
Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Data Acquisition
2.2. Publicly Available Annotations
2.3. RNA-Seq Reads Mapping and Transcriptome Assembly
2.4. Pipeline for lincRNA Identification
2.5. Differentially Expressed lincRNAs and mRNA Analysis
2.6. Prediction of PTGs of lincRNAs
2.7. Gene Ontology and Pathway Analysis
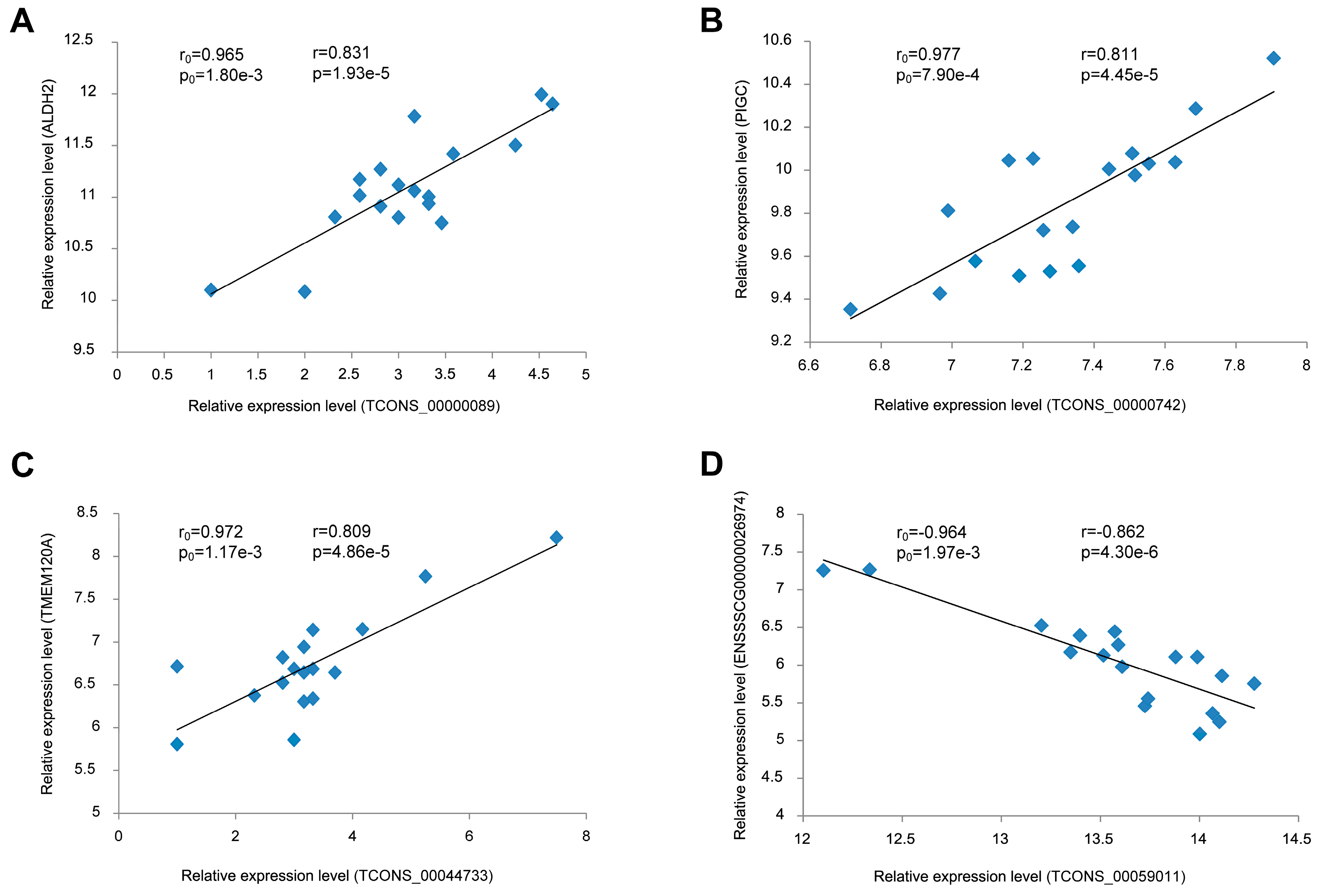
2.8. Correlation Validation between lincRNAs and Their PTGs
3. Results
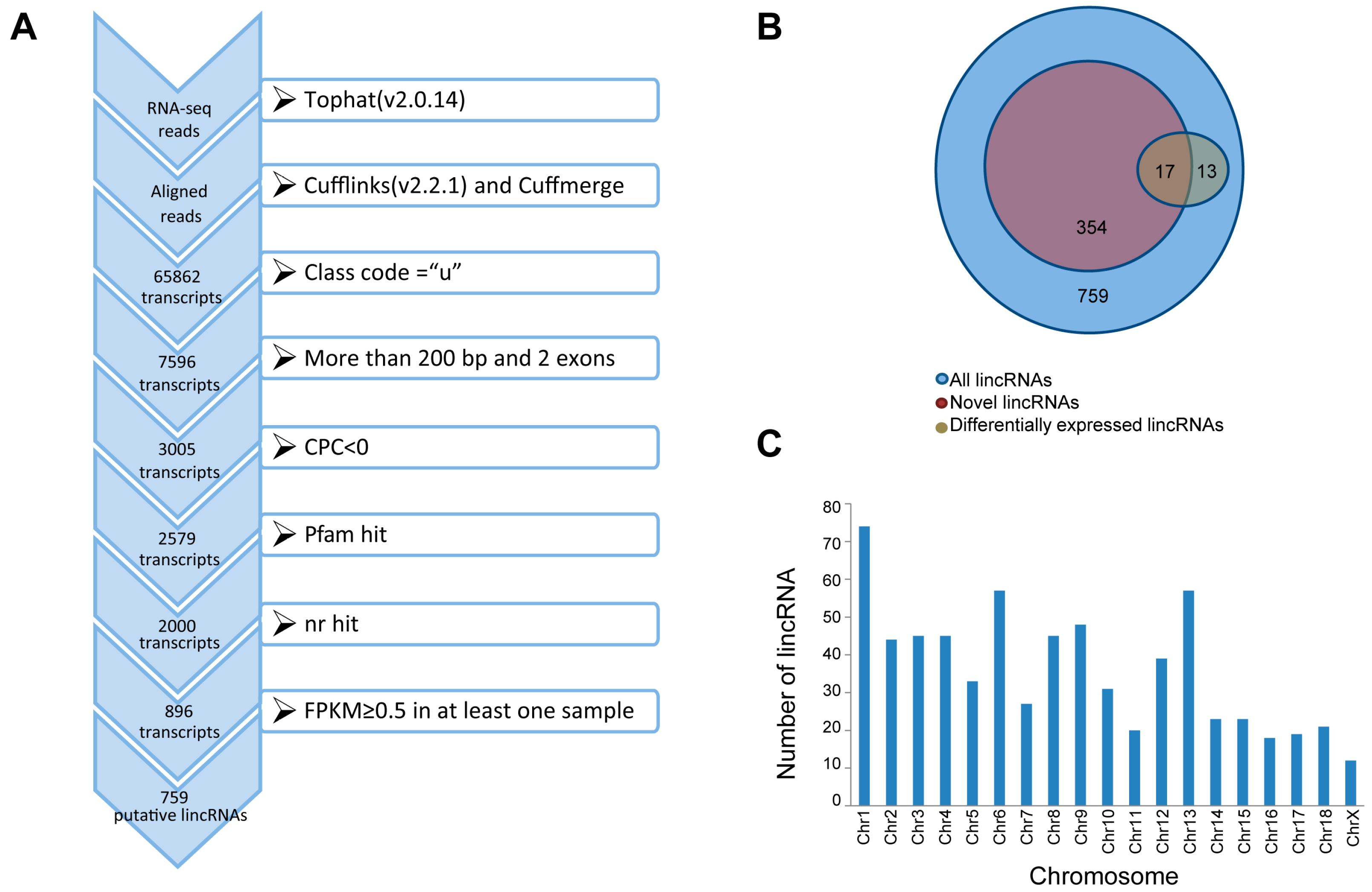
3.1. Transcripts Assembly and lincRNAs Identification
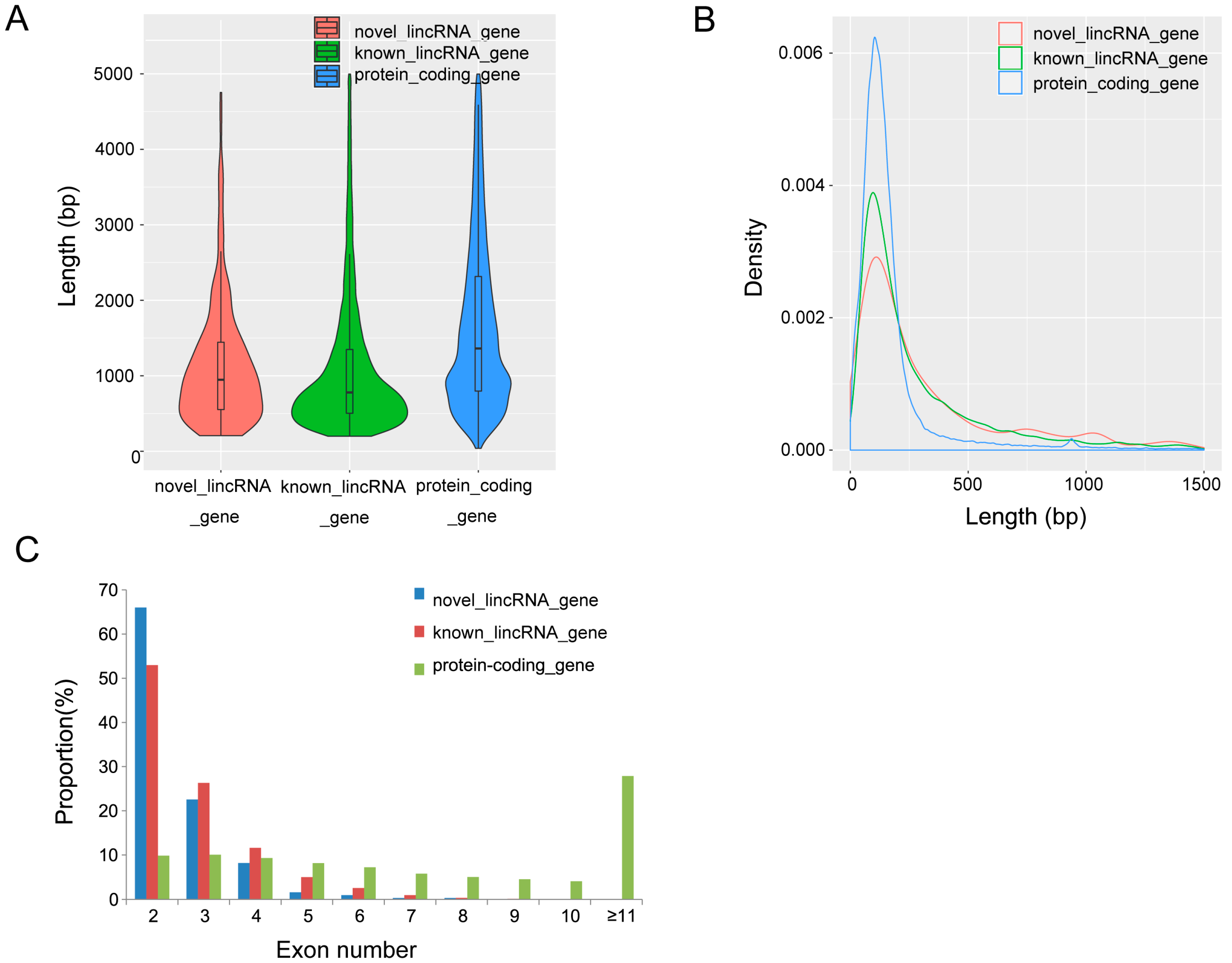
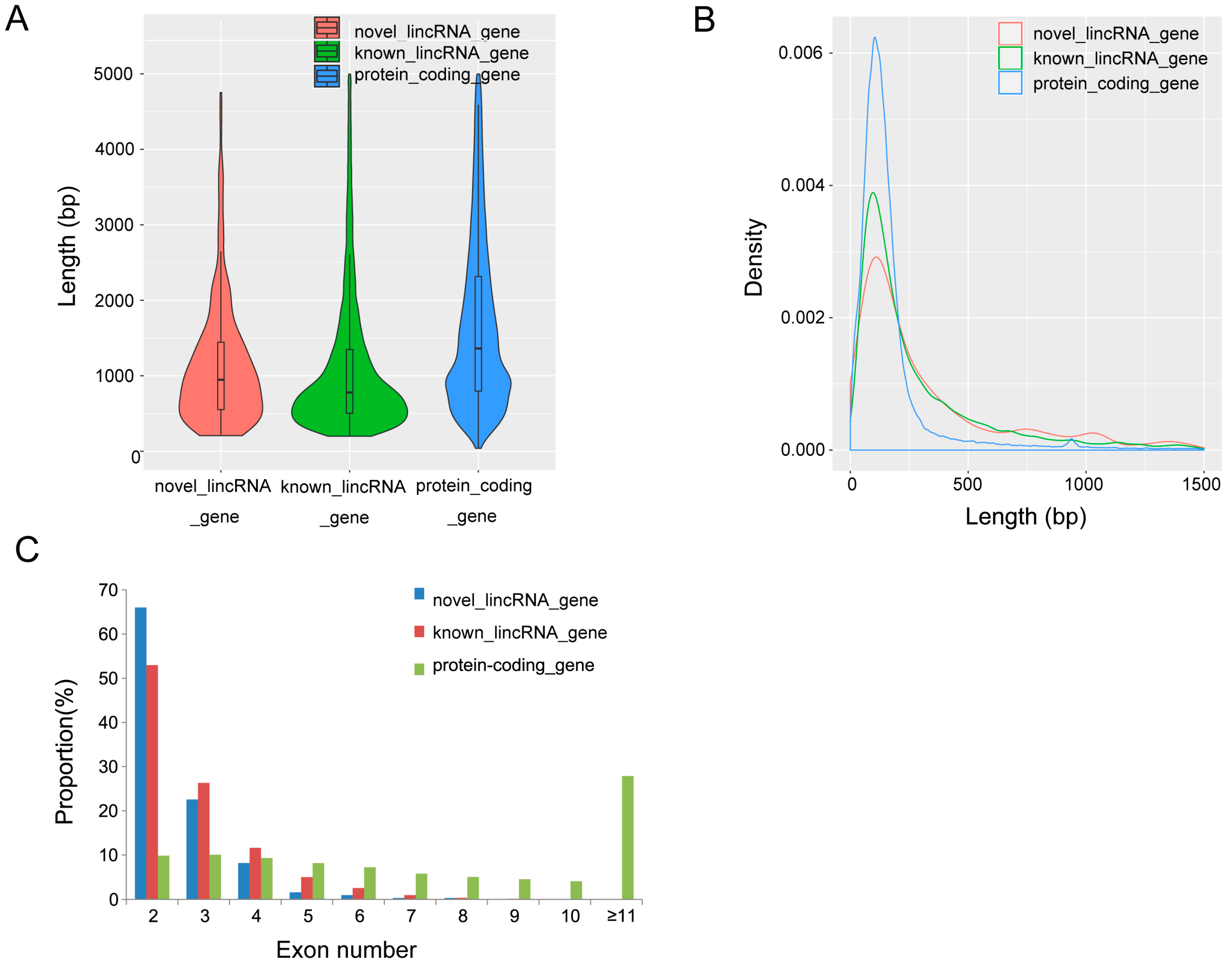
3.2. Characterization of Identified lincRNAs
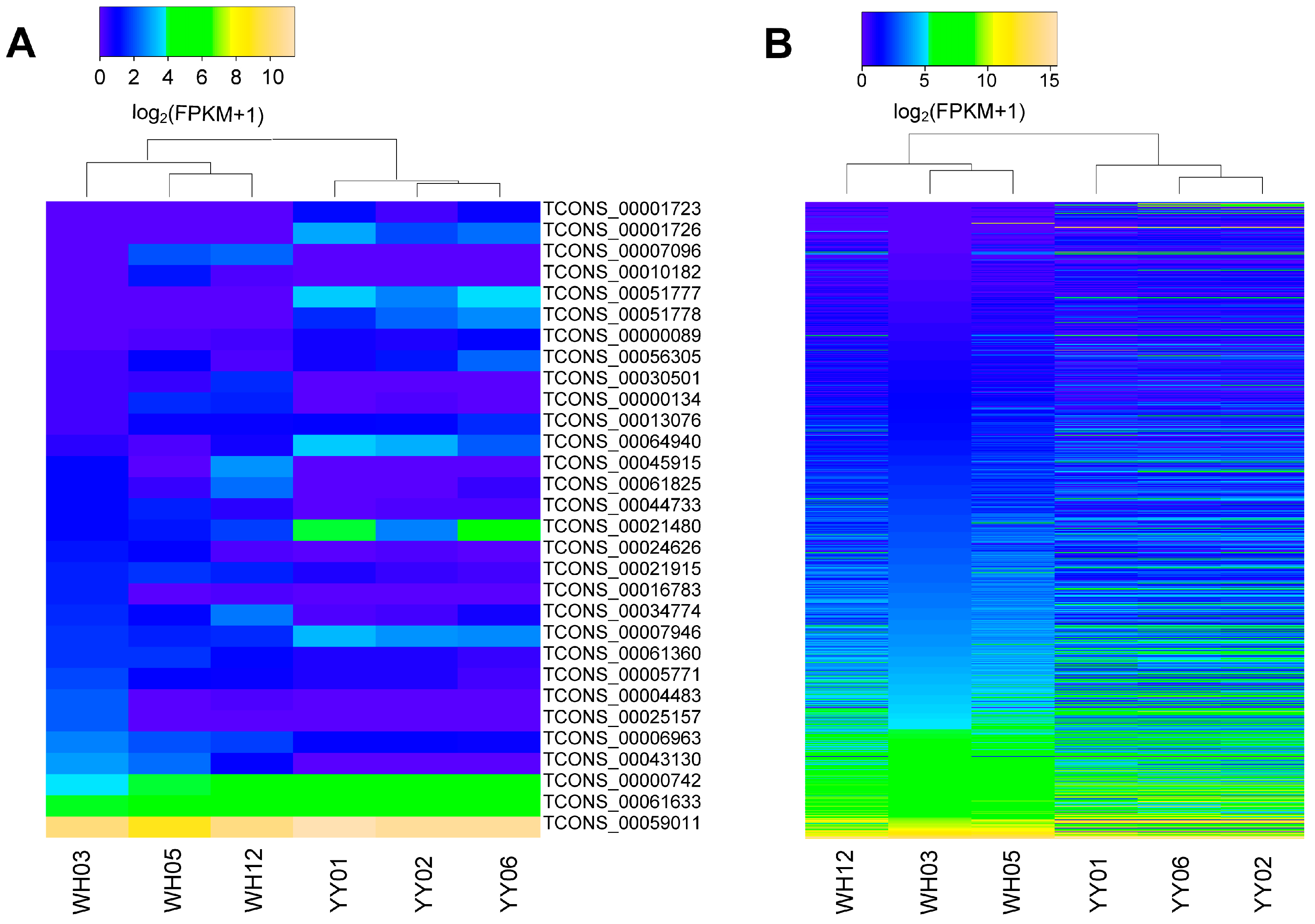
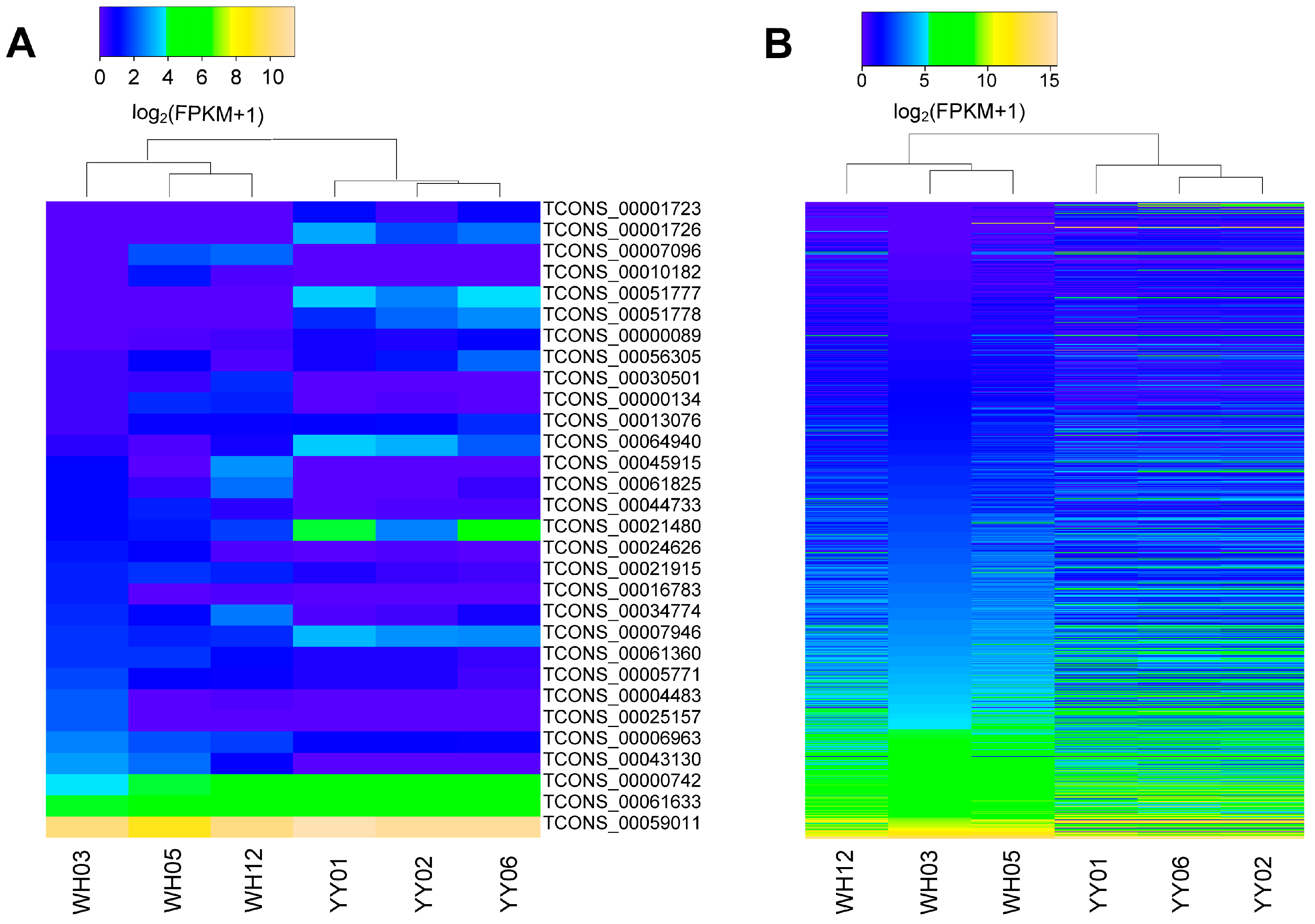
3.3. Differential Expression Analysis of lincRNAs and mRNA
3.4. Prediction of Differentially Expressed lincRNA Target Gene
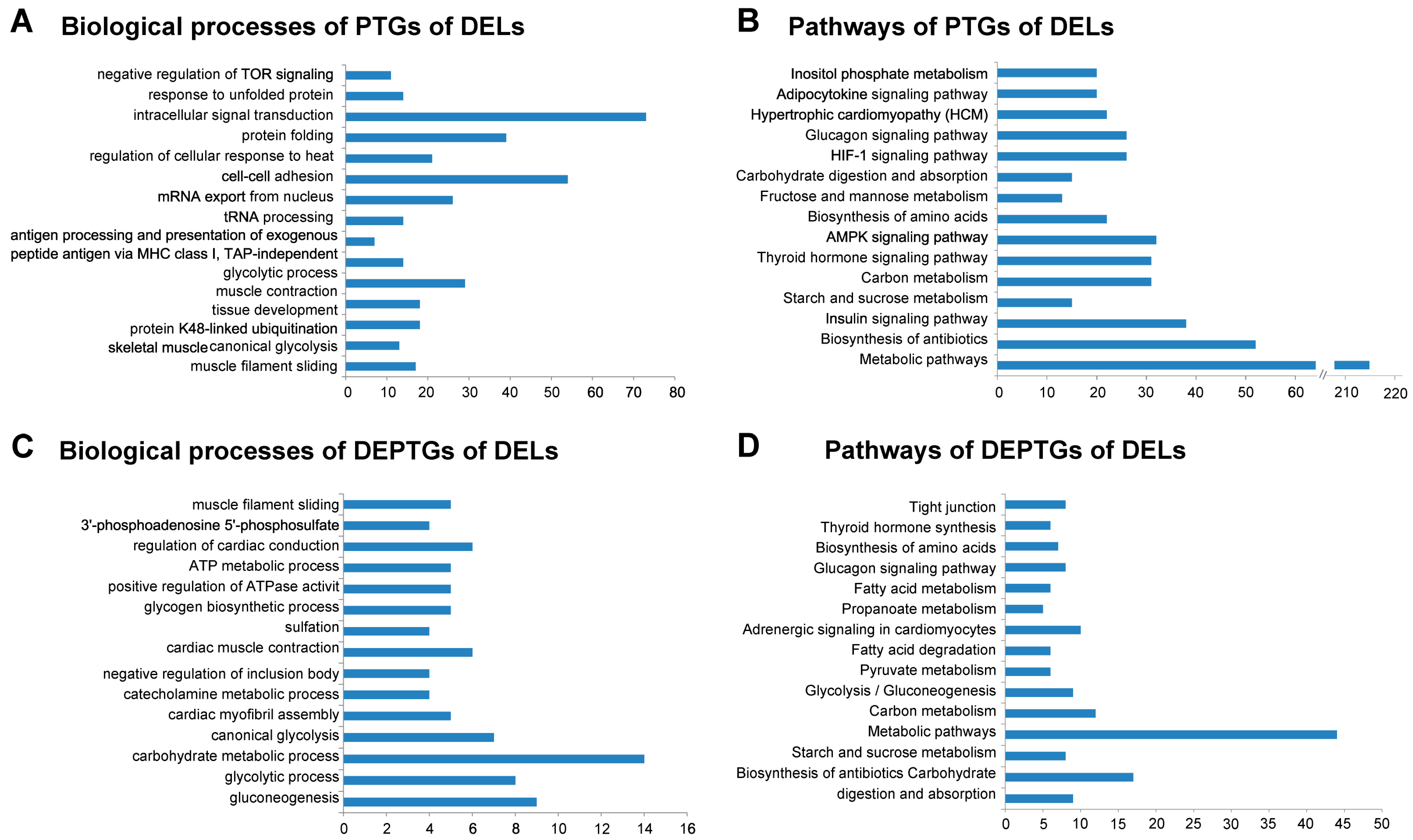
