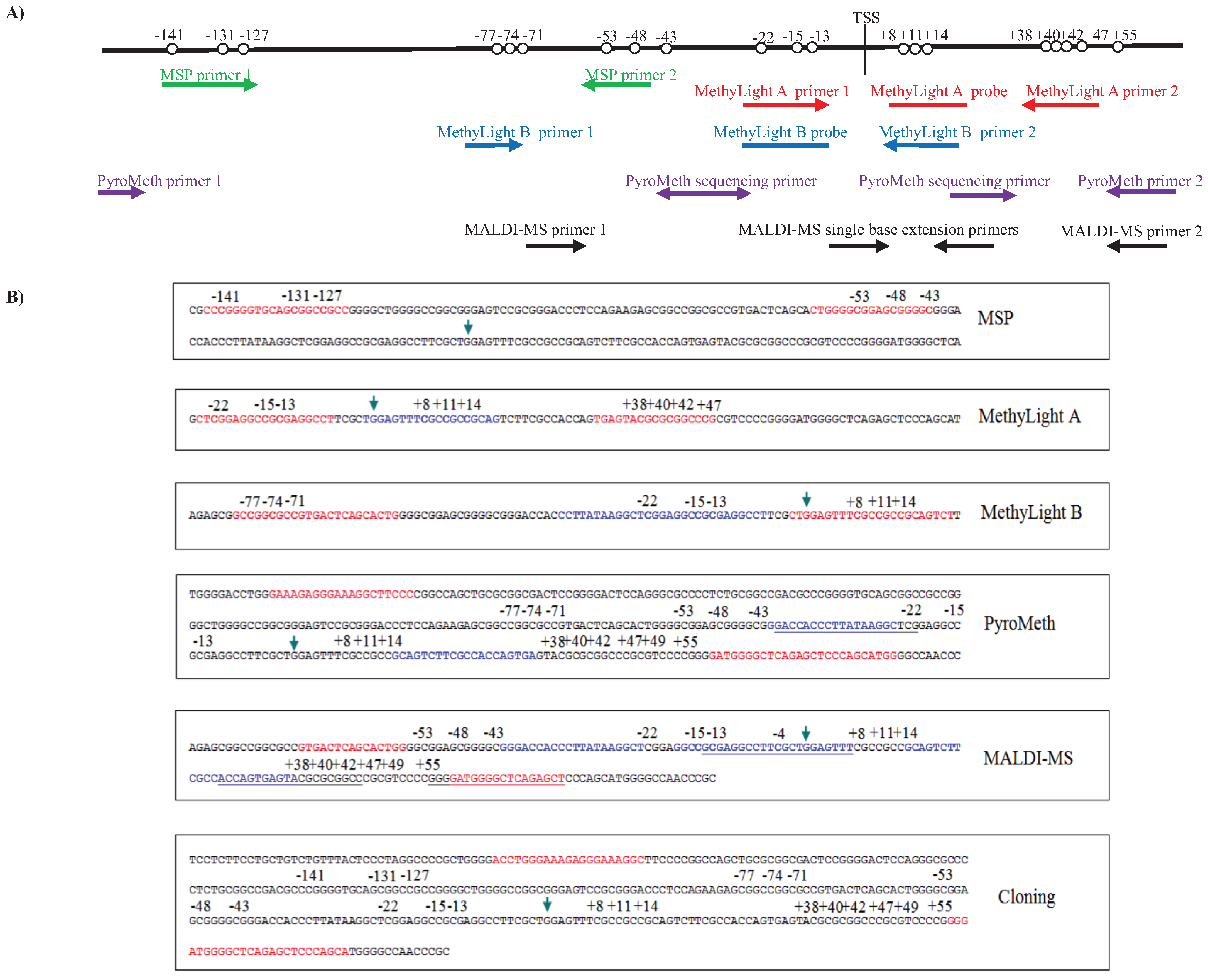
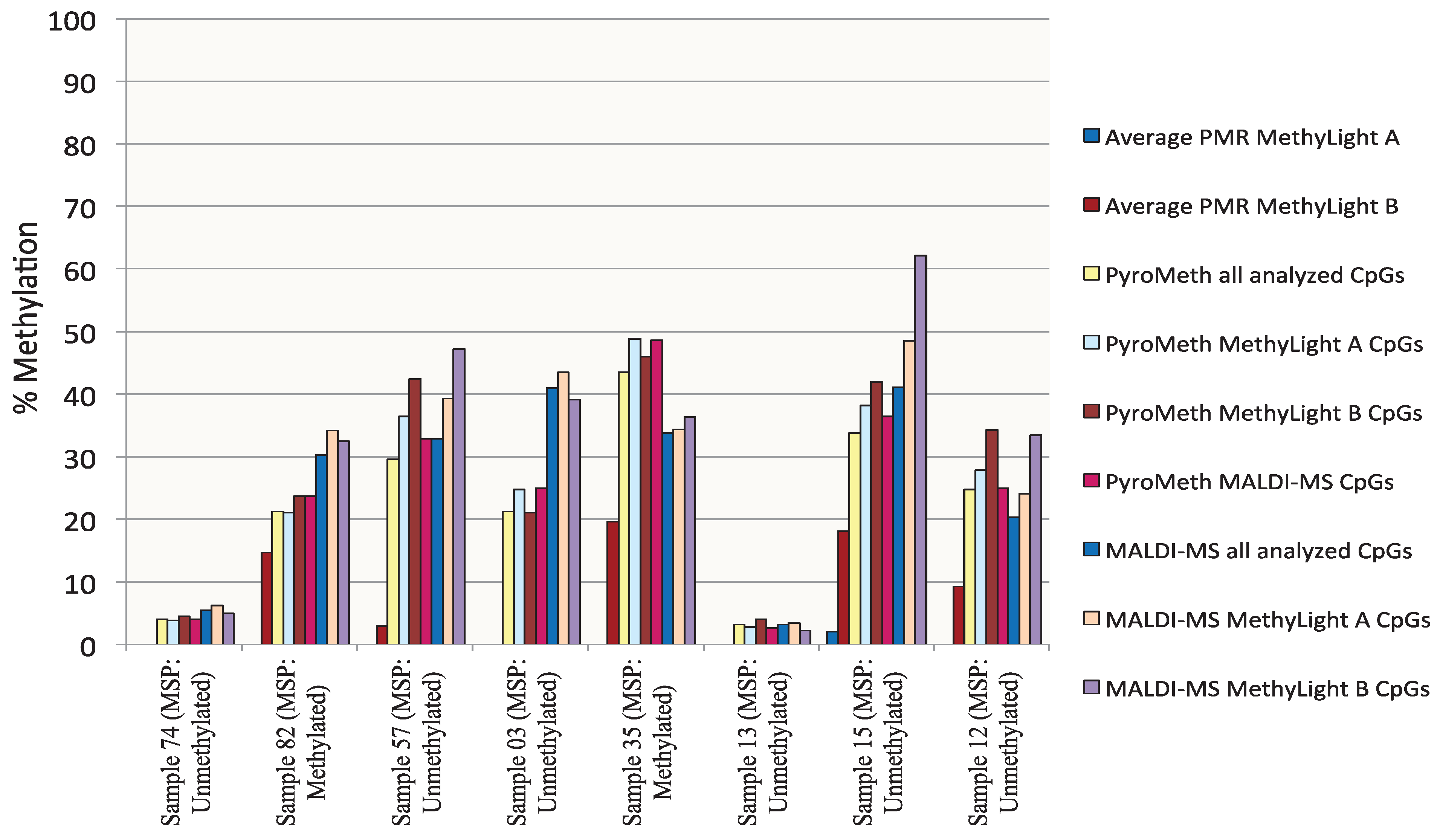
3.1. Analysis of the DNA Methylation Status of the GSTP1 Promoter Using MSP and MethyLight


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MethyLight A 1 | ||||||||||
| MethyLight B 2 | 0.0003 | |||||||||
| (n = 80) | ||||||||||
| PyroMeth-MethyLight A CpGs 3 | 4.0097 × 10−15 (n = 67) | |||||||||
| PyroMeth-MethyLight B CpGs 4 | 2.8522 × 10−11 (n = 67) | |||||||||
| PyroMeth - MALDI-MS CpGs 5 | ||||||||||
| PyroMeth-average of the analyzed CpGs 6 | 2.0858 × 10−16 (n = 66) | 5.3771 × 10−7 (n = 69) | ||||||||
| MALDI-MS-MethyLight A CpGs 7 | 5.4416 × 10−18 (n = 73) | 0.4639 | ||||||||
| (n = 70) | ||||||||||
| MALDI-MS-MethyLight B CpGs 8 | 1.8476 × 10−6 (n = 76) | 0.0929 | ||||||||
| (n = 68) | ||||||||||
| MALDI-MS-average of the analyzed CpGs 9 | 2.1717 × 10−17 (n = 73) | 1.3602 × 10−7 (n = 76) | 0.5727 | 0.8495 | ||||||
| (n = 68) | (n = 70) | |||||||||
| MSP 1 | 0.0011 | 0.9999 | 2.5759 × 10−6 | 7.7596 × 10−7 | ||||||
| (n = 71) | (n = 73) | (n = 63) | (n = 71) | |||||||
| MethyLight A 1 | MethyLight B 2 | PyroMeth-MethyLight A CpGs 3 | PyroMeth-MethyLight B CpGs 4 | PyroMeth-MALDI-MS CpGs 5 | PyroMeth-average of the analyzed CpGs 6 | MALDI-MS-MethyLight A CpGs 7 | MALDI-MS-MethyLight B CpGs 8 | MALDI-MS-average of the analyzed CpGs 9 | MSP 10 |
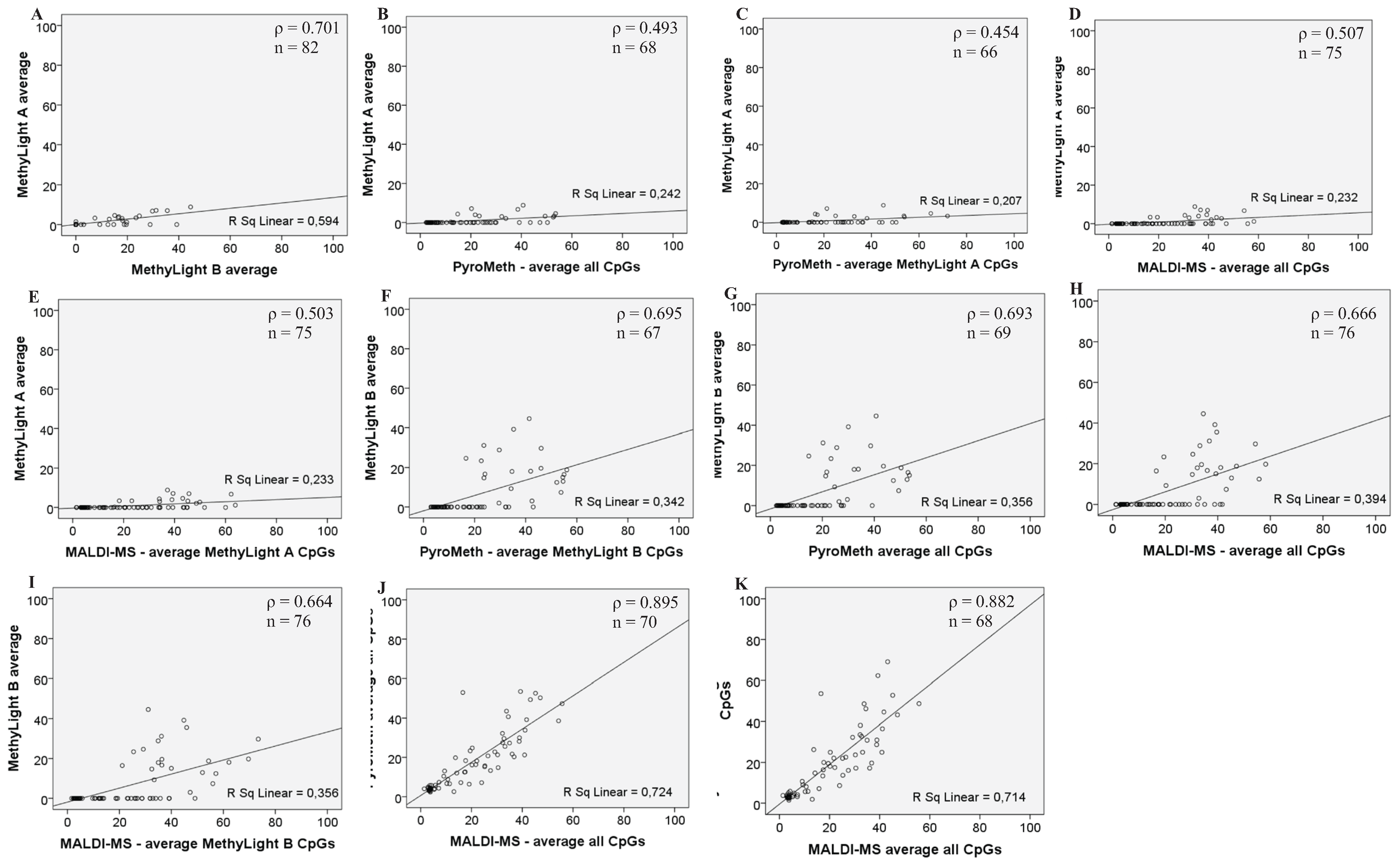
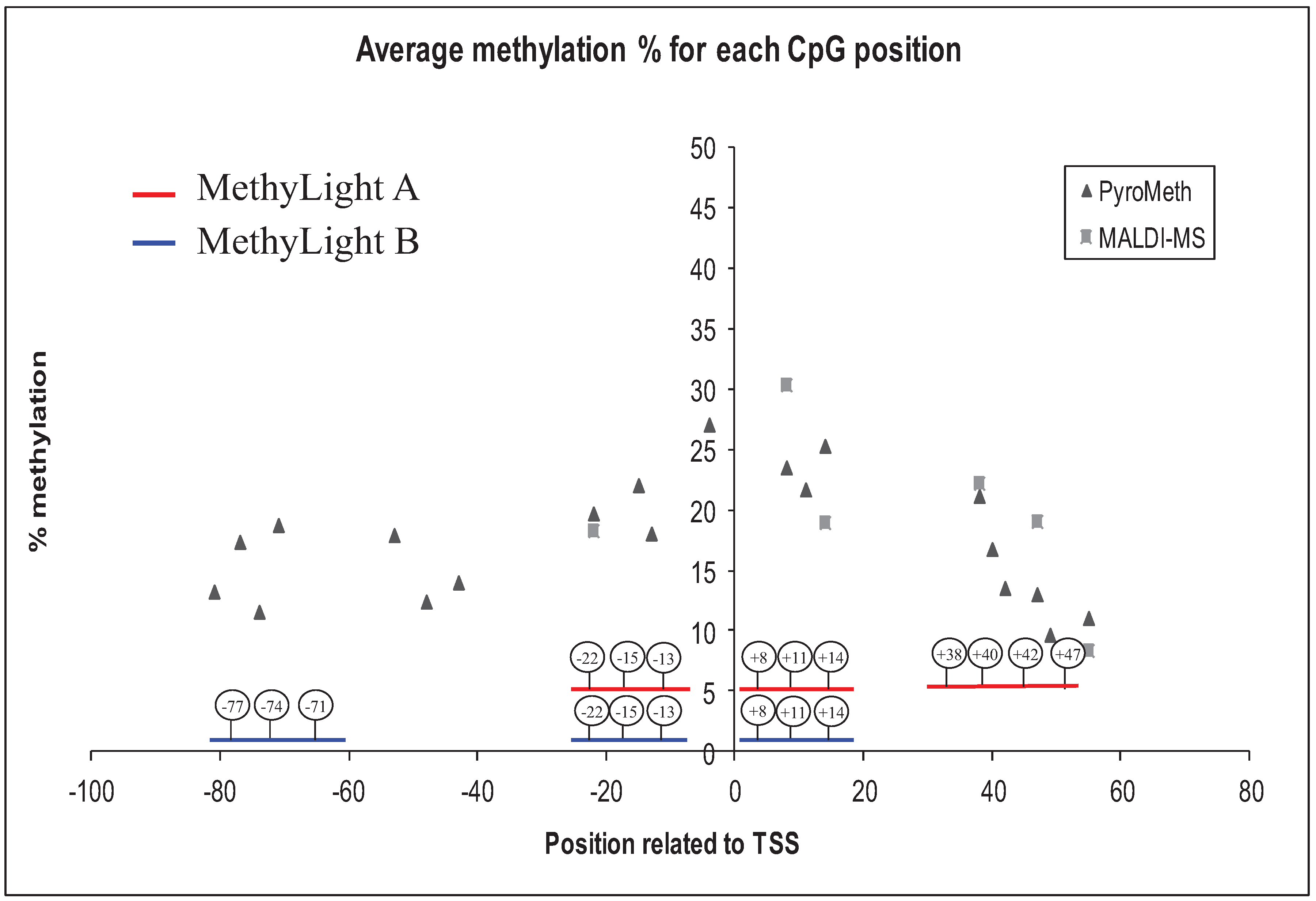
3.2. Analysis of the GSTP1 Promoter by Pyrosequencing and MALDI-MS-Based Epigenotyping
| Assay | ρ N | MethyLight A Average PMR | MethyLight B Average PMR | PyroMeth Average % of All Analyzed CpGs | PyroMeth Average % of MethyLight A CpGs | PyroMeth Average % of MethyLight B CpGs | PyroMeth Average % of MALDI-MS CpGs | MALDI-MS Average % of All Analyzed CpGs | MALDI-MS Average % of MethyLight A CpGs | MALDI-MS Average % of MethyLight B CpGs | MALDI-MS Average % of PyroMeth CpGs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MethyLight A average PMR | ρ | 1.000 | 0.071 | 0.493 | 0.454 | 0.480 | 0.445 | 0.507 | 0.503 | 0.482 | 0.507 |
