
Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Identification and Recruitment
2.2. DNA Isolation and Next Generation Sequencing
2.3. Polymerase Chain Reaction and Sanger Sequencing
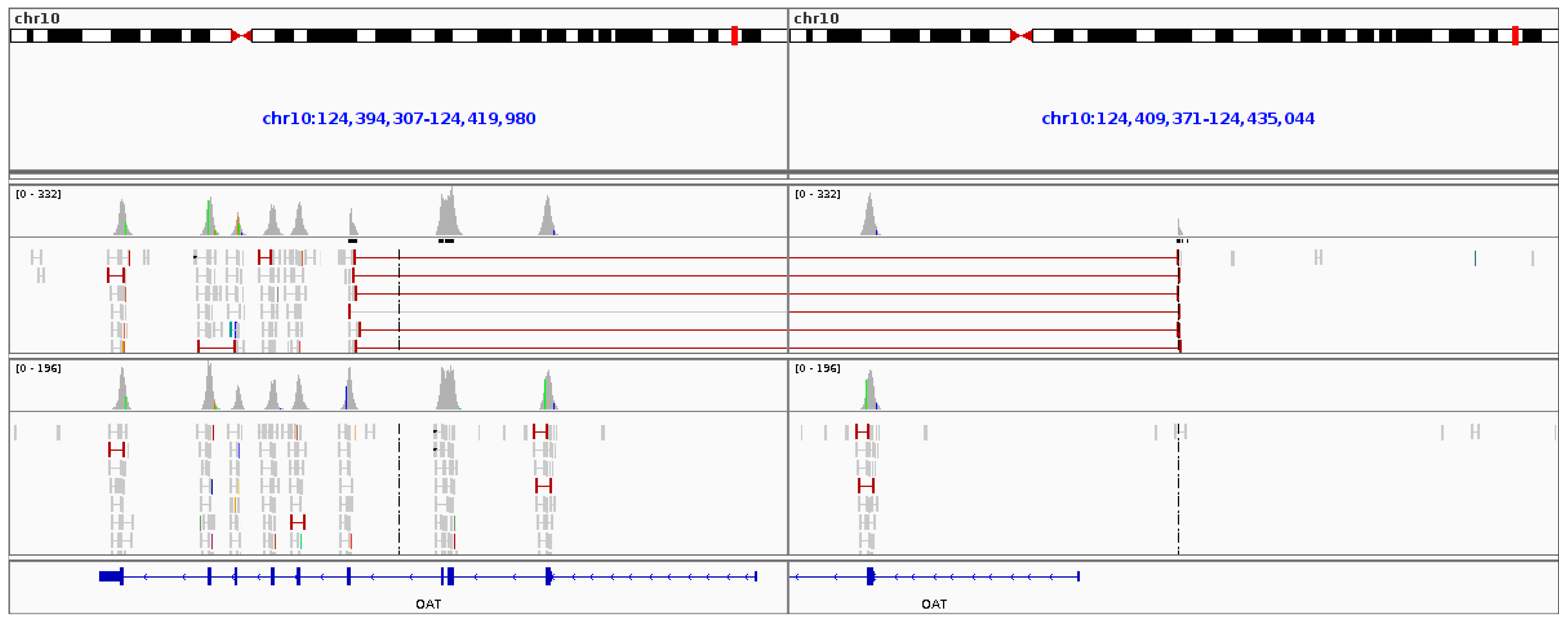
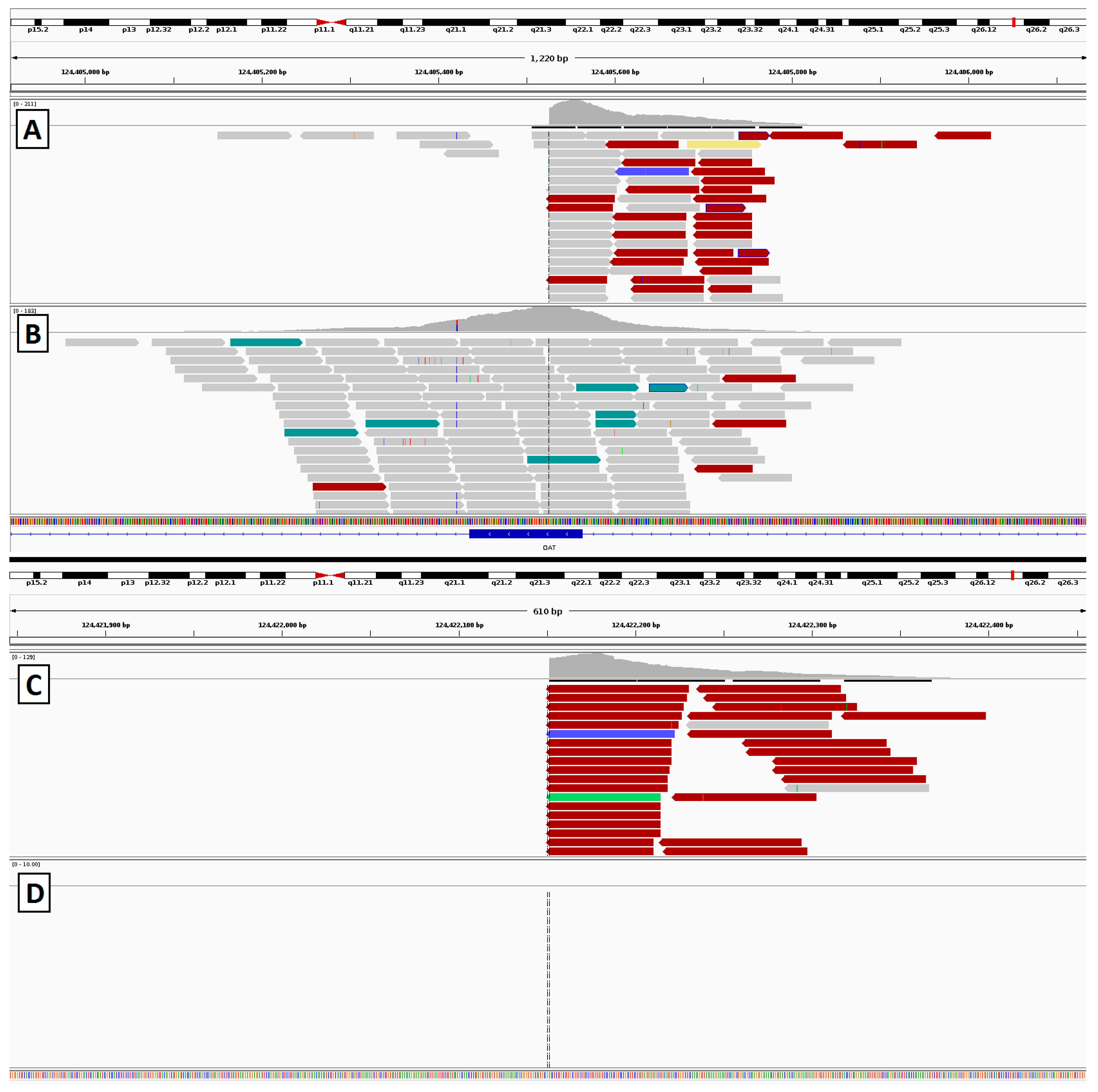
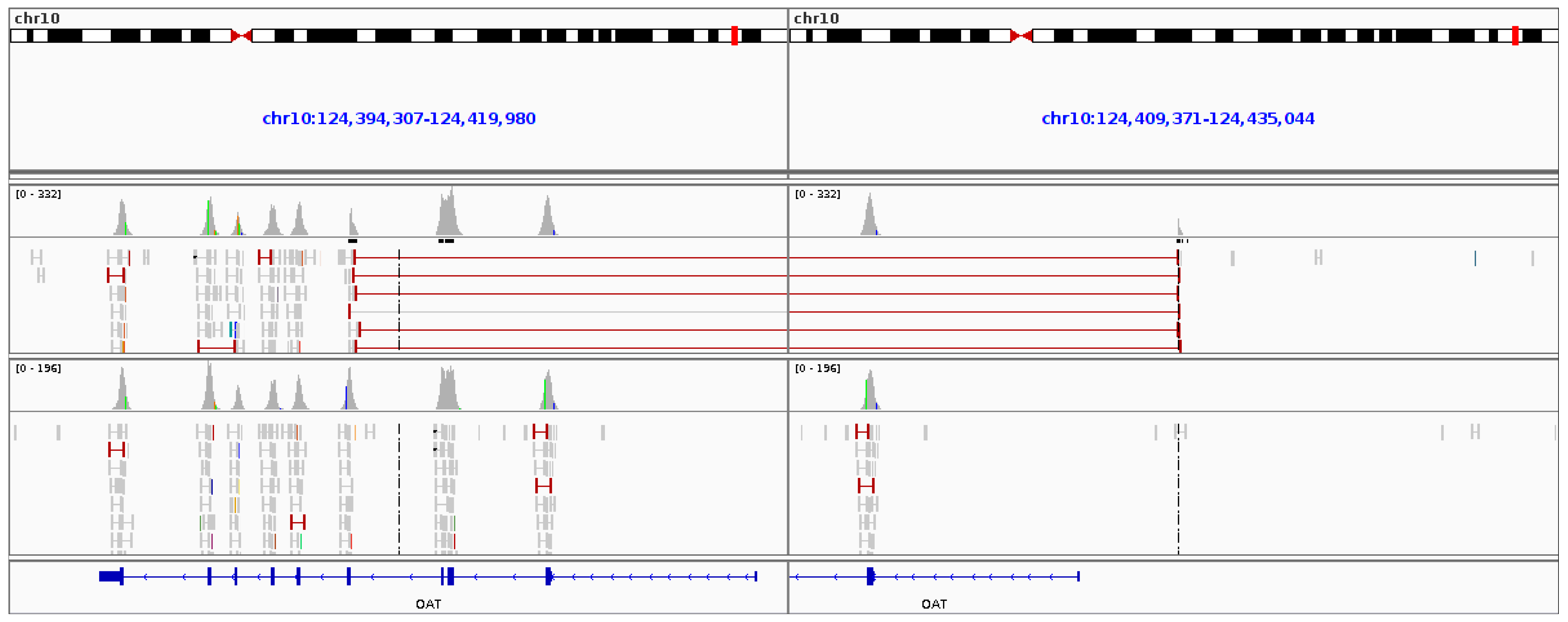
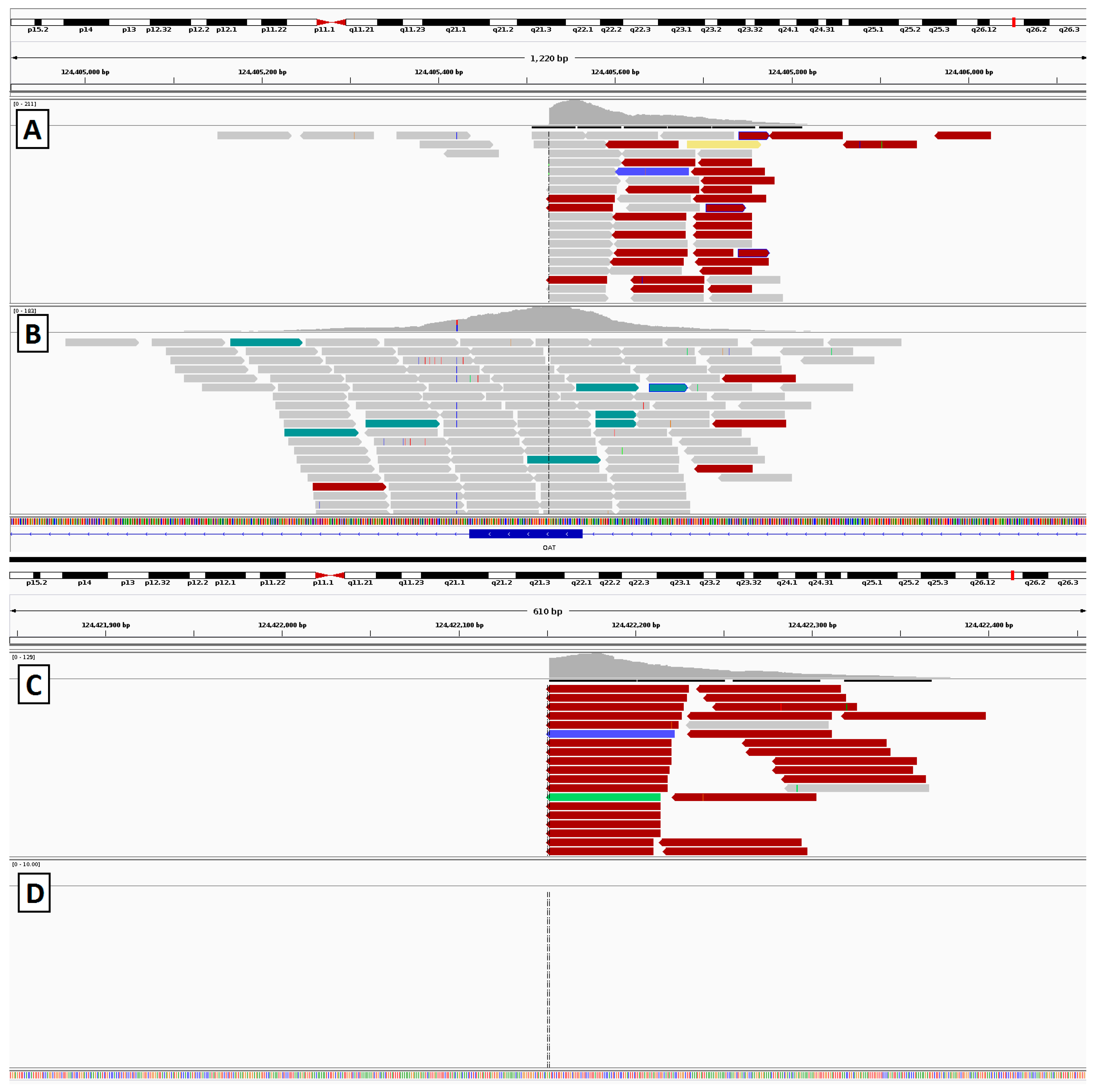
2.4. Gyrate Atrophy Inversion Confirmation
