
Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Mutation Detection of EDA, EDAR, EDARADD, and WNT10A
2.3. Splicing Analysis of EDAR
2.4. Bioinformatics Study
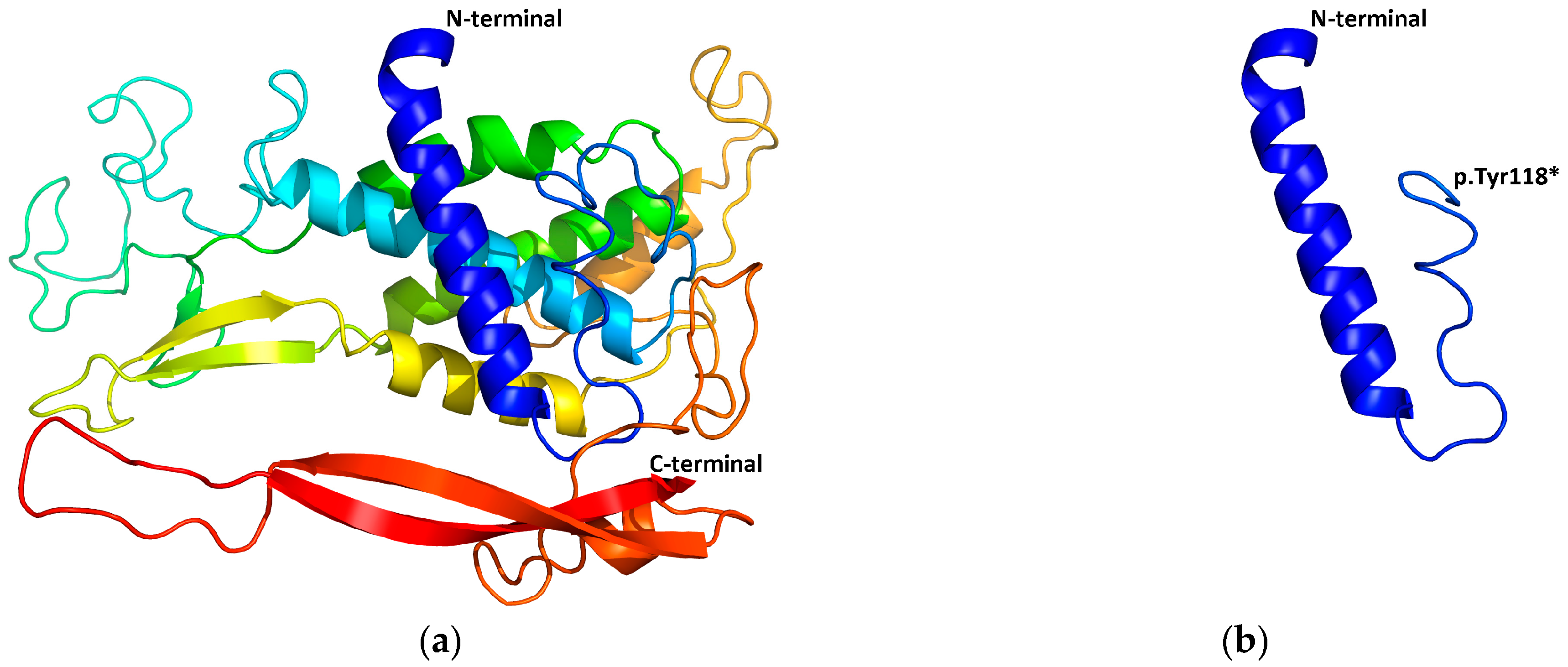
2.5. Structural Modeling
3. Results
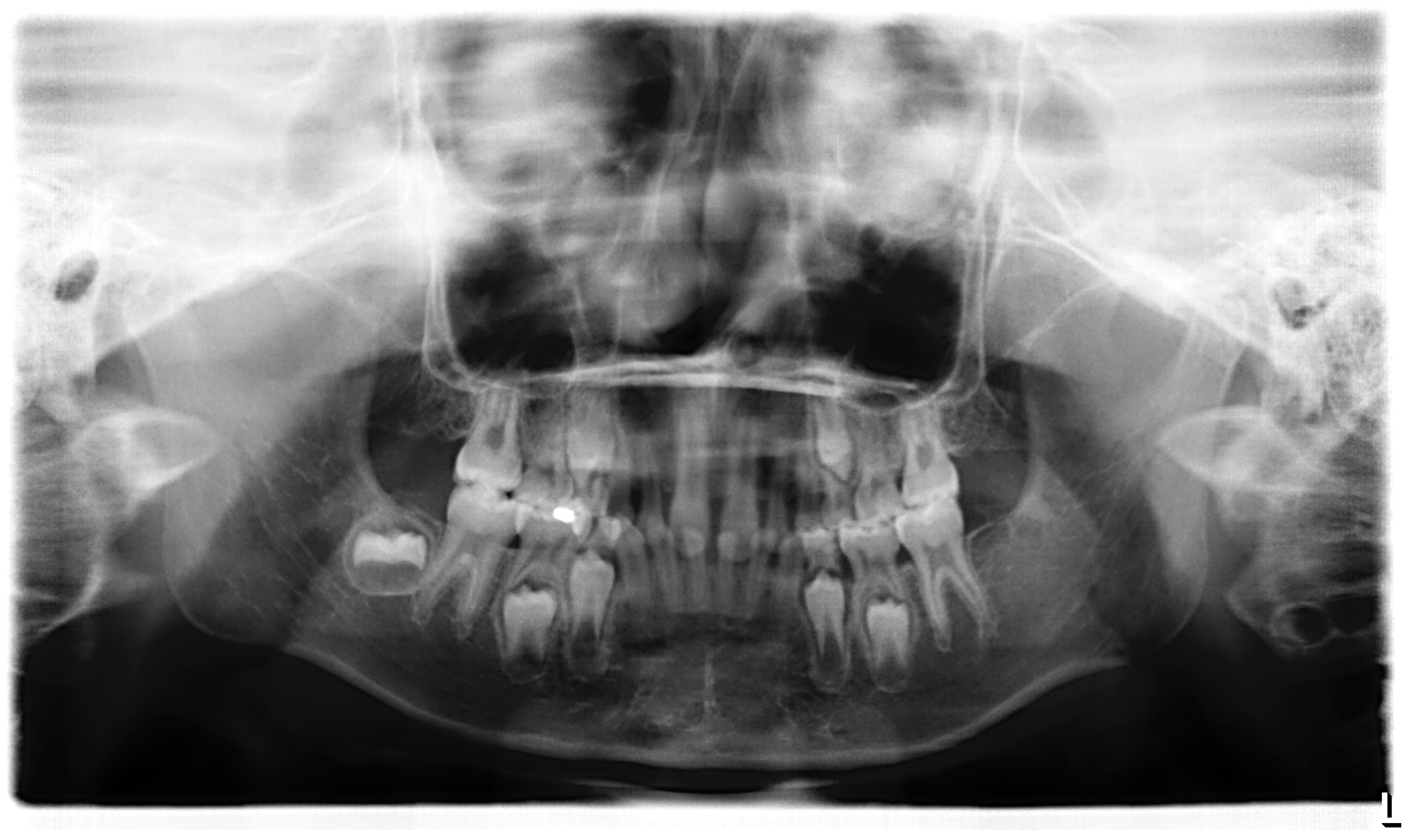
3.1. Clinical Report
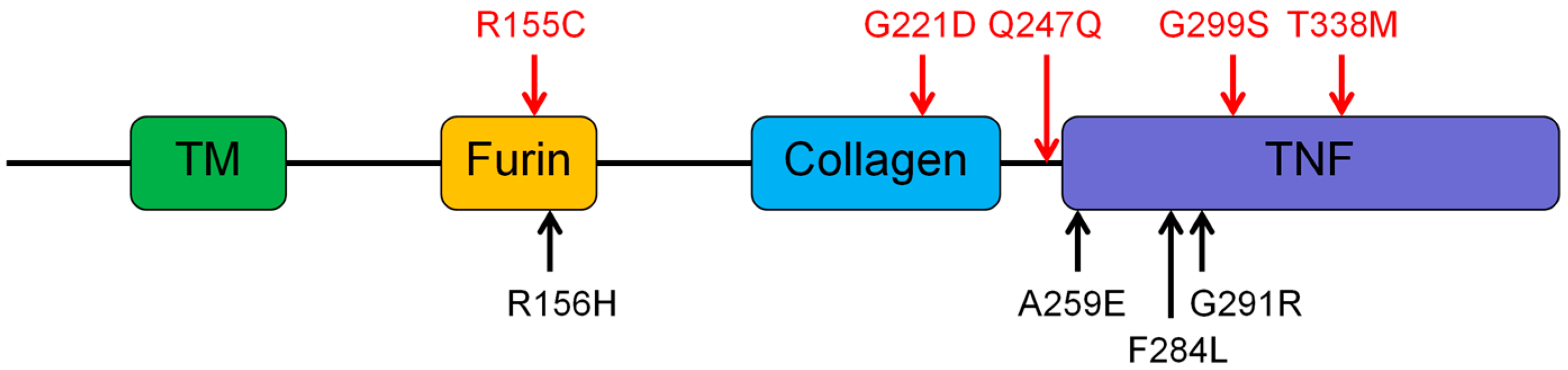
3.2. Genetic Findings of the EDA, EDAR, EDARADD, and WNT10A
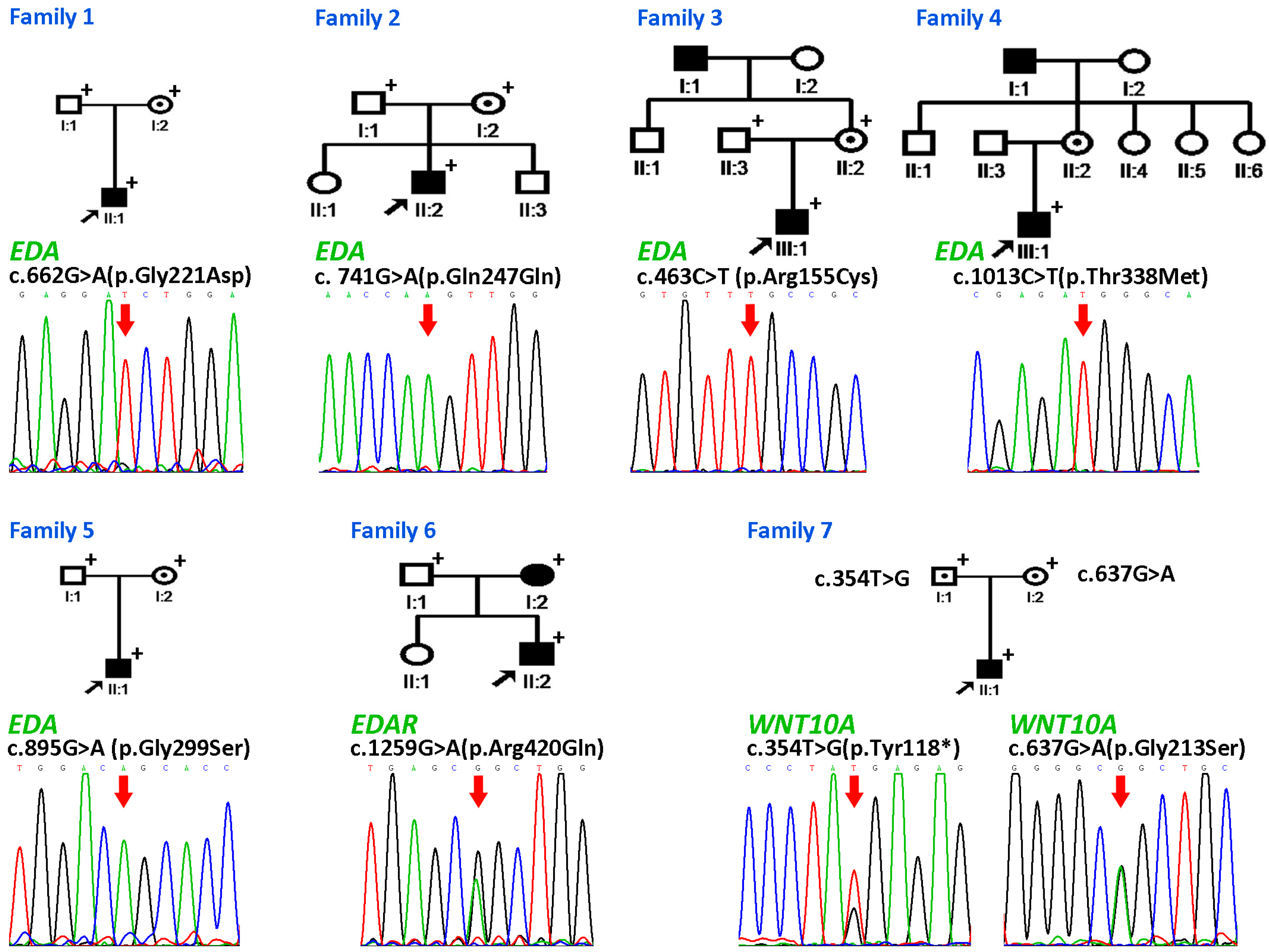
3.2.1. Family 1
3.2.2. Families 2–6
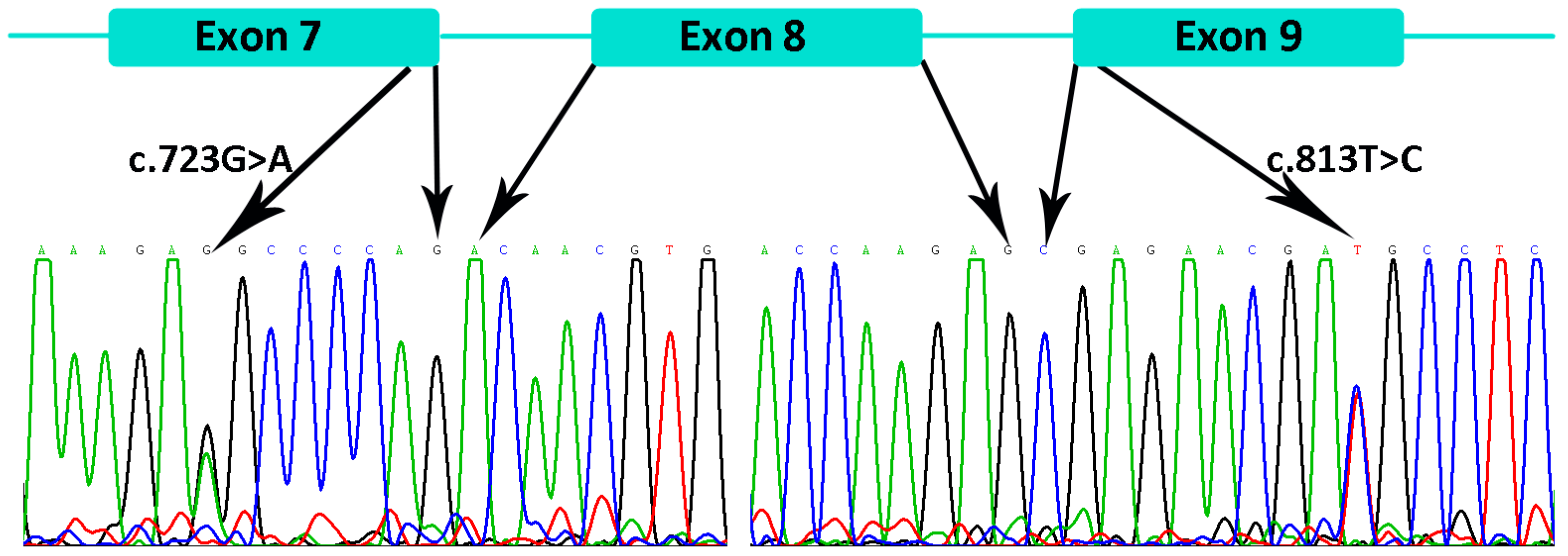
3.2.3. Family 7
3.2.4. Detection of the EDAR Variant rs3827760 (c.1109T>C, p.Val370Ala)