| N | 82 | 82 | 68 | 66 | 66 | 66 | 75 | 75 | 75 | 75 | |
| MethyLight B average PMR | ρ | 0.701 | 1.000 | 0.693 | 0.646 | 0.695 | 0.634 | 0.666 | 0.675 | 0.664 | 0.666 |
| N | 82 | 85 | 69 | 67 | 67 | 67 | 76 | 76 | 76 | 76 | |
| PyroMeth average % of all analyzed CpGs | ρ | 0.493 | 0.693 | 1.000 | 0.982 | 0.991 | 0.983 | 0.895 | 0.891 | 0.897 | 0.895 |
| N | 68 | 69 | 72 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | |
| PyroMeth average % of MethyLight A CpGs | ρ | 0.454 | 0.646 | 0.982 | 1.000 | 0.967 | 0.993 | 0.886 | 0.880 | 0.893 | 0.886 |
| N | 66 | 67 | 70 | 70 | 68 | 70 | 68 | 68 | 68 | 68 | |
| PyroMeth average % of MethyLight B CpGs | ρ | 0.480 | 0.695 | 0.991 | 0.967 | 1.000 | 0.966 | 0.902 | 0.900 | 0.911 | 0.902 |
| N | 66 | 67 | 70 | 68 | 70 | 68 | 68 | 68 | 68 | 68 | |
| PyroMeth average % of MALDI-MS CpGs | ρ | 0.445 | 0.634 | 0.983 | 0.993 | 0.966 | 1.000 | 0.882 | 0.876 | 0.882 | 0.882 |
| N | 66 | 67 | 70 | 70 | 68 | 70 | 68 | 68 | 68 | 68 | |
| MALDI-MS average % of all analyzed CpGs | ρ | 0.507 | 0.666 | 0.895 | 0.886 | 0.902 | 0.882 | 1.000 | 0.997 | 0.984 | 1.000 |
| N | 75 | 76 | 70 | 68 | 68 | 68 | 79 | 79 | 79 | 79 | |
| MALDI-MS average % of MethyLight A CpGs | ρ | 0.503 | 0.375 | 0.891 | 0.880 | 0.900 | 0.876 | 0.997 | 1.000 | 0.987 | 0.997 |
| N | 75 | 76 | 70 | 68 | 68 | 68 | 79 | 79 | 79 | 79 | |
| MALDI-MS average % of MethyLight B CpGs | ρ | 0.482 | 0.664 | 0.897 | 0.893 | 0.911 | 0.882 | 0.984 | 0.987 | 1.000 | 0.984 |
| N | 75 | 76 | 70 | 68 | 68 | 68 | 79 | 79 | 79 | 79 | |
| MALDI-MS average % of PyroMeth CpGs | ρ | 0.507 | 0.666 | 0.895 | 0.886 | 0.902 | 0.882 | 1.000 | 0.997 | 0.984 | 1.000 |