2.5. Data Analysis
2.6. Ethical Approval
3. Results
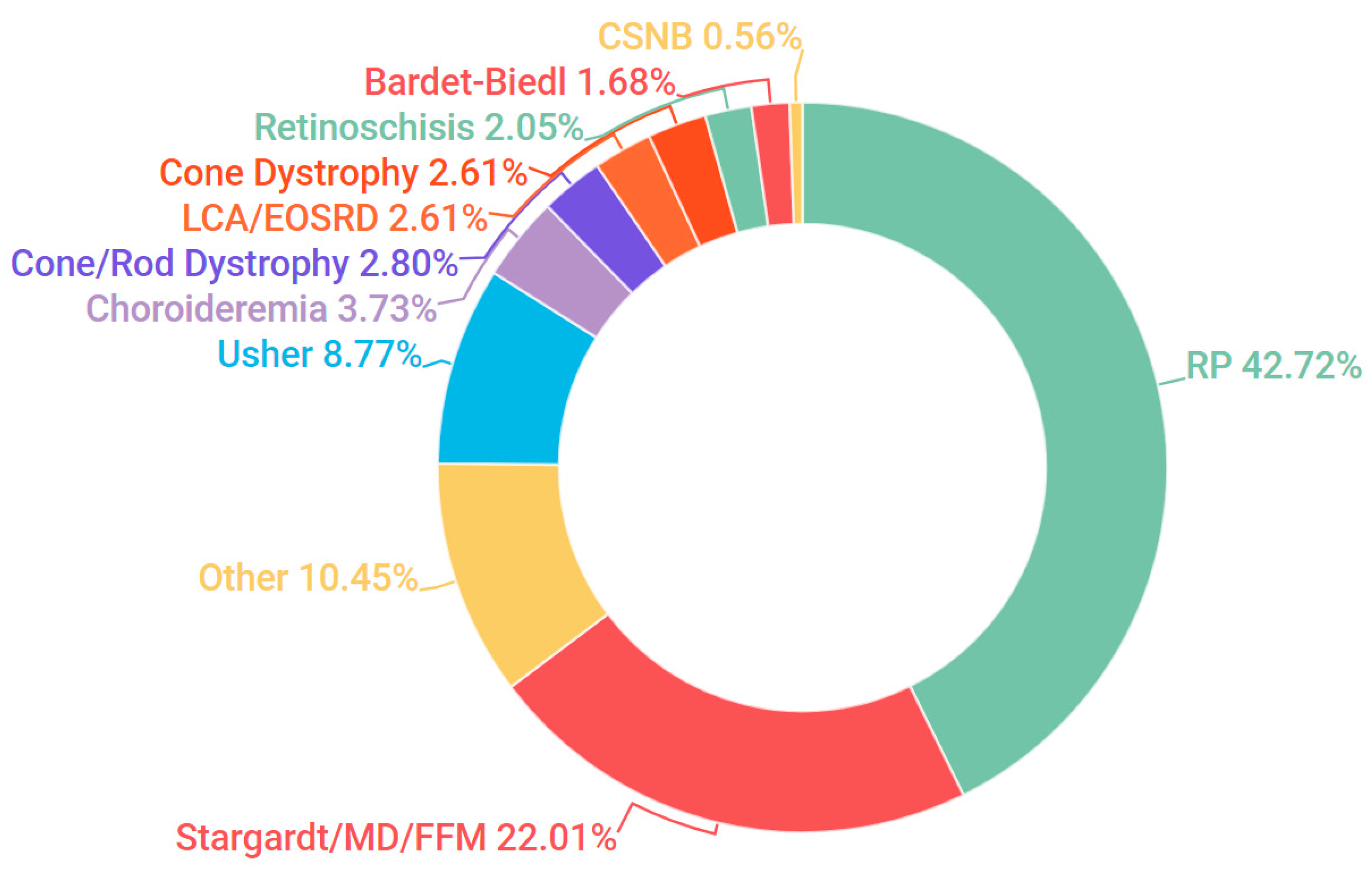
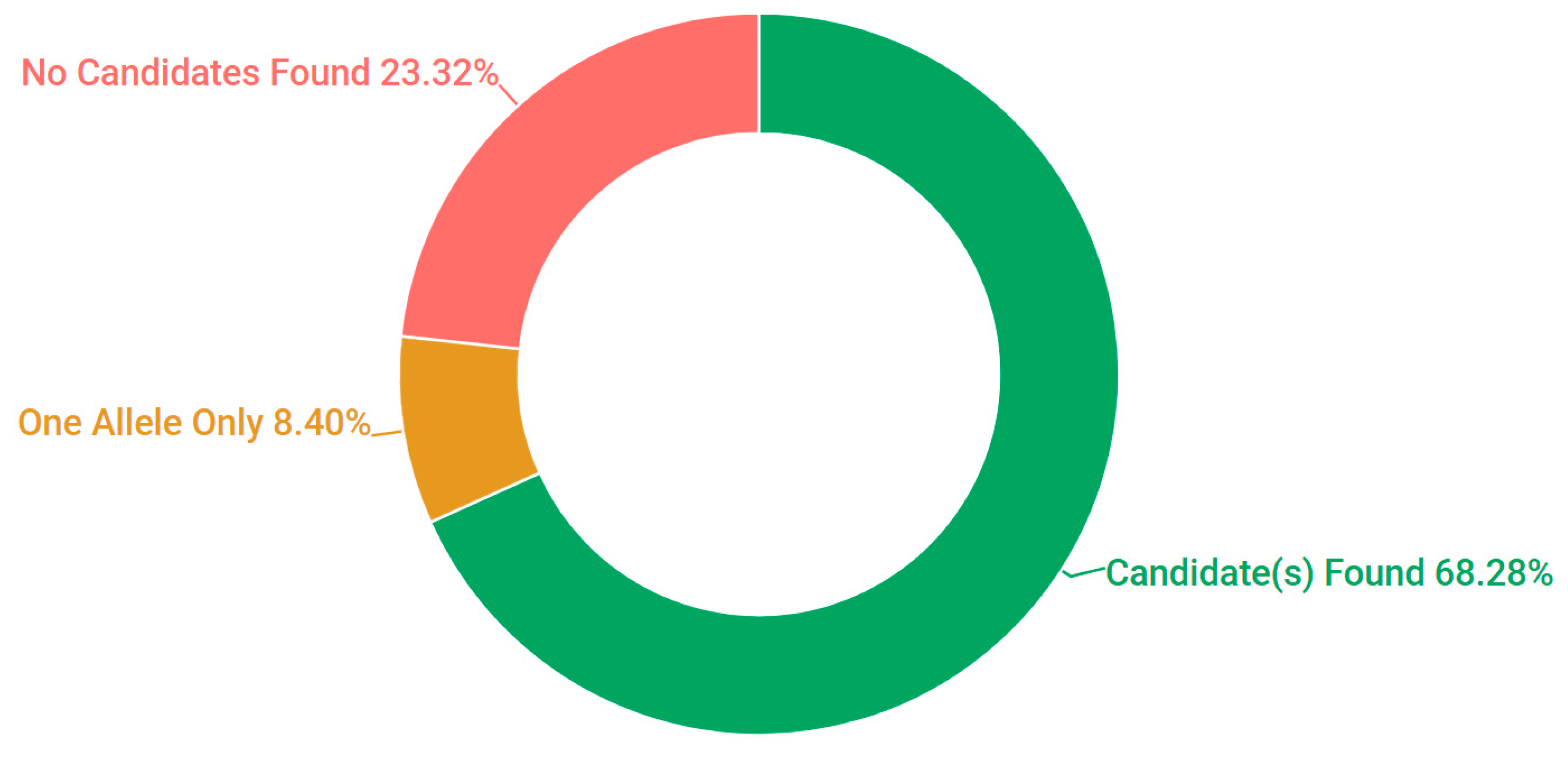
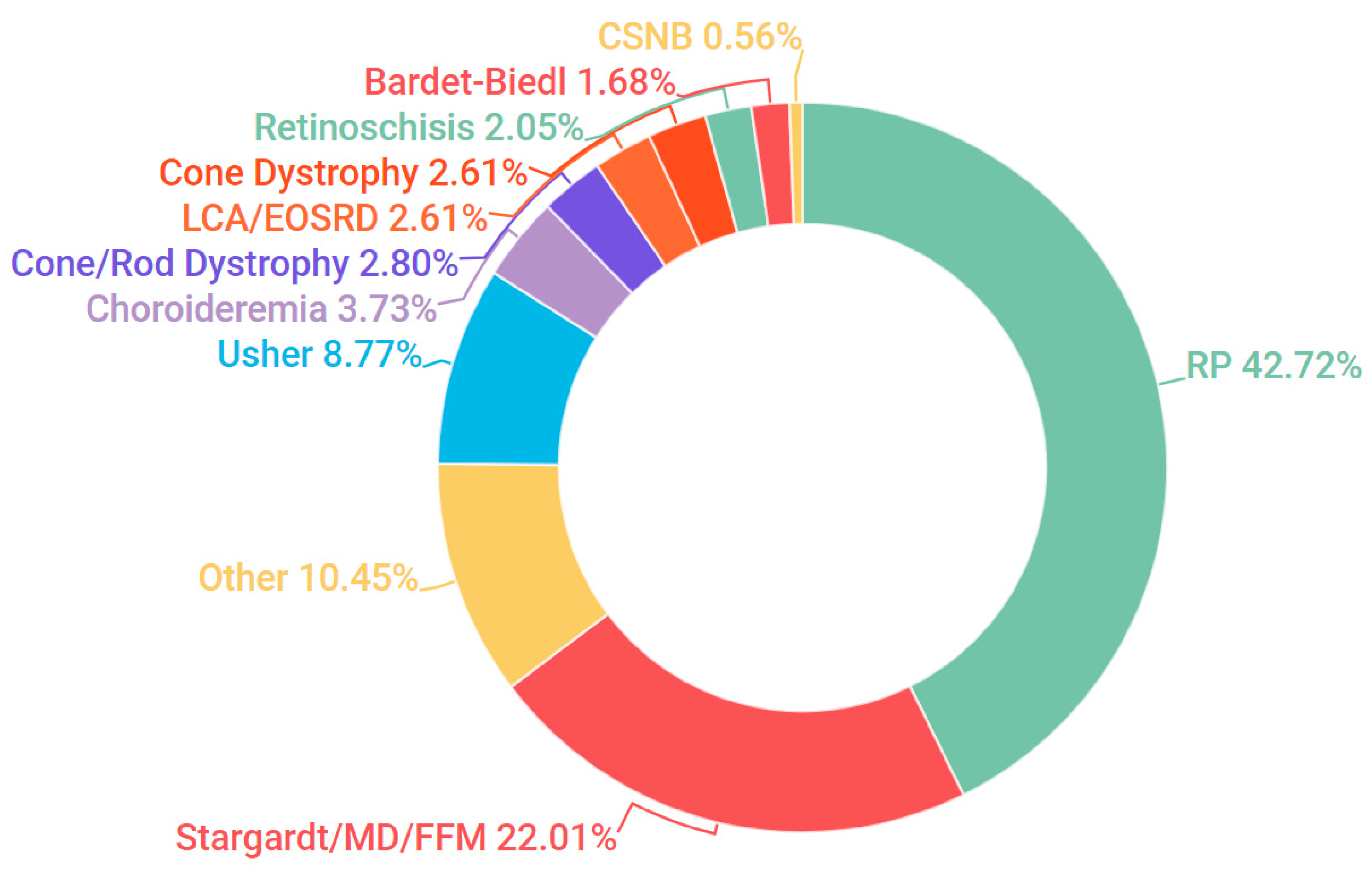
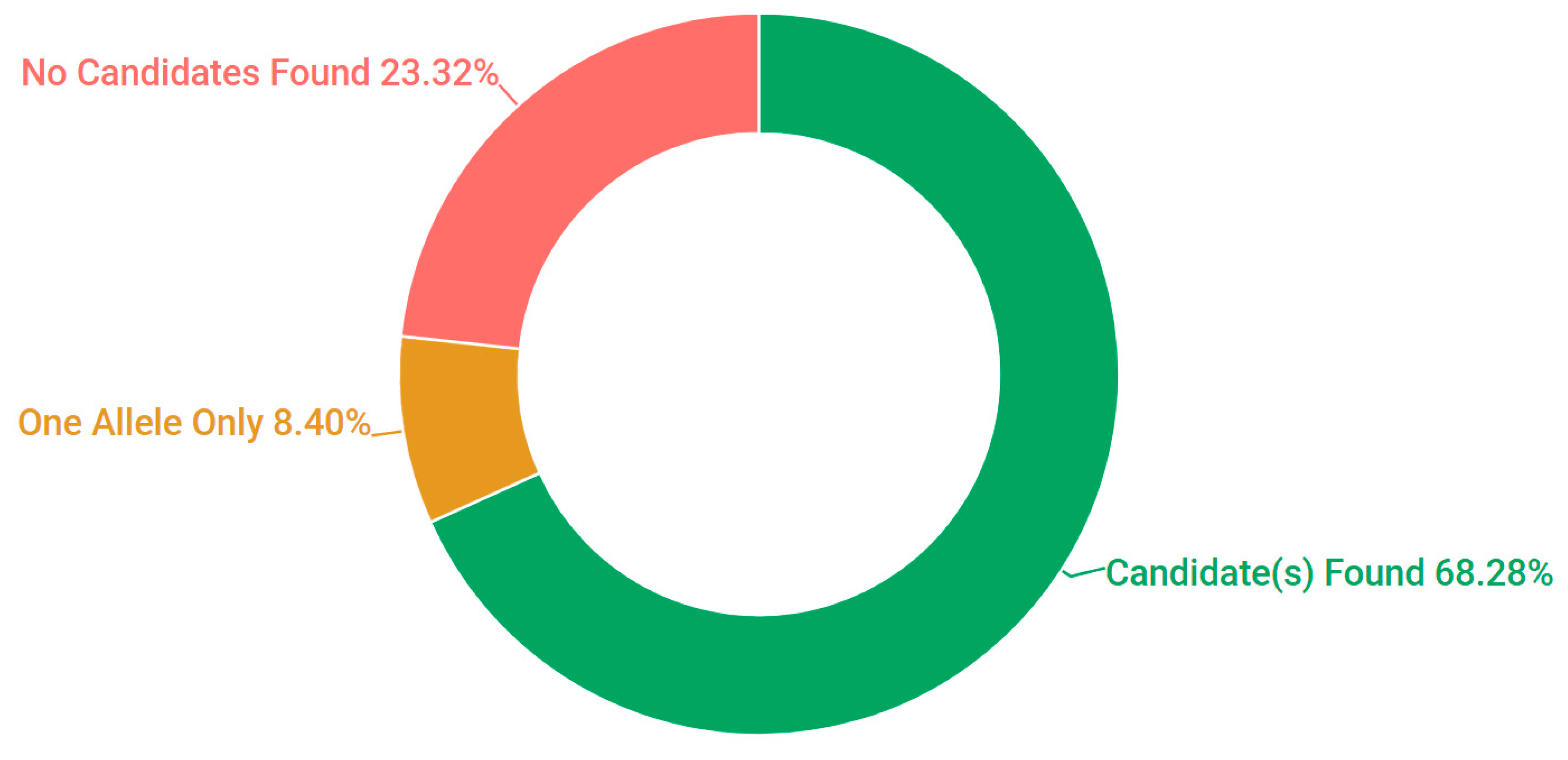
3.1. Presentation and Solve Rates
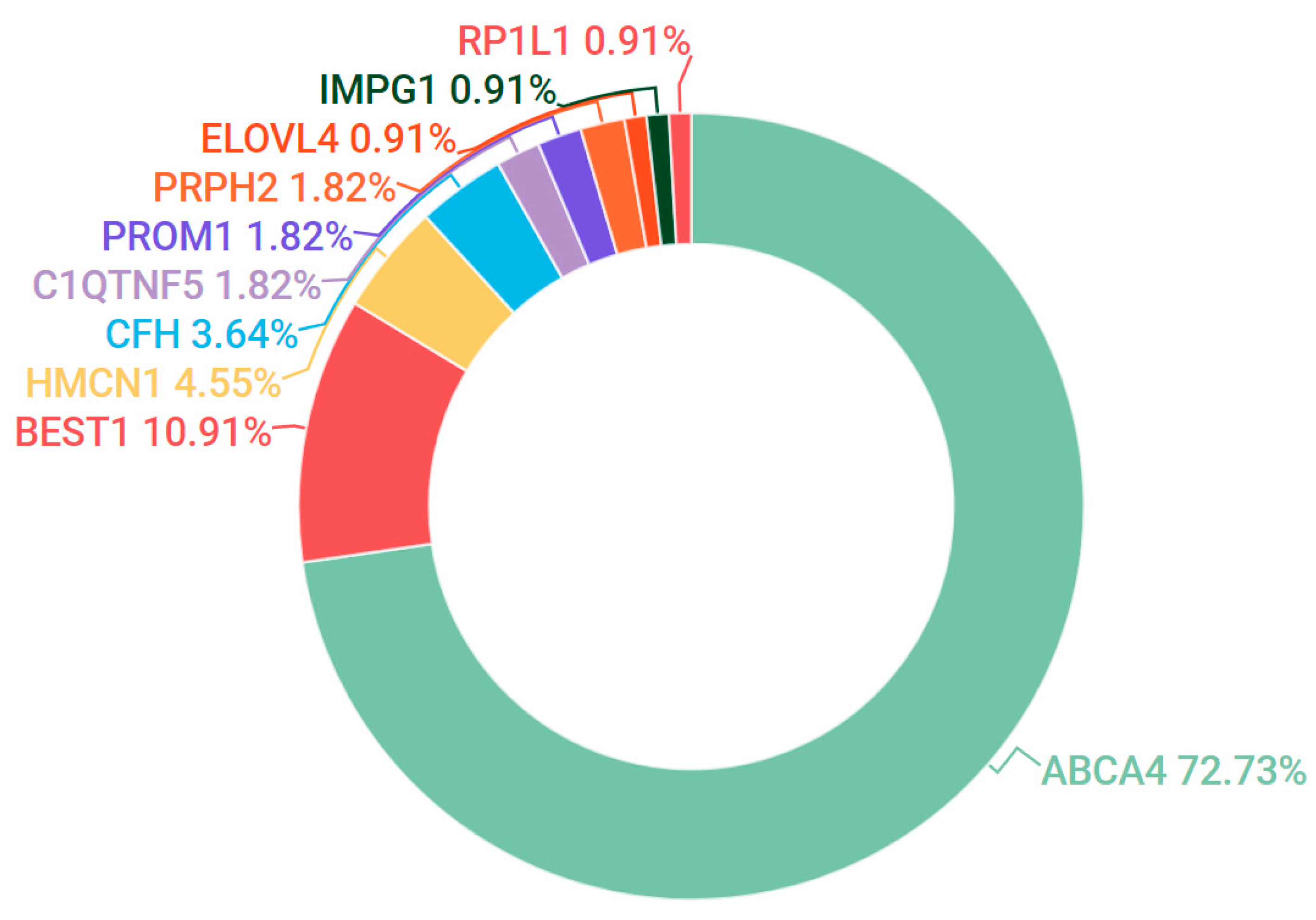
3.2. Stargardt Disease
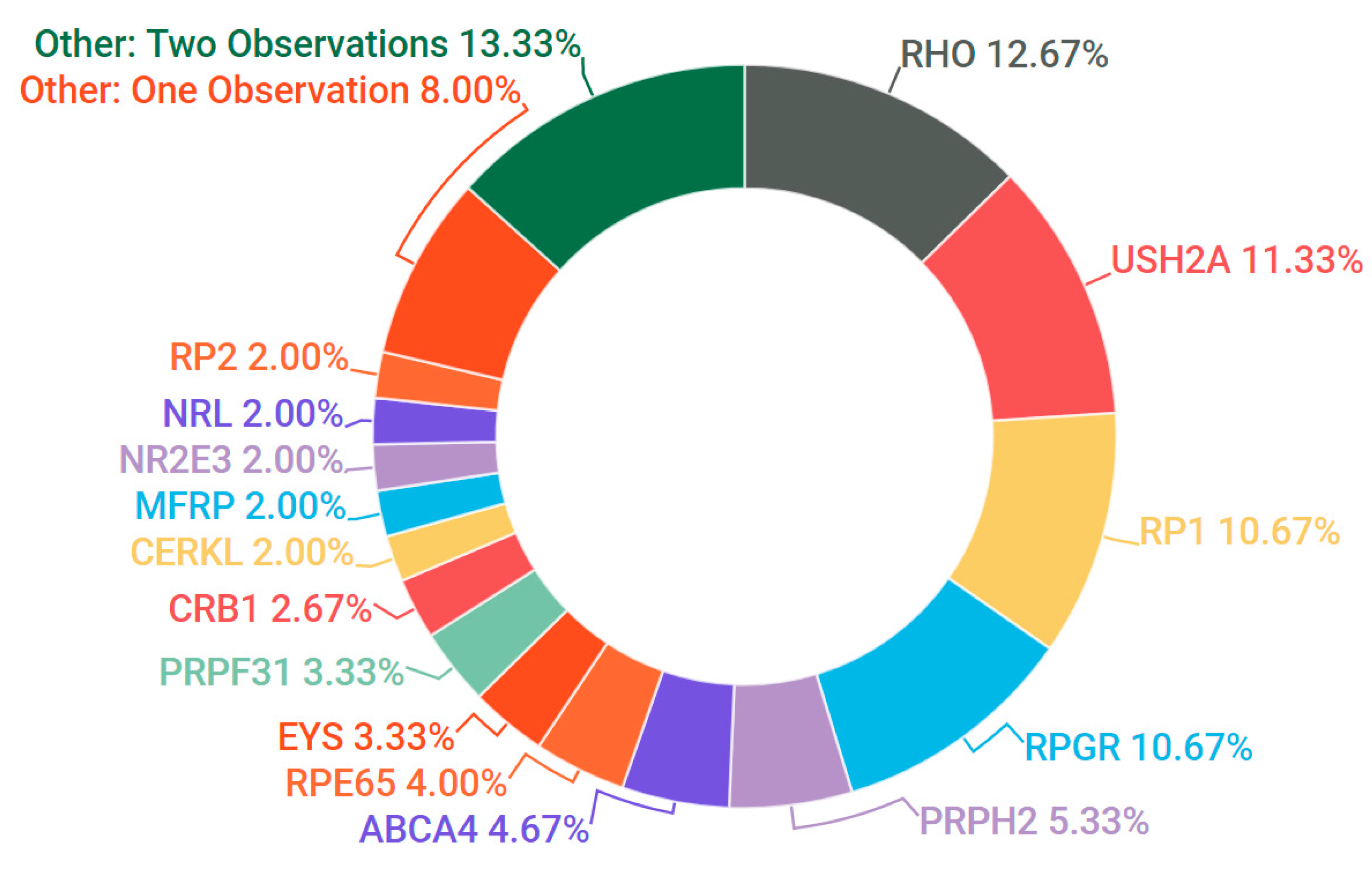
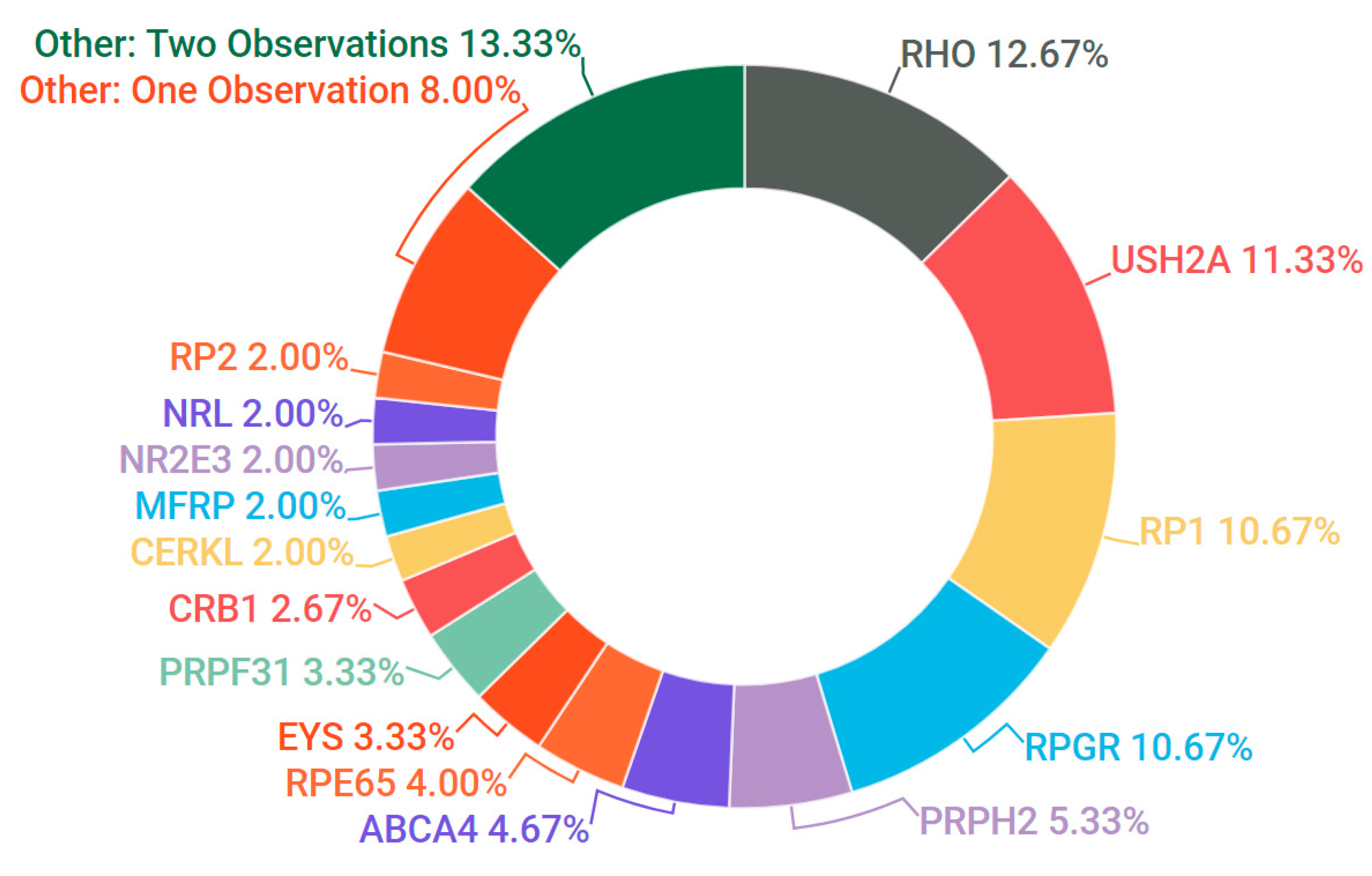