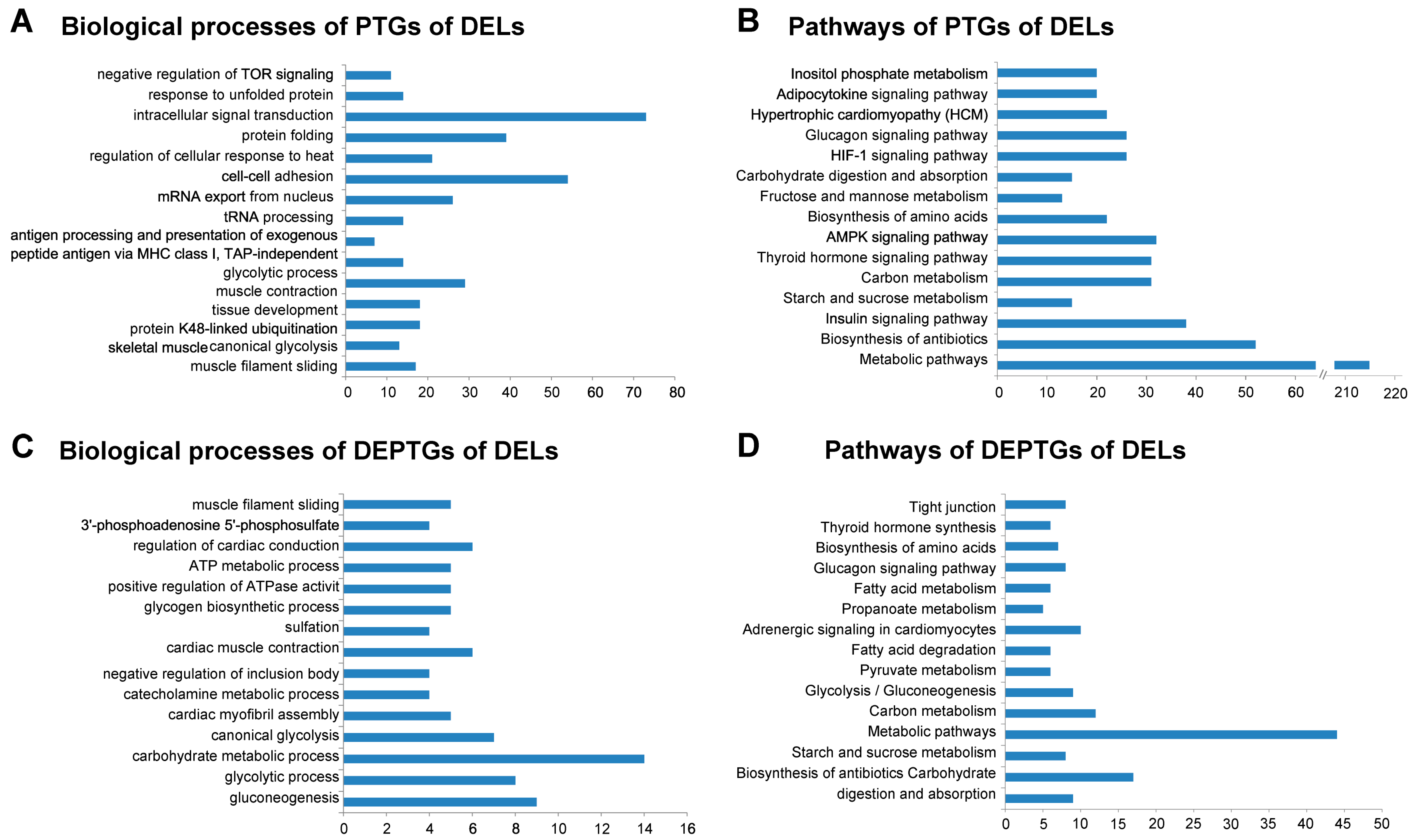
3.5. Gene Ontology and Pathway Analysis of PTGs and mRNA
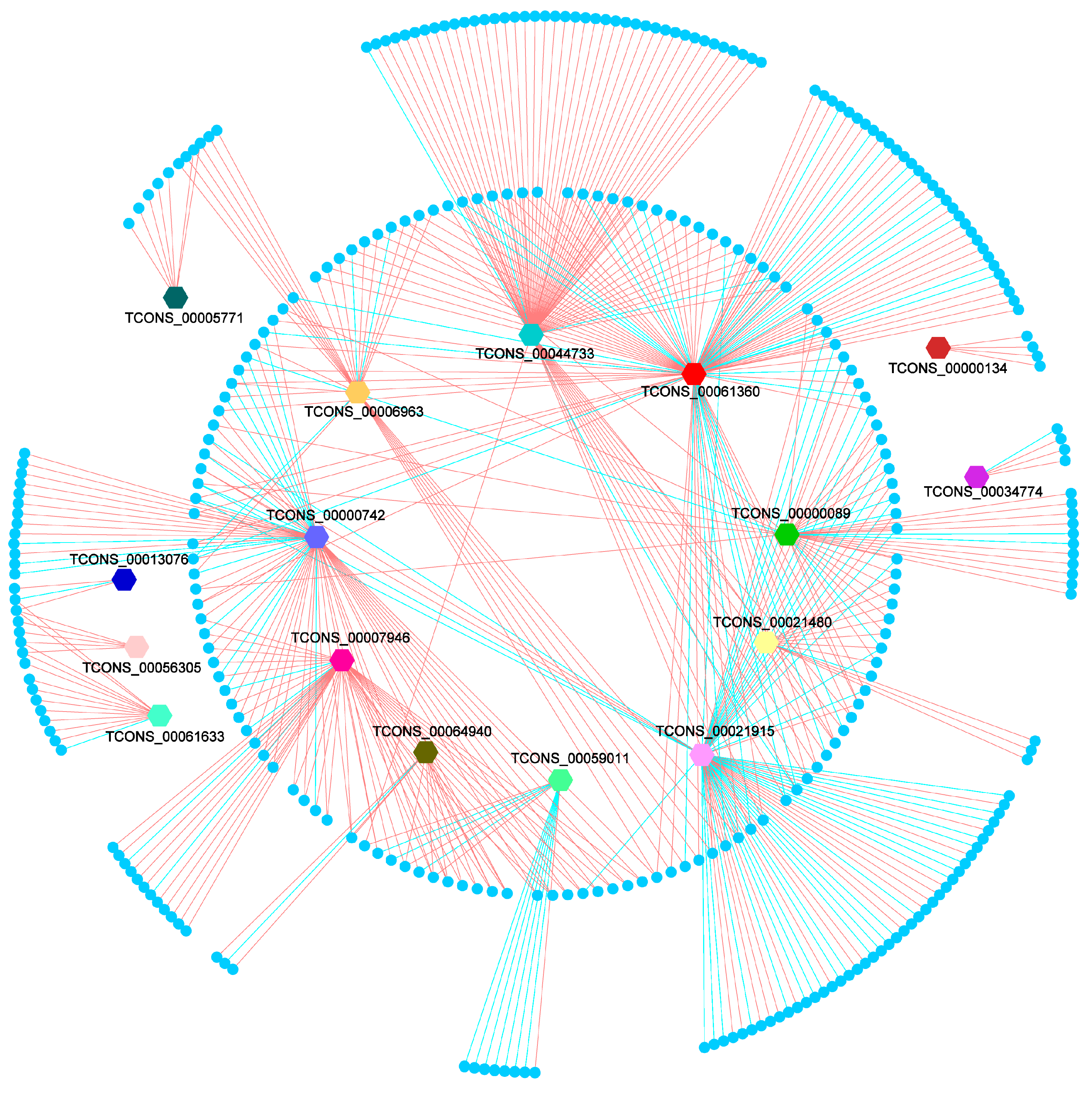
3.6. Expression Regulation Analysis of DELs and Their Differentially Expressed Potential Target Genes
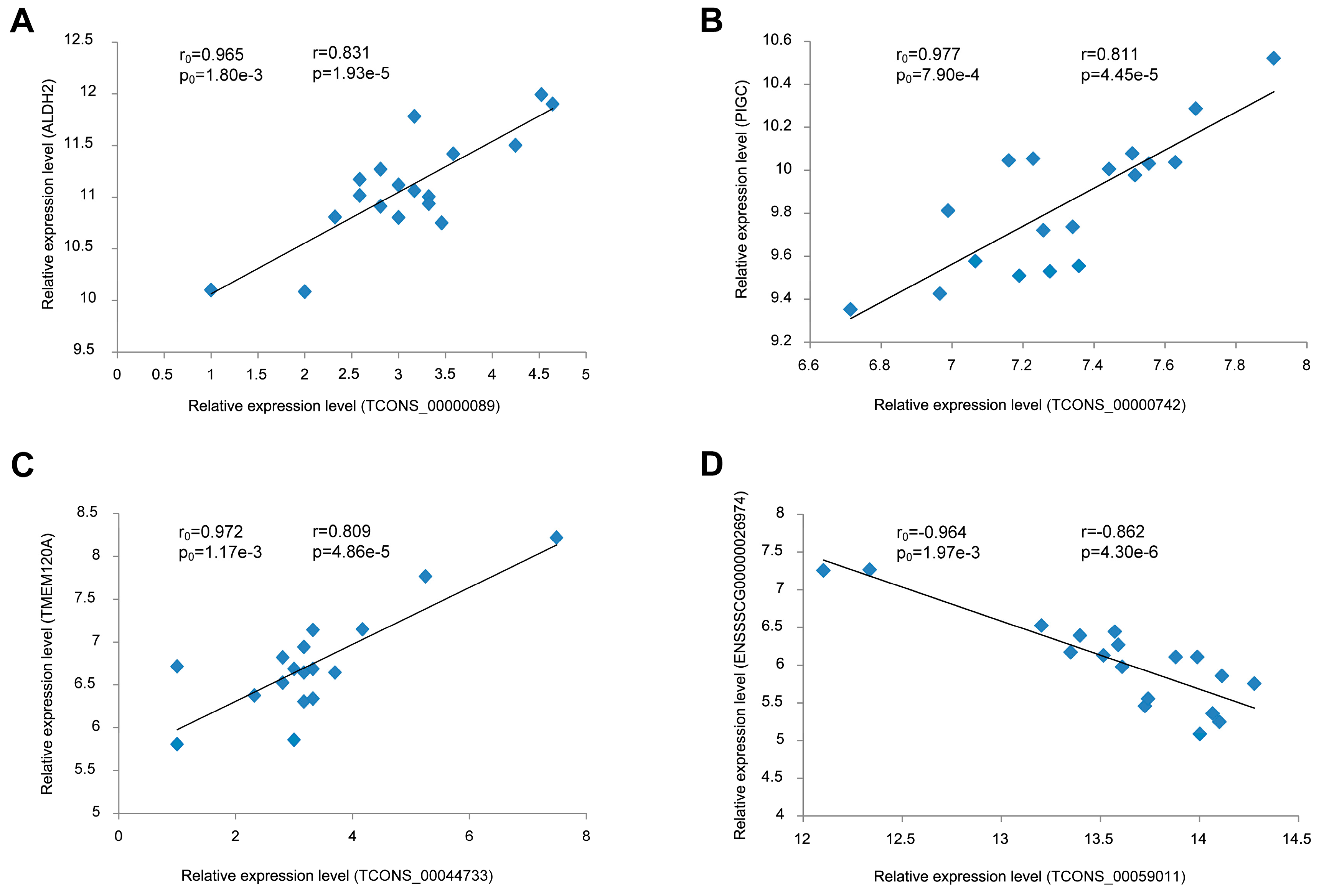
3.7. Validation of the Correlation between lincRNAs and Their Potential Target Genes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lefaucheur, L.; Ecolan, P.; Plantard, L.; Gueguen, N. New insights into muscle fiber types in the pig. J. Histochem. Cytochem. 2002, 50, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Zhou, J.; Liu, L.Q.; Qian, K.; Wang, C.L. Identification of genes in longissimus dorsi muscle differentially expressed between Wannanhua and Yorkshire pigs using RNA-sequencing. Anim. Genet. 2016, 47, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Patel, K. Skeletal muscle fibre plasticity in response to selected environmental and physiological stimuli. Histol. Histopathol. 2009, 24, 611–629. [Google Scholar] [PubMed]