| N | 75 | 76 | 70 | 68 | 68 | 68 | 79 | 79 | 79 | 79 |

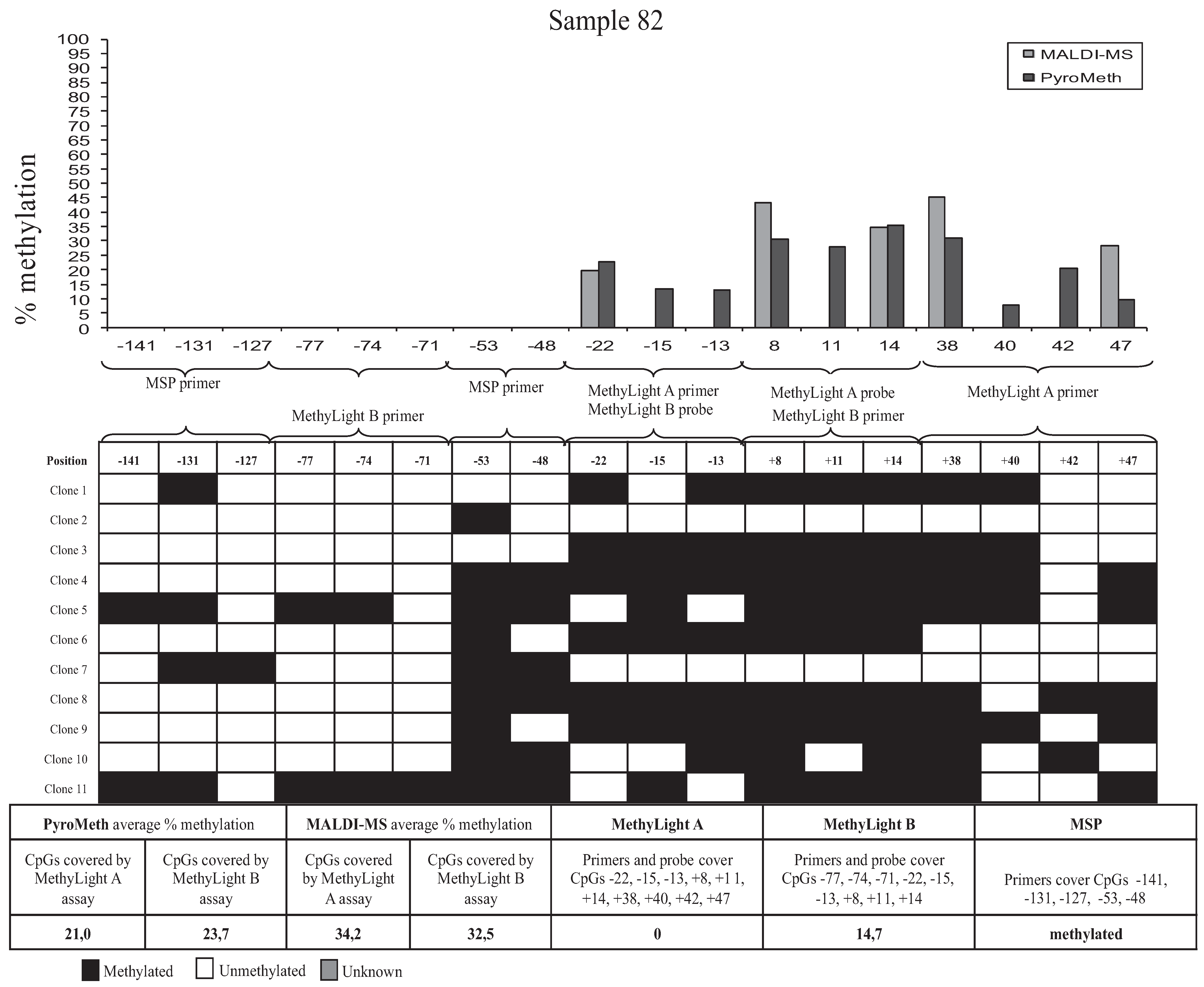
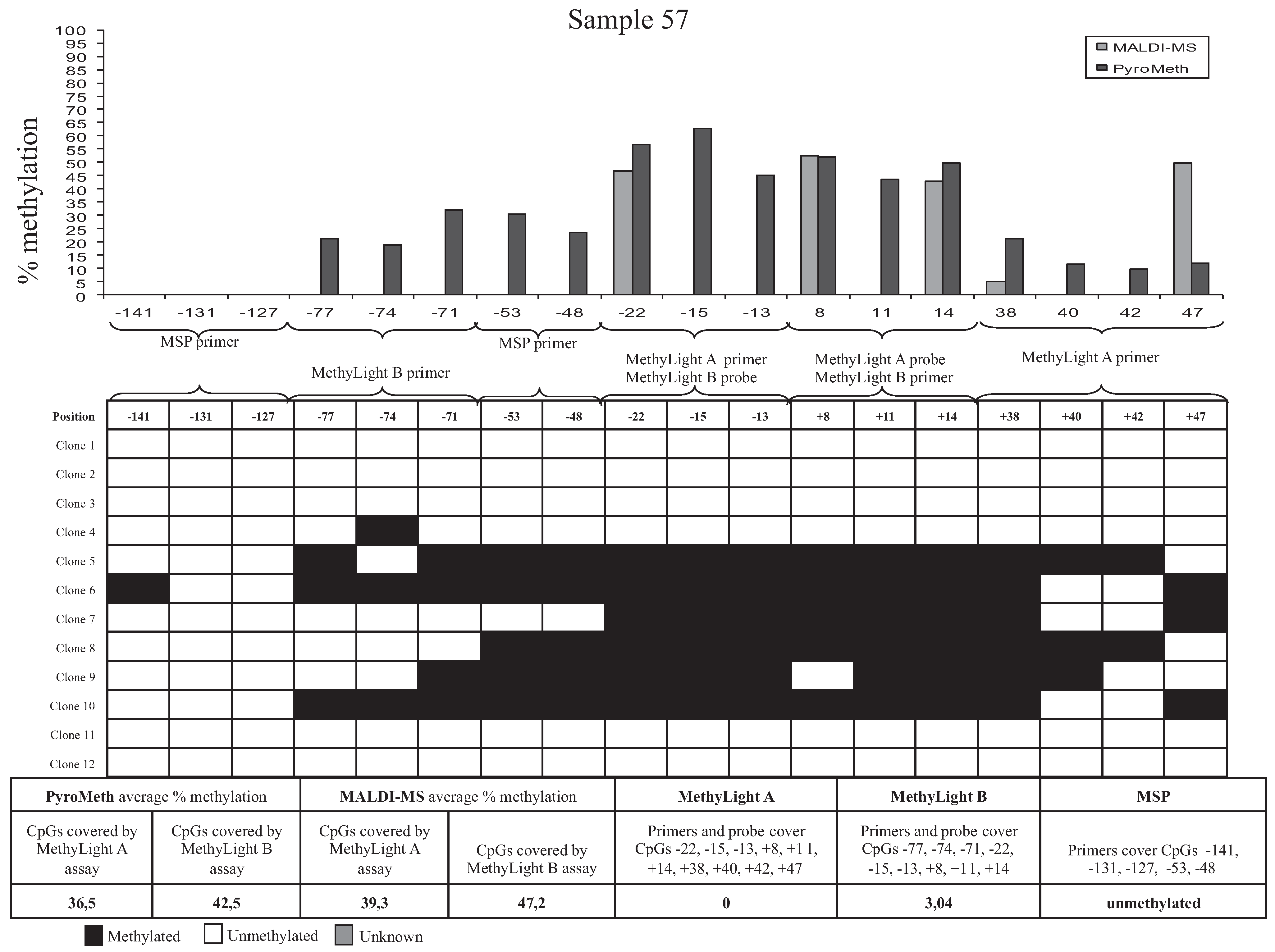
3.3. Bisulfite Sequencing of Single Molecules Reveals the Unexpected Presence of Heterogeneous DNA Methylation Patterns



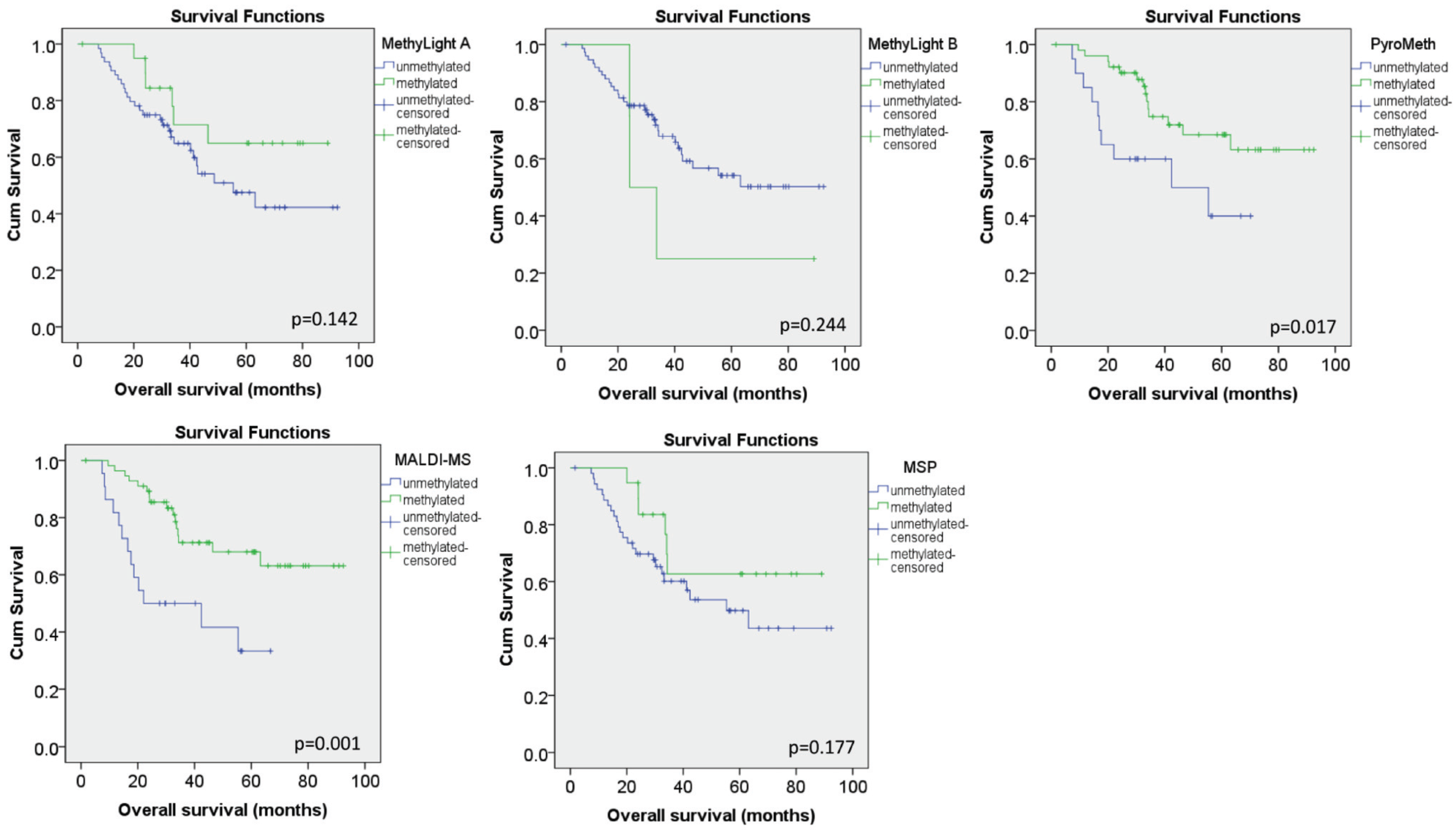
3.4. Methylation Call Rates Have a Significant Impact on the Correlation to Expression Levels and Clinical Parameters

3.5. Discussion
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tost, J. DNA methylation: An introduction to the biology and the disease-associated changes of a promising biomarker. Mol. Biotechnol. 2010, 44, 71–81. [Google Scholar] [CrossRef]
- Deng, D.; Liu, Z.; Du, Y. Epigenetic alterations as cancer diagnostic, prognostic, and predictive biomarkers. Adv. Genet. 2010, 71, 125–176. [Google Scholar]
- How Kit, A.; Nielsen, H.M.; Tost, J. DNA methylation based biomarkers: Practical considerations and applications. Biochimie 2012, 94, 2314–2337. [Google Scholar] [CrossRef] [PubMed]
- DeVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grutzmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.R. From discovery to the clinic: The novel DNA methylation biomarker mSEPT9 for the detection of colorectal cancer in blood. Epigenomics 2010, 2, 575–585. [Google Scholar] [CrossRef]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.G.; de Marzo, A.M.; Yegnasubramanian, S. Epigenetic alterations in human prostate cancers. Endocrinology 2009, 150, 3991–4002. [Google Scholar] [CrossRef] [PubMed]
- Chow, N.W.; Whang-Peng, J.; Kao-Shan, C.S.; Tam, M.F.; Lai, H.C.; Tu, C.P. Human glutathione S-transferases. The ha multigene family encodes products of different but overlapping substrate specificities. J. Biol. Chem. 1988, 263, 12797–12800. [Google Scholar] [PubMed]