3.3. Retinitis Pigmentosa
3.4. Bardet-Biedl Syndrome
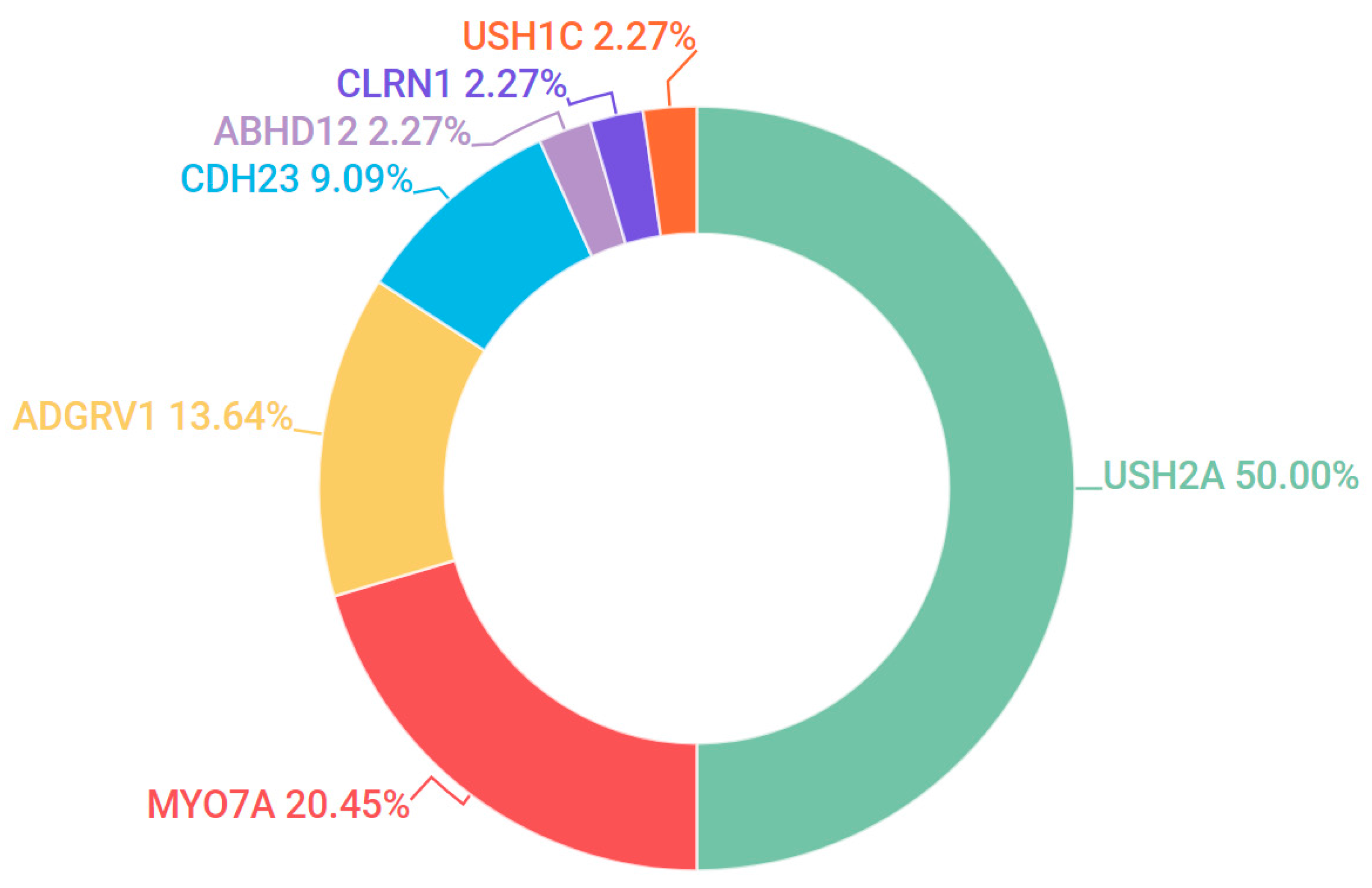
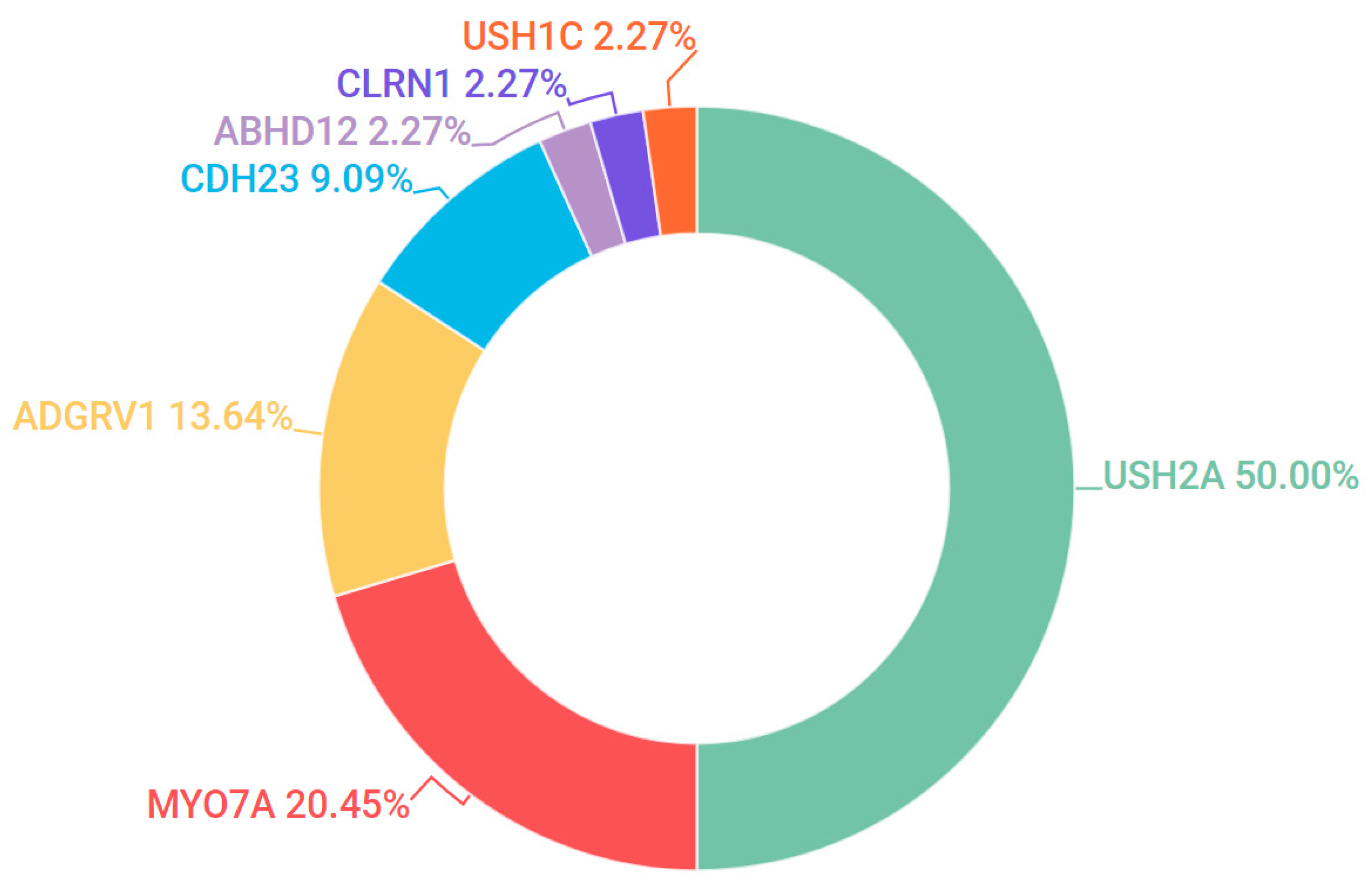
3.5. Usher Syndrome
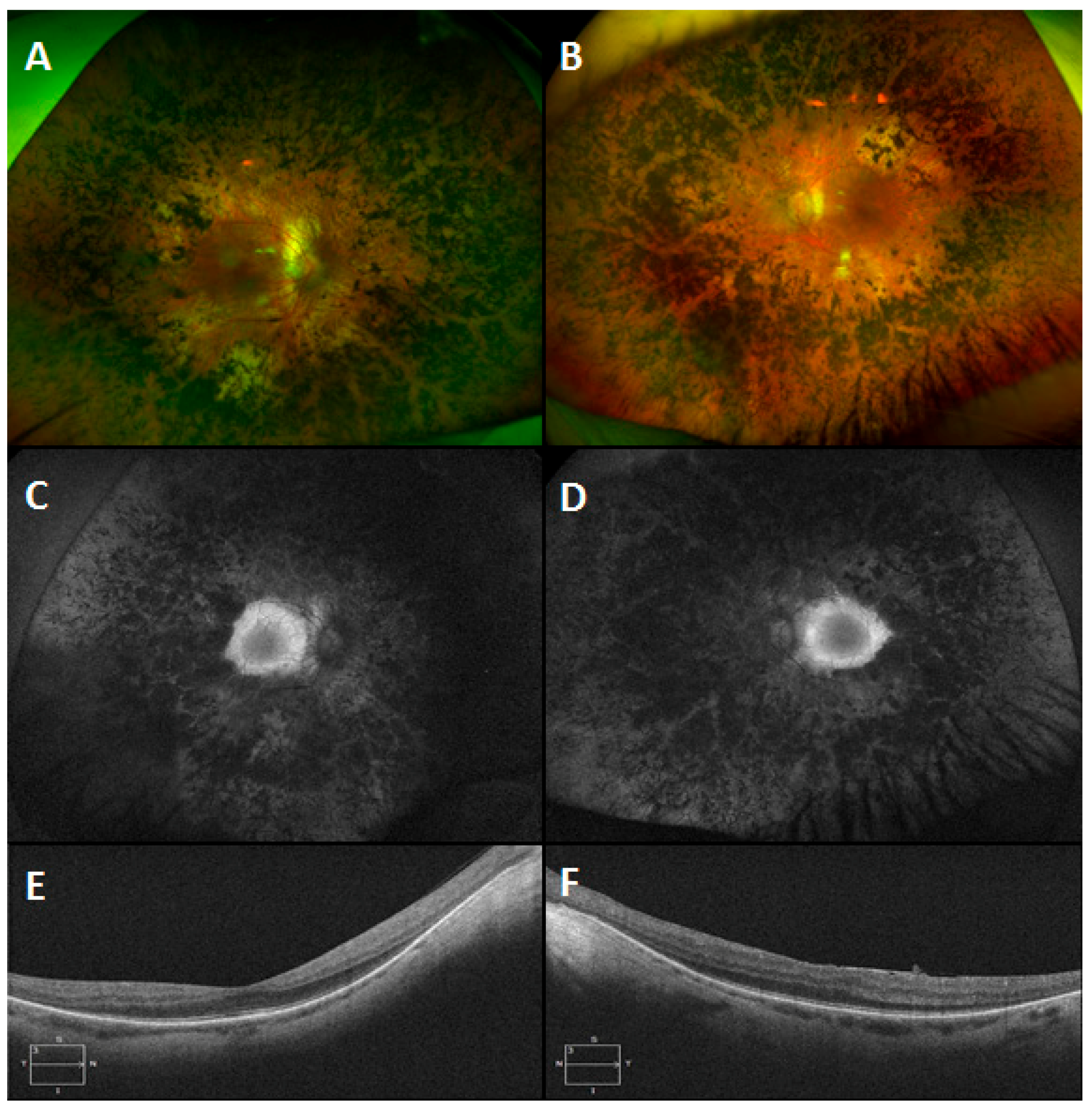
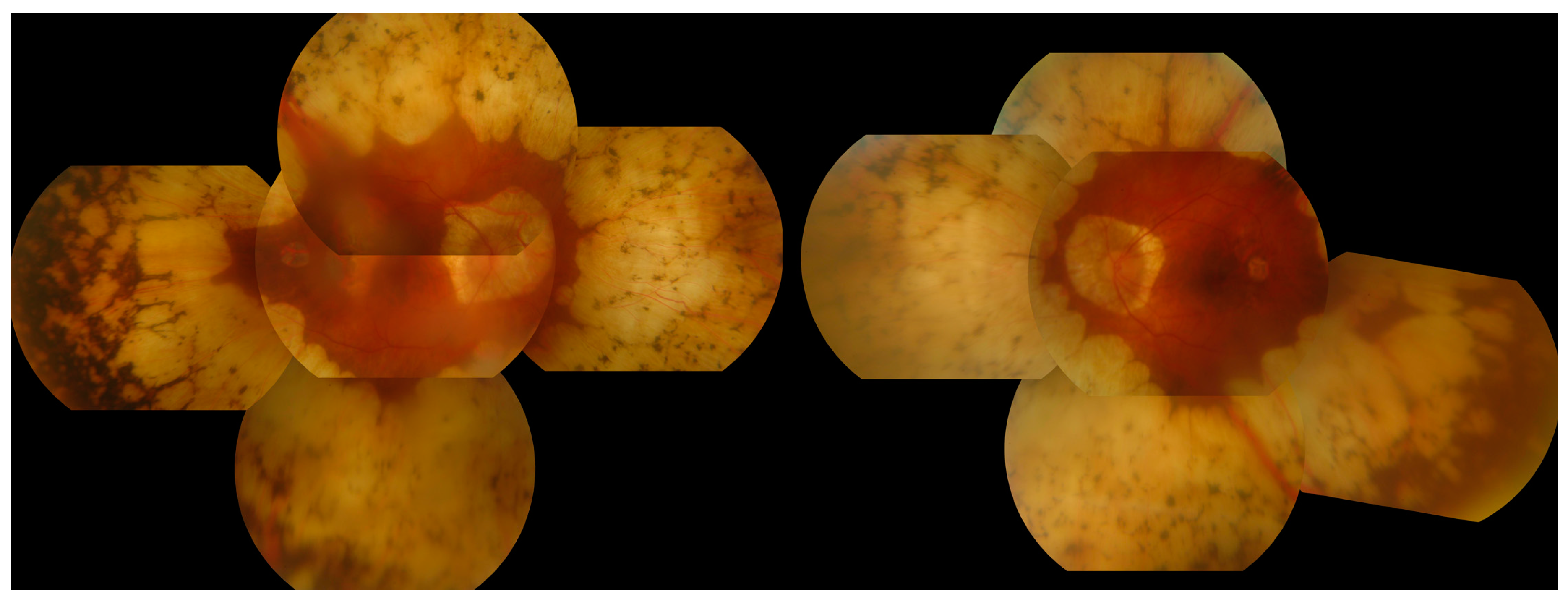
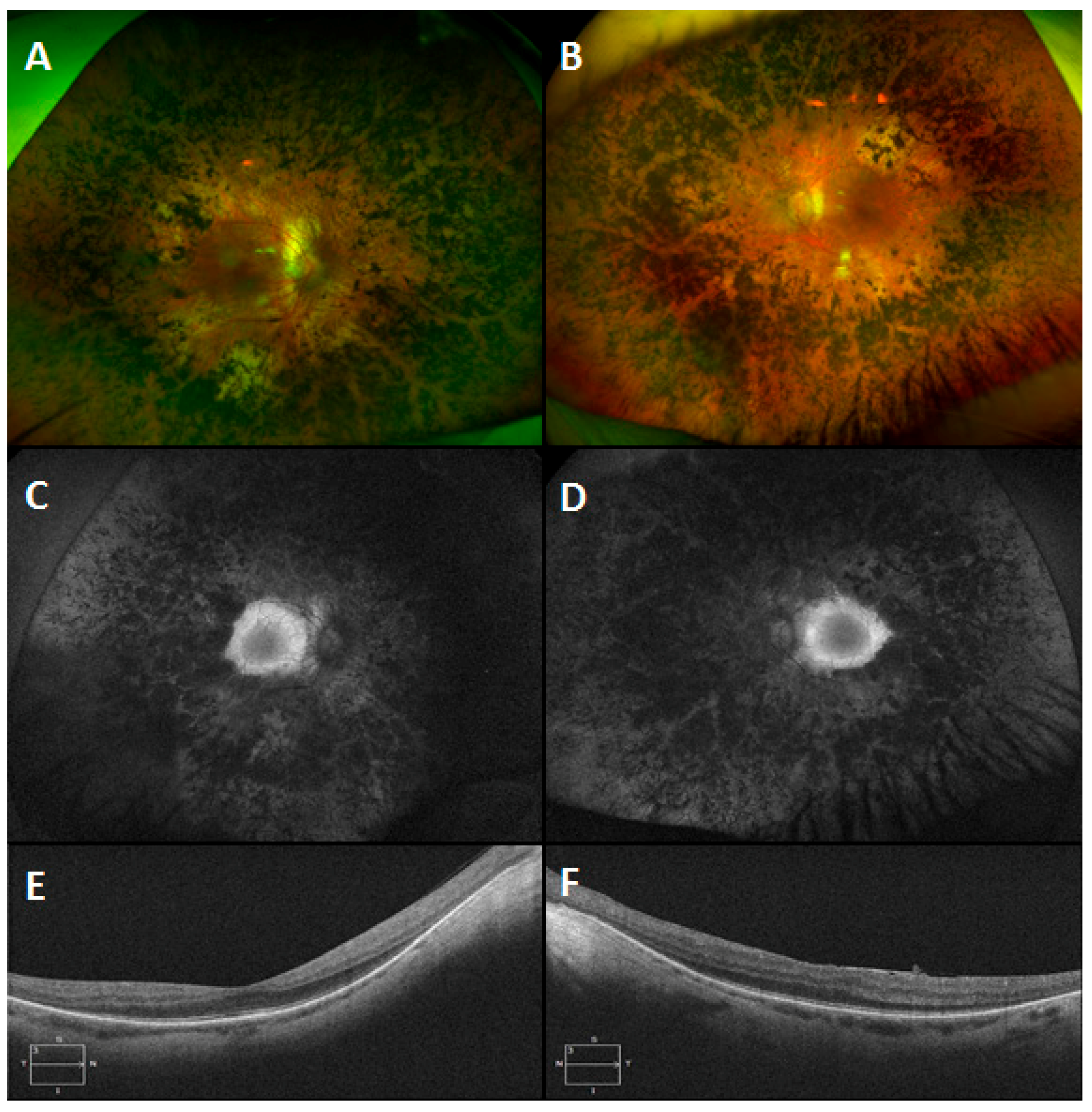
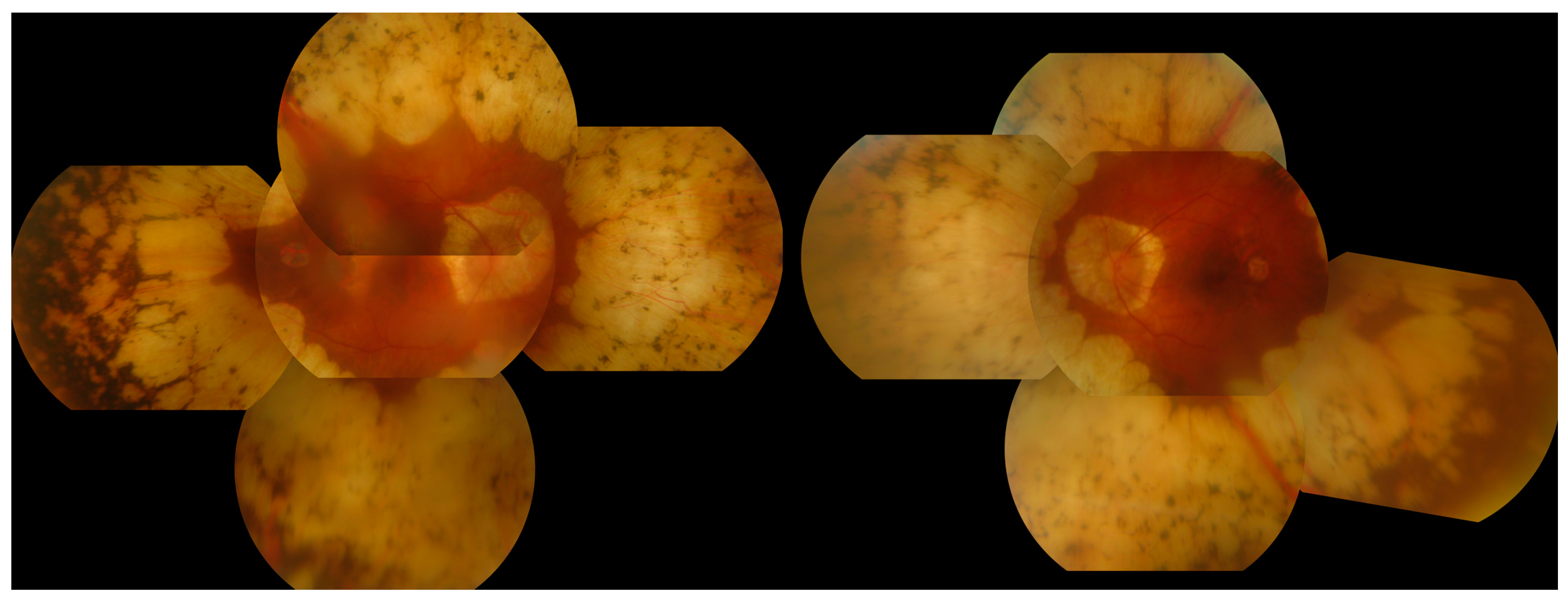
3.6. Gyrate Atrophy
3.7. Novel Variants