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Visinoni, A.F.; Lisboa-Costa, T.; Pagnan, N.A.; Chautard-Freire-Maia, E.A. Ectodermal dysplasias: clinical and molecular review. Am. J. Med. Genet. A 2009, 149A, 1980–2002. [Google Scholar] [CrossRef] [PubMed]
- Freire-Maia, N. Ectodermal dysplasias. Hum. Hered. 1971, 21, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Pagnan, N.A.; Visinoni, A.F. Update on ectodermal dysplasias clinical classification. Am. J. Med. Genet. A 2014, 164A, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, M.L. Molecular aspects of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 2009, 149A, 2031–2036. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Lu, H.; Xiao, X.; Zhou, L.; Lu, J.; Zhu, L.; Yu, D.; Zhao, W. Novel EDA mutation in X-linked hypohidrotic ectodermal dysplasia and genotype-phenotype correlation. Oral Dis. 2015, 21, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.; Prashanth, S. Ectodermal dysplasia: a genetic review. Int. J. Clin. Pediatr. Dent. 2012, 5, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Ferguson, B.M.; Headon, D.J.; Street, S.L.; Overbeek, P.A.; Zonana, J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 1999, 22, 366–369. [Google Scholar] [PubMed]
- Wisniewski, S.A.; Trzeciak, W.H. A new mutation resulting in the truncation of the TRAF6-interacting domain of XEDAR: a possible novel cause of hypohidrotic ectodermal dysplasia. J. Med. Genet. 2012, 49, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, S.A.; Trzeciak, W.H. A rare heterozygous TRAF6 variant is associated with hypohidrotic ectodermal dysplasia. Br. J. Dermatol. 2012, 166, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Li, R.; Hu, Y.; Hu, B.; Zhao, Q.; Liu, H.; Yuan, P.; Wang, Y. A novel mutation and a known mutation in the CLCN7 gene associated with relatively stable infantile malignant osteopetrosis in a Chinese patient. Gene 2016, 576, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zeng, B.; Yu, D.; Jing, X.; Hu, B.; Zhao, W.; Wang, Y. Complex dental anomalies in a belatedly diagnosed cleidocranial dysplasia patient. Imaging Sci. Dent. 2015, 45, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. Proceeding of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB '12), Orlando, FL, USA, 8–10 October 2012, 414–417.