- Schnekenburger, M.; Karius, T.; Diederich, M. Regulation of epigenetic traits of the glutathione S-transferase P1 gene: From detoxification toward cancer prevention and diagnosis. Front. Pharmacol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Takahashi, Y.; Takayama, T.; Kura, T.; Katahira, T.; Sakamaki, S.; Niitsu, Y. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996, 56, 3577–3582. [Google Scholar] [PubMed]
- Lee, W.P.; Lee, C.L.; Lin, H.C. Glutathione S-transferase and glutathione peroxidase are essential in the early stage of adriamycin resistance before P-glycoprotein overexpression in HOB1 lymphoma cells. Cancer Chemother. Pharmacol. 1996, 38, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Elwood, L.J.; Merino, M.; Masood, S.; Barnes, R.; Steinberg, S.M.; Lazarous, D.F.; Pierce, L.; d’Angelo, T.; Moscow, J.A.; et al. A pilot study of pi-class glutathione S-transferase expression in breast cancer: Correlation with estrogen receptor expression and prognosis in node-negative breast cancer. J. Clin. Oncol. 1993, 11, 49–58. [Google Scholar] [PubMed]
- Mulder, T.P.; Verspaget, H.W.; Sier, C.F.; Roelofs, H.M.; Ganesh, S.; Griffioen, G.; Peters, W.H. Glutathione S-transferase pi in colorectal tumors is predictive for overall survival. Cancer Res. 1995, 55, 2696–2702. [Google Scholar] [PubMed]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Arai, T.; Miyoshi, Y.; Kim, S.J.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of GSTP1 CpG islands hypermethylation with poor prognosis in human breast cancers. Breast Cancer Res. Treat. 2006, 100, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Dejeux, E.; Ronneberg, J.A.; Solvang, H.; Bukholm, I.; Geisler, S.; Aas, T.; Gut, I.G.; Borresen-Dale, A.L.; Lonning, P.E.; Kristensen, V.N.; et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Ronneberg, J.A.; Tost, J.; Solvang, H.K.; Alnaes, G.I.; Johansen, F.E.; Brendeford, E.M.; Yakhini, Z.; Gut, I.G.; Lonning, P.E.; Borresen-Dale, A.L.; et al. Gstp1 promoter haplotypes affect DNA methylation levels and promoter activity in breast carcinomas. Cancer Res. 2008, 68, 5562–5571. [Google Scholar] [CrossRef] [PubMed]