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- RetNet. Available online: http://www.sph.uth.tmc.edu/RetNet/ (accessed on 1 Novemeber 2017).
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 2011, 12, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.U.; Rahman, M.S.U.; Cao, J.; Yuan, P.X. Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. 3 Biotech 2017, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- Bangal, S.; Bhandari, A.; Dhaytadak, P.; Gogri, P. Gyrate atrophy of choroid and retina with myopia, cataract and systemic proximal myopathy: A rare case report from rural India. Australas. Med. J. 2012, 5, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. International Society for Clinical Electrophysiology of Vision (ISCEV) Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.J.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, M.; De Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; De Jaegere, S.; De Ravel, T.; De Rademaeker, M.; et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Le Quesne Stabej, P.; Lenassi, E.; Luxon, L.M.; Claustres, M.; Roux, A.-F.; Webster, A.R.; Bitner-Glindzicz, M. Screening for duplications, deletions and a common intronic mutation detects 35% of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J. Rare Dis. 2013, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Maxeiner, S.; Luo, F.; Tan, A.; Schmitz, F.; Südhof, T.C. How to make a synaptic ribbon: RIBEYE deletion abolishes ribbons in retinal synapses and disrupts neurotransmitter release. EMBO J. 2016, 35, 1098–1114. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 1 Novemeber 2017).