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Gambardella, S.; Bulli, C.; Rinaldi, F.; Di Marino, D.; Falconi, M.; Bramanti, P.; Desideri, A.; Novelli, G. Screening of EDA1 gene in X-linked anhidrotic ectodermal dysplasia using DHPLC: identification of 14 novel mutations in Italian patients. Genet. Test. 2008, 12, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Cheng, J.; Zhou, B.; Tong, X.; Dong, X.; Wang, Z.; Hu, Q.; Chen, M.; Hua, Z.C. Non-syndromic tooth agenesis in two Chinese families associated with novel missense mutations in the TNF domain of EDA (ectodysplasin A). PLoS ONE 2008, 3, e2396. [Google Scholar] [CrossRef] [PubMed]
- Plaisancie, J.; Bailleul-Forestier, I.; Gaston, V.; Vaysse, F.; Lacombe, D.; Holder-Espinasse, M.; Abramowicz, M.; Coubes, C.; Plessis, G.; Faivre, L.; et al. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am. J. Med. Genet. A 2013, 161A, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Megarbane, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Megarbane, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Schopf, E.; Schulz, H.J.; Passarge, E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects Orig. Artic. Ser. 1971, 7, 219–221. [Google Scholar] [PubMed]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [PubMed]
- Vink, C.P.; Ockeloen, C.W.; Ten, K.S.; Koolen, D.A.; Ploos, V.A.J.; Kuijpers-Jagtman, A.M.; van Heumen, C.C.; Kleefstra, T.; Carels, C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014, 22, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Wedgeworth, E.K.; Nagy, N.; White, J.M.; Pembroke, A.C.; McGrath, J.A. Intra-familial variability of ectodermal defects associated with WNT10A mutations. Acta Derm. Venereol. 2011, 91, 346–347. [Google Scholar] [PubMed]
- Mou, C.; Thomason, H.A.; Willan, P.M.; Clowes, C.; Harris, W.E.; Drew, C.F.; Dixon, J.; Dixon, M.J.; Headon, D.J. Enhanced ectodysplasin-A receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Hum. Mutat. 2008, 29, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Gaczkowska, A.; Abdalla, E.M.; Dowidar, K.M.; Elhady, G.M.; Jagodzinski, P.P.; Mostowska, A. De novo EDA mutations: Variable expression in two Egyptian families. Arch. Oral Biol. 2016, 68, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.K.; Sankar, V.H.; Bashyam, M.D. A novel large deletion that encompasses EDA and the downstream gene AWAT2 causes X-linked hypohidrotic/anhidrotic ectodermal dysplasia. J. Dermatol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaard, M.J.; Creton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; Ploos, V.A.H. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Biedziak, B.; Zadurska, M.; Dunin-Wilczynska, I.; Lianeri, M.; Jagodzinski, P.P. Nucleotide variants of genes encoding components of the Wnt signalling pathway and the risk of non-syndromic tooth agenesis. Clin. Genet. 2013, 84, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 2014, 164A, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Bonds, J.; Xiang, L.; Vieira, A.R.; Seymen, F.; Klein, O.; D'Souza, R.N. The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am. J. Med. Genet. A 2014, 164A, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O'Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xu, R.; Huang, F.; Wang, B.; Tao, Y.; Jiang, Z.; Li, H.; Yao, J.; Xu, P.; Wu, X.; et al. A novel missense mutation in collagenous domain of EDA gene in a Chinese family with X-linked hypohidrotic ectodermal dysplasia. J. Genet. 2015, 94, 115–119. [Google Scholar] [CrossRef] [PubMed]