- Muggerud, A.A.; Ronneberg, J.A.; Warnberg, F.; Botling, J.; Busato, F.; Jovanovic, J.; Solvang, H.; Bukholm, I.; Borresen-Dale, A.L.; Kristensen, V.N.; et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Klajic, J.; Fleischer, T.; Dejeux, E.; Edvardsen, H.; Warnberg, F.; Bukholm, I.; Lonning, P.E.; Solvang, H.; Borresen-Dale, A.L.; Tost, J.; et al. Quantitative DNA methylation analyses reveal stage dependent DNA methylation and association to clinico-pathological factors in breast tumors. BMC Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. Methylight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, e32. [Google Scholar] [CrossRef] [PubMed]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Tost, J.; Schatz, P.; Schuster, M.; Berlin, K.; Gut, I.G. Analysis and accurate quantification of CpG methylation by MALDI mass spectrometry. Nucleic Acids Res. 2003, 31, e50. [Google Scholar] [CrossRef] [PubMed]
- Klajic, J.; Busato, F.; Edvardsen, H.; Touleimat, N.; Fleischer, T.; Bukholm, I.; Borresen-Dale, A.L.; Lonning, P.E.; Tost, J.; Kristensen, V.N. DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors. Clin. Cancer Res. 2014, 20, 6357–6366. [Google Scholar] [CrossRef] [PubMed]
- Cairns, P.; Esteller, M.; Herman, J.G.; Schoenberg, M.; Jeronimo, C.; Sanchez-Cespedes, M.; Chow, N.H.; Grasso, M.; Wu, L.; Westra, W.B.; et al. Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin. Cancer Res. 2001, 7, 2727–2730. [Google Scholar] [PubMed]
- Eads, C.A.; Lord, R.V.; Wickramasinghe, K.; Long, T.I.; Kurumboor, S.K.; Bernstein, L.; Peters, J.H.; DeMeester, S.R.; DeMeester, T.R.; Skinner, K.A.; et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001, 61, 3410–3418. [Google Scholar] [PubMed]