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012; arXiv:1207.3907. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; NHLBI Exome Sequencing Project; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.W.; Ho, A.; Muñoz, B.; Cideciyan, A.V.; Sahel, J.-A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Kenna, P.; Redmond, R.; Shiels, D.; McWilliam, P.; Humphries, M.M.; Sharp, E.M.; Jordan, S.; Kumar-Singh, R.; Humphries, P. Autosomal dominant retinitis pigmentosa: A mutation in codon 178 of the rhodopsin gene in two families of Celtic origin. Genomics 1991, 11, 1170–1171. [Google Scholar] [CrossRef]
- Farrar, G.J.; Findlay, J.B.; Kumar-Singh, R.; Kenna, P.; Humphries, M.M.; Sharpe, E.; Humphries, P. Autosomal dominant retinitis pigmentosa: A novel mutation in the rhodopsin gene in the original 3q linked family. Hum. Mol. Genet. 1992, 1, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81. [Google Scholar] [CrossRef]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Quinn, T.; Meehan, T.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, S.; Abid, A.; Ismail, M.; Hameed, A.; Mohyuddin, A.; Lall, P.; Aziz, A.; Anwar, K.; Mehdi, S.Q. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2005, 42, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.J.; Law, J.P.; Lai, T.Y.Y.; Chiang, S.W.Y.; Tam, P.O.S.; Chu, K.Y.; Wang, N.; Zhang, M.; Pang, C.P. Differential pattern of RP1 mutations in retinitis pigmentosa. Mol. Vis. 2010, 16, 1353–1360. [Google Scholar] [PubMed]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sánchez, S.; Álvarez-Satta, M.; Cortón, M.; Guillén, E.; Ayuso, C.; Valverde, D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J. Med. Genet. 2015, 52, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.F.; Kerr, N.C.; Kedrov, M.; Nishimura, D.; Jennings, B.J.; Stone, E.M.; Sheffield, V.C.; Iannaccone, A. Phenotypic expression of Bardet-Biedl syndrome in patients homozygous for the common M390R mutation in the BBS1 gene. Vis. Res. 2012, 75, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Moloney, T.P.; O’Hagan, S.; Lee, L. Ultrawide-field fundus photography of the first reported case of gyrate atrophy from Australia. Clin. Ophthalmol. 2014, 8, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; McClatchey, A.I.; Ramesh, N.; Benoit, L.A.; Berson, E.L.; Shih, V.E.; Gusella, J.F. Molecular basis of ornithine aminotransferase deficiency in B-6-responsive and -nonresponsive forms of gyrate atrophy. Proc. Natl. Acad. Sci. USA 1988, 85, 3777–3780. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.; Weiner, C.; Nasie, I.; Anikster, Y.; Landau, Y.; Koren, T.; Pokroy, R.; Abulafia, A.; Pras, E. Reversal of cystoid macular edema in gyrate atrophy patients. Ophthalmic Genet. 2017, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Humphries, P.; Palfi, A.; Kenna, P.F.; Farrar, G.J. A novel homozygous truncating GNAT1 mutation implicated in retinal degeneration. Br. J. Ophthalmol. 2016, 100, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, J.; Feng, Y.; Xu, M.; Chen, R.; Zou, X.; Sui, R.; Chang, E.Y.; Lewis, R.A.; Zhang, V.W.; et al. Improved Diagnosis of Inherited Retinal Dystrophies by High-Fidelity PCR of ORF15 followed by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-F.; Wu, J.; Lv, J.-N.; Zhang, X.; Jin, Z.-B. Identification of false-negative mutations missed by next-generation sequencing in retinitis pigmentosa patients: A complementary approach to clinical genetic diagnostic testing. Genet. Med. 2015, 17, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Kabir, F.; Ullah, I.; Ali, S.; Gottsch, A.D.H.; Naeem, M.A.; Assir, M.Z.; Khan, S.N.; Akram, J.; Riazuddin, S.; Ayyagari, R.; et al. Loss of function mutations in RP1 are responsible for retinitis pigmentosa in consanguineous familial cases. Mol. Vis. 2016, 22, 610–625. [Google Scholar] [PubMed]
- Méndez-Vidal, C.; Bravo-Gil, N.; González-Del Pozo, M.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antiñolo, G. Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet. 2014, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, M.; Abu Safieh, L.; Alkuraya, H.; Aldahmesh, M.A.; Alzahrani, J.; Diya, M.; Hashem, M.; Hardcastle, A.J.; Al-Hazzaa, S.A.F.; Alkuraya, F.S. RP1 and retinitis pigmentosa: Report of novel mutations and insight into mutational mechanism. Br. J. Ophthalmol. 2012, 96, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Boulanger-Scemama, E.; Lancelot, M.-E.; Antonio, A.; Démontant, V.; Condroyer, C.; Letexier, M.; Saraiva, J.-P.; Mohand-Saïd, S.; Sahel, J.-A.; et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. Biomed. Res. Int. 2015, 2015, 485624. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.H.J.; Hoefsloot, L.H.; van den Born, L.I.; Phan, M.; Klevering, B.J.; Westeneng-van Haaften, C.; Braun, T.A.; et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.J.; Goercharn-Ramlal, A.S.A.; den Engelsman-van Dijk, A.H.A.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In Silico Functional Meta-Analysis of 5962 ABCA4 Variants in 3928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Miró, M.; Tonda, R.; Escudero-Ferruz, P.; Andrés, R.; Mayor-Lorenzo, A.; Castro, J.; Ciccioli, M.; Hidalgo, D.A.; Rodríguez-Ezcurra, J.J.; Farrando, J.; et al. Novel Candidate Genes and a Wide Spectrum of Structural and Point Mutations Responsible for Inherited Retinal Dystrophies Revealed by Exome Sequencing. PLoS ONE 2016, 11, e0168966. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. 2017, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Bernardis, I.; Chiesi, L.; Tenedini, E.; Artuso, L.; Percesepe, A.; Artusi, V.; Simone, M.L.; Manfredini, R.; Camparini, M.; Rinaldi, C.; et al. Unravelling the Complexity of Inherited Retinal Dystrophies Molecular Testing: Added Value of Targeted Next-Generation Sequencing. Biomed. Res. Int. 2016, 2016, 6341870. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Navarro, R.; Ruiz-Nogales, S.; Méndez, P.; Burés-Jelstrup, A.; Corcóstegui, B.; Pomares, E. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci. Rep. 2017, 7, 42078. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed]
- Savoie, B.T.; Ferrone, P.J. Complicated Congenital Retinoschisis. Retinal Cases Brief Rep. 2017, 11 (Suppl. 1), S202–S210. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, I.S.; Freund, P.R.; Knowles, J.A.; MacDonald, I.M. The Natural History of Full-Field Stimulus Threshold Decline in Choroideremia. Retina 2017. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; Pott, J.-W.R.; Collin, R.W.J.; Kroes, H.Y.; Verheij, J.B.G.M.; Blokland, E.A.W.; de Castro Miró, M.; Hoyng, C.B.; Klaver, C.C.W.; Koenekoop, R.K.; et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3646–3652. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Condition | Transcript ID | Genomic Location | Nucleotide Change | Protein Change | MetaLR Score | Spidex Z-Core | Observed with: |
|---|---|---|---|---|---|---|---|---|
| ABCA4 | Stargardt Disease | NM_000350.2 | g.1:94007721 | c.5917delG | p.Val1973fs | None | None | p.Arg290Trp |
| ABCA4 | Stargardt Disease | NM_000350.2 | g.1:94098827 | c.735T>G | p.Tyr245 * | None | −3.542 | p.Asn1868Ile |
| ABCA4 | Stargardt Disease | NM_000350.2 | g.1:94030459 | c.4320delT | p.Phe1440fs | None | None | p.Asn1868Ile |
| ADGRV1 | USH Type II | NM_032119.3 | g.1:91153391 | c.18795delA | p.Leu6265fs | None | None | p.Arg5772 * |
| BBS1 | Bardet-Biedl | NM_024649.4 | g.11:66531658 | c.1614delC | p.Leu539fs | None | None | p.Met390Arg |
| CNGA3 | Cone Dystrophy | NM_001298.2 | g.2:98396398 | c.1228C>T | p.Arg410Trp | 0.9441 | None | p.Arg410Trp |
| ELOVL4 | Stargardt Disease | NM_022726.3 | g.6:79916758 | c.789delTAACTTinsAACT | p.Phe265fs_Asn264fs | None | None | / |
| MYO7A | USH Type I | NM_000260.3 | g.11:77202351 | c.5095C>T | p.Gln1699 * | None | −3.017 | p.Ala2009fs |
| MYO7A | USH Type I | NM_000260.3 | g.11:77208775 | c.6025delG | p.Ala2009fs | None | None | p.Gln1699 * |
| NR2E3 | Retinitis Pigmentosa (Recessive) | NM_014249.3 | g.15:71813635 | c.994G>T | p.Glu332 * | None | None | p.119-2A>C |
| NYX | Congenital Stationary Night Blindness | NM_022567.2 | g.X:41474475 | c.1022A>C | p.Asp341Ala | 0.7224 | None | / |
| OPA1 | Optic Atrophy | NM_130837.2 | g.3:193614740 | c.53_62delTGAAACACAG | p.Val18fs | None | None | / |
| PRPF31 | Retinitis Pigmentosa (Dominant) | NM_015629.3 | g.19:54123748 | c.528-1G>T | / | None | −2.131 | / |
| PRPF8 | Retinitis Pigmentosa (Dominant) | NM_006445.3.2 | g.17:1653571 | c.6337_6339delAAG | p.Lys2113del | None | None | / |
| RHO | Retinitis Pigmentosa (Dominant) | NM_000539.3 | g.3:139531026 | c.512C>A | p.Pro171Gln | 0.4368 | 0.875 | / |
| RP1 | Retinitis Pigmentosa (Recessive) | NM_006269.1 | g.8:54621124 | c.160delG | p.Val54fs | None | None | p.Val54fs |
| RP2 | Retinitis Pigmentosa (X-Linked) | NM_006915.2 | g.X:46853795 | c.425delA | p.Asn142fs | None | None | / |
| RPGR | Retinitis Pigmentosa (X-Linked) | NM_001034853.1 | g.X:38286761 | c.2236_2237delGA | p.Glu746fs | None | None | / |
| RPGRIP1 | Leber Congenital Amaurosis | NM_020366.3 | g.14:21328427 | c.2899C>T | p.Gln967 * | None | −3.491 | p.Gln967 * |
| RS1 | Retinoschisis | NM_000330.3 | g.X:18644535 | c.416delA | p.Gln139fs | None | None | / |
| RS1 | Retinoschisis | NM_000330.3 | g.X:18647190 | c.326+1G>A | / | None | −2.194 | / |
| USH2A | Retinitis Pigmentosa (Recessive) | NM_206933.2 | g.1:215647520 | c.14791+2T>A | / | None | −2.921 | p.Pro4090Thr |
| USH2A | USH Type II | NM_206933.2 | g.2015680269 | c.12172_12172delCTGinsTAAA | p.Leu4058fs | None | None | p.Glu767fs |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. https://doi.org/10.3390/genes8110304
Dockery A, Stephenson K, Keegan D, Wynne N, Silvestri G, Humphries P, Kenna PF, Carrigan M, Farrar GJ. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes. 2017; 8(11):304. https://doi.org/10.3390/genes8110304
Chicago/Turabian StyleDockery, Adrian, Kirk Stephenson, David Keegan, Niamh Wynne, Giuliana Silvestri, Peter Humphries, Paul F. Kenna, Matthew Carrigan, and G. Jane Farrar. 2017. "Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations" Genes 8, no. 11: 304. https://doi.org/10.3390/genes8110304