- Ozawa, S.; Mitsuhashi, T.; Mitsumoto, M.; Matsumoto, S.; Itoh, N.; Itagaki, K.; Kohno, Y.; Dohgo, T. The characteristics of muscle fiber types of longissimus thoracis muscle and their influences on the quantity and quality of meat from Japanese black steers. Meat Sci. 2000, 54, 65–70. [Google Scholar] [CrossRef]
- Lee, S.H.; Joo, S.T.; Ryu, Y.C. Skeletal muscle fiber type and myofibrillar proteins in relation to meat quality. Meat Sci. 2010, 86, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Larzul, C.; Lefaucheur, L.; Ecolan, P.; Gogue, J.; Talmant, A.; Sellier, P.; Le Roy, P.; Monin, G. Phenotypic and genetic parameters for longissimus muscle fiber characteristics in relation to growth, carcass, and meat quality traits in large white pigs. J. Anim. Sci. 1997, 75, 3126–3137. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qian, K.; Wang, C. Discovery of porcine miRNA-196a/b may influence porcine adipogenesis in longissimus dorsi muscle by miRNA sequencing. Anim. Genet. 2017, 48, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, X.; Zhang, P.J.; Xu, Y. Characterization of muscle-regulatory gene, MyoD, from flounder (Paralichthys olivaceus) and analysis of its expression patterns during embryogenesis. Mar. Biotechnol. 2006, 8, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Fang, Q.; Yin, J.; Li, D.; Li, W. S-adenosylmethionine stimulates fatty acid metabolism-linked gene expression in porcine muscle satellite cells. Mol. Biol. Rep. 2010, 37, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Cao, J.H.; Li, X.Y.; Zhao, S.H. Inhibition of miR-214 expression represses proliferation and differentiation of C2C12 myoblasts. Cell Biochem. Funct. 