- Weisenberger, D.J.; Campan, M.; Long, T.I.; Kim, M.; Woods, C.; Fiala, E.; Ehrlich, M.; Laird, P.W. Analysis of repetitive element DNA methylation by methylight. Nucleic Acids Res. 2005, 33, 6823–6836. [Google Scholar] [CrossRef] [PubMed]
- Ronneberg, J.A.; Fleischer, T.; Solvang, H.K.; Nordgard, S.H.; Edvardsen, H.; Potapenko, I.; Nebdal, D.; Daviaud, C.; Gut, I.; Bukholm, I.; et al. Methylation profiling with a panel of cancer related genes: Association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol. Oncol. 2011, 5, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, S.E.; Distler, J.; Goodman, N.S.; Mooney, S.H.; Kluth, A.; Olek, A.; Schwope, I.; Tetzner, R.; Ziebarth, H.; Berlin, K. A real-time pcr assay for DNA-methylation using methylation-specific blockers. Nucleic Acids Res. 2004, 32, e10. [Google Scholar] [CrossRef] [PubMed]
- Zeschnigk, M.; Bohringer, S.; Price, E.A.; Onadim, Z.; Masshofer, L.; Lohmann, D.R. A novel real-time pcr assay for quantitative analysis of methylated alleles (QAMA): Analysis of the retinoblastoma locus. Nucleic Acids Res. 2004, 32, e125. [Google Scholar] [CrossRef] [PubMed]
- Vinarskaja, A.; Goering, W.; Ingenwerth, M.; Schulz, W.A. Id4 is frequently downregulated and partially hypermethylated in prostate cancer. World J. Urol. 2012, 30, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Everhard, S.; Tost, J.; El Abdalaoui, H.; Criniere, E.; Busato, F.; Marie, Y.; Gut, I.G.; Sanson, M.; Mokhtari, K.; Laigle-Donadey, F.; et al. Identification of regions correlating mgmt promoter methylation and gene expression in glioblastomas. Neuro Oncol. 2009, 11, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Baylin, S.B.; Herman, J.G. P15(ink4b) cpg island methylation in primary acute leukemia is heterogeneous and suggests density as a critical factor for transcriptional silencing. Blood 1999, 94, 2445–2451. [Google Scholar] [PubMed]