- HGMD public version. Available online: http://www.hgmd.cf.ac.uk/ac/all.php (accessed on 3 August 2016).
- Vincent, M.C.; Biancalana, V.; Ginisty, D.; Mandel, J.L.; Calvas, P. Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2001, 9, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Van der Hout, A.H.; Oudesluijs, G.G.; Venema, A.; Verheij, J.B.; Mol, B.G.; Rump, P.; Brunner, H.G.; Vos, Y.J.; van Essen, A.J. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008, 16, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements. J. Hum. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Kinkorova, J. [Horizon 2020, new EU Framework programme for research and innovation, 2014-2020]. Cas. Lek. Cesk. 2014, 153, 254–256. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
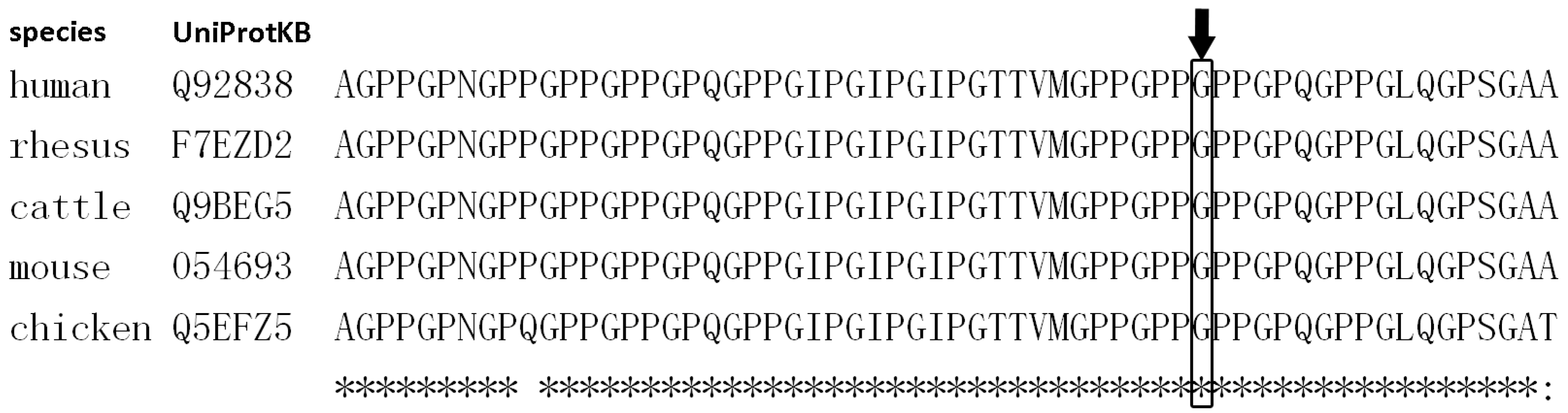{kind=link}
{kind=link}
{kind=link}
| Family | Patient | Age and Gender | Gene Involved | Nucleotide Change | Amino Acid Change | Domain | Mode of Inheritance | n Missing Primary Teeth | n Missing Permanent Teeth † |
|---|---|---|---|---|---|---|---|---|---|
| 1 | II:1 | 4y, M | EDA | c.662G>A | p.Gly221Asp | Collagen | X-linked | 18 | ND |
| 2 | II:2 | 8y, M | EDA | c. 741G>A | p.Gln247Gln | X-linked | 18 | 26 | |
| 3 | III:1 | 6y, M | EDA | c.463C>T | p.Arg155Cys | Furin | X-linked | 5 | 19 |
| 4 | III:1 | 11y, M | EDA | c.1013C>T | p.Thr338Met | TNF | X-linked | ND | 14 |
| 5 | II:1 | 7y, M | EDA | c.895G>A | p.Gly299Ser | TNF | X-linked | 20 | ND |
| 6 | II:2 | 8y, M | EDAR | c.1259G>A | p.Arg420Gln | DD | AD | ND | 7 |
| I:2 | 28y, F | EDAR | c.1259G>A | p.Arg420Gln | DD | AD | ND | 4 | |
| 7 | II:1 | 11y, M | WNT10A | c.354T>G | p.Tyr118* | AR | 0 | 15 | |
| WNT10A | c.637G>A | p.Gly213Ser |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, B.; Xiao, X.; Li, S.; Lu, H.; Lu, J.; Zhu, L.; Yu, D.; Zhao, W. Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients. Genes 2016, 7, 65. https://doi.org/10.3390/genes7090065
Zeng B, Xiao X, Li S, Lu H, Lu J, Zhu L, Yu D, Zhao W. Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients. Genes. 2016; 7(9):65. https://doi.org/10.3390/genes7090065
Chicago/Turabian StyleZeng, Binghui, Xue Xiao, Sijie Li, Hui Lu, Jiaxuan Lu, Ling Zhu, Dongsheng Yu, and Wei Zhao. 2016. "Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients" Genes 7, no. 9: 65. https://doi.org/10.3390/genes7090065
APA StyleZeng, B., Xiao, X., Li, S., Lu, H., Lu, J., Zhu, L., Yu, D., & Zhao, W. (2016). Eight Mutations of Three Genes (EDA, EDAR, and WNT10A) Identified in Seven Hypohidrotic Ectodermal Dysplasia Patients. Genes, 7(9), 65. https://doi.org/10.3390/genes7090065