2011, 29, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, Z.; Li, X.; Ning, X.; Li, Y.; Yang, G. Biological role of microRNA-103 based on expression profile and target genes analysis in pigs. Mol. Biol. Rep. 2011, 38, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Prickett, A.R.; Oakey, R.J. A survey of tissue-specific genomic imprinting in mammals. Mol. Genet. Genom. 2012, 287, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Mu, Y.; Ma, L.; Wang, C.; Tang, Z.; Yang, S.; Zhou, R.; Hu, X.; Li, M.H.; Li, K. Systematic identification and characterization of long intergenic non-coding RNAs in fetal porcine skeletal muscle development. Sci. Rep. 2015, 5, 8957. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Wang, Y.; Xu, R.X.; Wang, G.Q.; Xiong, Y.; Yu, T.Y.; Yang, G.S.; Pang, W.J. PU.1 antisense lncRNA against its mRNA translation promotes adipogenesis in porcine preadipocytes. Anim. Genet. 2015, 46, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ensembl Database. Available online: ftp://ftp.ensembl.org/pub/release-87/gtf/sus_scrofa (accessed on 8 December 2016).
- Luo, H.; Sun, S.; Li, P.; Bu, D.; Cao, H.; Zhao, Y. Comprehensive characterization of 10,571 mouse large intergenic noncoding RNAs from whole transcriptome sequencing. PLoS ONE 2013, 8, e70835. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Li, A.M.; Adeola, A.C.; Liu, Y.H.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Genome-Wide identification of long intergenic noncoding RNA genes and their potential association with domestication in pigs. Genome Biol. Evol. 2014, 6, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Available online: ftp://ftp.ncbi.nih.gov/blast/db/ (accessed on 17 August 2016).
- Ensembl Database. Available online: ftp://ftp.ensembl.org/pub/release-89/fasta/homo_sapiens/ncrna/ (accessed on 7 May 2017).
- Ensembl Database. Available online: ftp://ftp.ensembl.org/pub/release-89/fasta/mus_musculus/ncrna/ (accessed on 7 May 2017).
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. Cpc: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, X.; Li, Q. Genome-Wide identification and characterization of long intergenic noncoding RNAs and their potential association with larval development in the Pacific oyster. Sci. Rep. 2016, 6, 20796. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fu, L.; Koganti, P.P.; Wang, L.; Hand, J.M.; Ma, H.; Yao, J. Identification and functional prediction of large intergenic noncoding RNAs (lincRNAs) in rainbow trout (Oncorhynchus mykiss). Mar. Biotechnol. 2016, 18, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-Scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Ensembl Database. Available online: http://asia.ensembl.org/biomart/martview/265b18e4080d32cc7326dc26cfc3bdf0 (accessed on 7 May 2017).