- Parkinson, J.F.; Wheeler, H.R.; Clarkson, A.; McKenzie, C.A.; Biggs, M.T.; Little, N.S.; Cook, R.J.; Messina, M.; Robinson, B.G.; McDonald, K.L. Variation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation in serial samples in glioblastoma. J. Neurooncol. 2008, 87, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Peng, E.; Gum, J.; Terdiman, J.; Sleisenger, M.; Kim, Y.S. Methylation of HMLH1 promoter correlates with the gene silencing with a region-specific manner in colorectal cancer. Br. J. Cancer 2002, 86, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Schuffler, P.; Mikeska, T.; Waha, A.; Lengauer, T.; Bock, C. Methmarker: User-friendly design and optimization of gene-specific DNA methylation assays. Genome Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Van Vlodrop, I.J.; Niessen, H.E.; Derks, S.; Baldewijns, M.M.; van Criekinge, W.; Herman, J.G.; van Engeland, M. Analysis of promoter CpG island hypermethylation in cancer: Location, location, location! Clin. Cancer Res. 2011, 17, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barriere, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S. GSTP1 promoter hypermethylation is an early event in breast carcinogenesis. Virchows Arch. 2007, 450, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Shen, J.; Gammon, M.D.; Zhang, Y.J.; Wang, Q.; Gonzalez, K.; Xu, X.; Bradshaw, P.T.; Teitelbaum, S.L.; Garbowski, G.; et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res. Treat. 2012, 131, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Parrella, P.; Poeta, M.L.; Gallo, A.P.; Prencipe, M.; Scintu, M.; Apicella, A.; Rossiello, R.; Liguoro, G.; Seripa, D.; Gravina, C.; et al. Nonrandom distribution of aberrant promoter methylation of cancer-related genes in sporadic breast tumors. Clin. Cancer Res. 2004, 10, 5349–5354. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Kohler, C.; Haghighi, M.M.; Fan, A.X.; Holzgreve, W.; Zhong, X.Y. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene 2009, 28, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, L.; Bedeir, A.; Ringquist, S.; Styche, A.; Bhargava, R.; Trucco, G. Quantification of CpG island methylation in progressive breast lesions from normal to invasive carcinoma. Cancer Lett. 2007, 257, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.D.; Cairns, P.; Shore, R.E.; Klein, C.B.; Wirgin, I.; Afanasyeva, Y.; Zeleniuch-Jacquotte, A. DNA methylation in pre-diagnostic serum samples of breast cancer cases: Results of a nested case-control study. Cancer Epidemiol. 2010, 34, 717–723. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnaes, G.I.G.; Ronneberg, J.A.; Kristensen, V.N.; Tost, J. Heterogeneous DNA Methylation Patterns in the GSTP1 Promoter Lead to Discordant Results between Assay Technologies and Impede Its Implementation as Epigenetic Biomarkers in Breast Cancer. Genes 2015, 6, 878-900. https://doi.org/10.3390/genes6030878
Alnaes GIG, Ronneberg JA, Kristensen VN, Tost J. Heterogeneous DNA Methylation Patterns in the GSTP1 Promoter Lead to Discordant Results between Assay Technologies and Impede Its Implementation as Epigenetic Biomarkers in Breast Cancer. Genes. 2015; 6(3):878-900. https://doi.org/10.3390/genes6030878
Chicago/Turabian StyleAlnaes, Grethe I. Grenaker, Jo Anders Ronneberg, Vessela N. Kristensen, and Jörg Tost. 2015. "Heterogeneous DNA Methylation Patterns in the GSTP1 Promoter Lead to Discordant Results between Assay Technologies and Impede Its Implementation as Epigenetic Biomarkers in Breast Cancer" Genes 6, no. 3: 878-900. https://doi.org/10.3390/genes6030878