- Pilcher, C.M.; Jones, C.K.; Schroyen, M.; Severin, A.J.; Patience, J.F.; Tuggle, C.K.; Koltes, J.E. Transcript profiles in longissimus dorsi muscle and subcutaneous adipose tissue: A comparison of pigs with different postweaning growth rates. J. Anim. Sci. 2015, 93, 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Kwenda, S.; Birch, P.R.; Moleleki, L.N. Genome-Wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genom. 2016, 17, 614. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, Z.; Wang, X.; Liu, H.; Zhang, Y.; Liu, Z. Identification and functional analysis of long intergenic noncoding RNA genes in porcine pre-implantation embryonic development. Sci. Rep. 2016, 6, 38333. [Google Scholar] [CrossRef] [PubMed]
- Billerey, C.; Boussaha, M.; Esquerre, D.; Rebours, E.; Djari, A.; Meersseman, C.; Klopp, C.; Gautheret, D.; Rocha, D. Identification of large intergenic non-coding RNAs in bovine muscle using next-generation transcriptomic sequencing. BMC Genom. 2014, 15, 499. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, S.L.; Dowell, R.D.; Grisafi, P.; Gifford, D.K.; Fink, G.R. Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc. Natl. Acad. Sci. USA 2009, 106, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Rapicavoli, N.A.; Poth, E.M.; Zhu, H.; Blackshaw, S. The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev. 2011, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Fan, C.; Cheng, T.; Su, Y.; Wei, Q.; Li, G. Genome-Wide identification, characterization and evolutionary analysis of long intergenic noncoding RNAs in cucumber. PLoS ONE 2015, 10, e0121800. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA hotair reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Bean, C.; Facchinello, N.; Faulkner, G.; Lanfranchi, G. The effects of Ankrd2 alteration indicate its involvement in cell cycle regulation during muscle differentiation. Biochim. Biophys. Acta 2008, 1783, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.S.; Lopez, M.A.; Cox, G.A.; Boriek, A.M. Ankyrin repeat domain protein 2 and inhibitor of DNA binding 3 cooperatively inhibit myoblast differentiation by physical interaction. J. Biol. Chem. 2013, 288, 24560–24568. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Miskovic, D.; Tonouchi, M.; Lemieux, K.; Wilson, M.C.; Marette, A.; Halestrap, A.P. Abundance and subcellular distribution of MCT1 and MCT4 in heart and fast-twitch skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1067–E1077. [Google Scholar] [PubMed]
- Wilson, M.C.; Jackson, V.N.; Heddle, C.; Price, N.T.; Pilegaard, H.; Juel, C.; Bonen, A.; Montgomery, I.; Hutter, O.F.; Halestrap, A.P. Lactic acid efflux from white skeletal muscle is catalyzed by the monocarboxylate transporter isoform MCT3. J. Biol. Chem. 1998, 273, 15920–15926. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.R.; Swatland, H.J. A review of the relationships of pH with physical aspects of pork quality. Meat Sci. 1988, 24, 85–126. [Google Scholar] [CrossRef]
- Pilegaard, H.; Terzis, G.; Halestrap, A.; Juel, C. Distribution of the lactate/H+ transporter isoforms MCT1 and MCT4 in human skeletal muscle. Am. J. Physiol. 1999, 276, E843–E848. [Google Scholar] [PubMed]
- Choe, J.H.; Choi, Y.M.; Lee, S.H.; Shin, H.G.; Ryu, Y.C.; Hong, K.C.; Kim, B.C. The relation between glycogen, lactate content and muscle fiber type composition, and their influence on postmortem glycolytic rate and pork quality. Meat Sci. 2008, 80, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Oh, W.; Kordari, P.; Wakil, S.J. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. USA 2003, 100, 10207–10212. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Accession Number | Raw Reads | Clean Reads | Mapped Reads | Mapping Ratio |
|---|---|---|---|---|---|
| WH03 | SRR2919657 | 64,584,126 | 61,821,920 | 49,030,410 | 79.3% |
| WH05 | SRR2919658 | 64,327,738 | 61,484,062 | 48,584,612 | 79.0% |
| WH12 | SRR2919659 | 59,244,502 | 56,584,368 | 43,757,304 | 77.3% |
| YY01 | SRR2919660 | 57,969,134 | 55,324,162 | 41,665,620 | 75.3% |
| YY02 | SRR2919661 | 68,254,168 | 64,955,554 | 49,480,266 | 76.2% |
| YY06 | SRR2919662 | 72,298,142 | 68,884,390 | 50,825,220 | 73.8% |
| DELs | Number | DELs | Number | ||||
|---|---|---|---|---|---|---|---|
| DEPTGs | UpRegulated PTGs | DownRegulated PTGs | DEPTGs | UpRegulated PTGs | DownRegulated PTGs | ||
| TCONS_00000089 | 41 | 34 | 7 | TCONS_00021915 | 71 | 27 | 44 |
| TCONS_00000134 | 4 | 4 | 0 | TCONS_00034774 | 4 | 4 | 0 |
| TCONS_00000742 | 53 | 37 | 16 | TCONS_00044733 | 80 | 76 | 4 |
| TCONS_00005771 | 7 | 7 | 0 | TCONS_00056305 | 5 | 5 | 0 |
| TCONS_00006963 | 31 | 26 | 5 | TCONS_00059011 | 14 | 3 | 11 |
| TCONS_00007946 | 46 | 42 | 4 | TCONS_00061360 | 110 | 79 | 31 |
| TCONS_00013076 | 3 | 2 | 1 | TCONS_00061633 | 13 | 13 | 0 |
| TCONS_00021480 | 12 | 11 | 1 | TCONS_00064940 | 11 | 10 | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, C.; Li, S.; Deng, L.; Guan, Y.; Chen, D.; Yuan, X.; Xia, T.; He, X.; Shan, Y.; Li, C. Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig. Genes 2017, 8, 203. https://doi.org/10.3390/genes8080203
Zou C, Li S, Deng L, Guan Y, Chen D, Yuan X, Xia T, He X, Shan Y, Li C. Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig. Genes. 2017; 8(8):203. https://doi.org/10.3390/genes8080203
Chicago/Turabian StyleZou, Cheng, Sha Li, Lulu Deng, Yang Guan, Dake Chen, Xiongkun Yuan, Tianrui Xia, Xinglin He, Yawei Shan, and Changchun Li. 2017. "Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig" Genes 8, no. 8: 203. https://doi.org/10.3390/genes8080203
APA StyleZou, C., Li, S., Deng, L., Guan, Y., Chen, D., Yuan, X., Xia, T., He, X., Shan, Y., & Li, C. (2017). Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig. Genes, 8(8), 203. https://doi.org/10.3390/genes8080203