Comprehensive Transcriptome Profiling and Identification of Potential Genes Responsible for Salt Tolerance in Tall Fescue Leaves under Salinity Stress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth Conditions
2.2. Salt Treatment
2.3. Total RNA Extraction and Library Construction
2.4. Sequencing Data Filtering and De Novo Assembly
2.5. Unigene Annotation and Coding DNA Sequence Forecast
2.6. Unigene’s Transcription Factors Coding Capacity Prediction, Simple Sequence Repeats, and Single Nucleotide Polymorphism Test
2.7. Unigene Expression Calculation and Differentially Expressed Gene (DEG) Detection
2.8. Functional Analysis of Differentially Expressed Genes
2.9. qPCR Validation
3. Results
3.1. Sequencing Statistics
3.2. De Novo Assembly and Unigene Annotation
3.3. Unigene’s Transcription Factors Coding Capacity Prediction and Unigene’s Coding DNA Sequence Forecast
3.4. Unigene’s SSR and SNPs Test
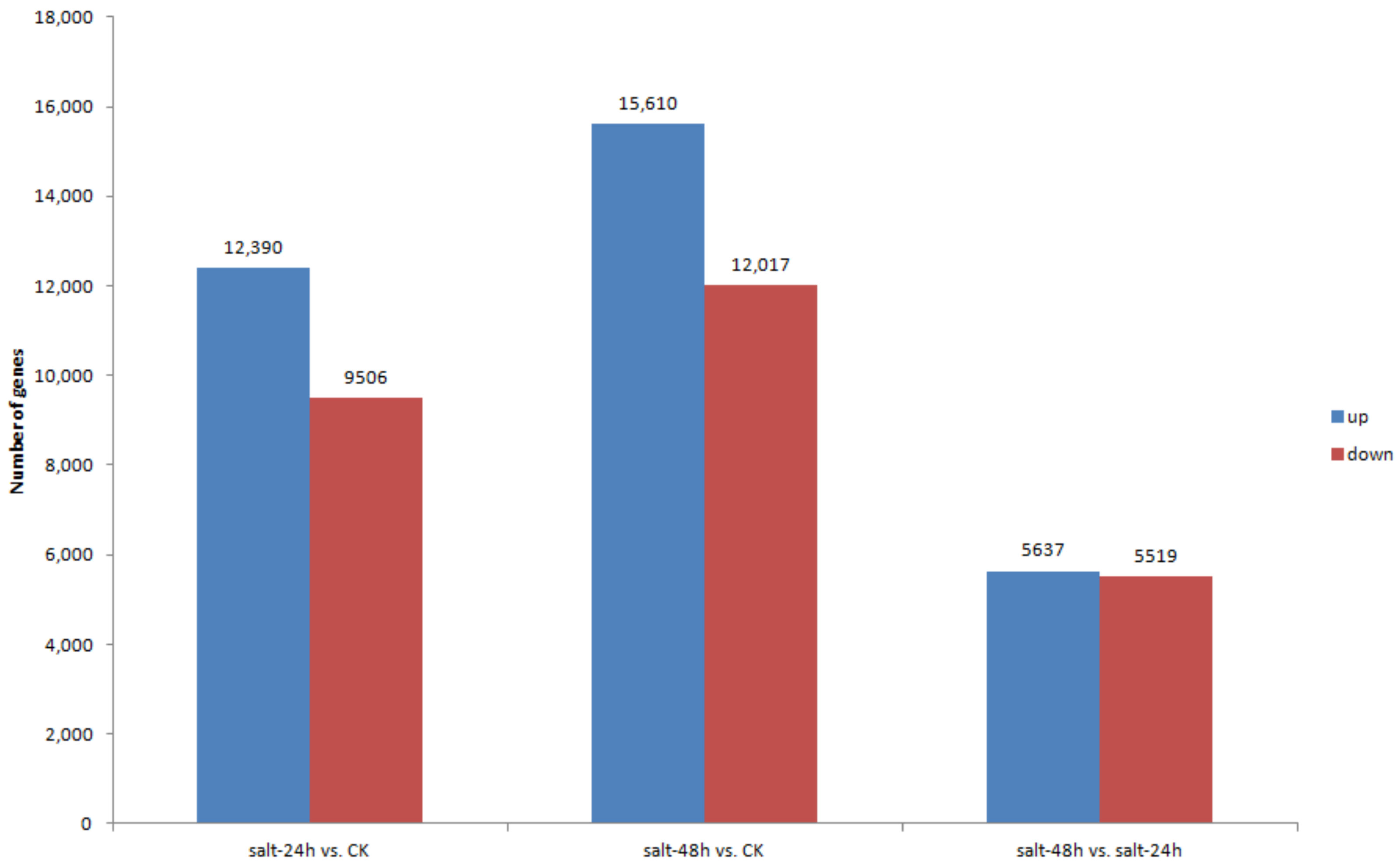
3.5. Differential Gene Expression and Distribution in Samples
3.6. Functional Analysis of Differentially Expressed Genes
3.7. Differential Protein Interaction Analysis
3.8. qPCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheeseman, J.M. Mechanisms of salinity tolerance in plants. Plant Physiol. 1988, 87, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Munns, R. Comparative physiology of salt and water stress. Plant Cell Environ. 2002, 25, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Du, Z.; Ren, J.; Amombo, E.; Hu, T.; Fu, J. Association of SSR markers with functional traits from heat stress in diverse tall fescue accessions. BMC Plant Biol. 2015, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Alshammary, S.F.; Qian, Y.L.; Wallner, S.J. Growth response of four turfgrass species to salinity. Agric. Water Manag. 2004, 66, 97–111. [Google Scholar] [CrossRef]
- Watkins, E.; Fei, S.; Gardner, D.; Stier, J.; Bughrara, S.; Li, D.; Bigelow, C.; Schleicher, L.; Horgan, L.; Diesburg, K. Low-input turfgrass species for the north-central United States. Appl. Turfgrass Sci. 2011, 8. [Google Scholar] [CrossRef]
- Amombo, E.; Li, H.; Fu, J. Research advances on tall fescue salt tolerance: From root signaling to molecular and metabolic adjustment. J. Am. Soc. Hortic. Sci. 2017, 142, 337–345. [Google Scholar] [CrossRef]
- Gao, Y.; Li, D.; Chen, Y. Differentiation of carbonate, chloride, and sulfate salinity responses in tall fescue. Sci. Hortic. 2012, 139, 1–7. [Google Scholar] [CrossRef]
- Gao, Y.; Li, D. Growth responses of tall fescue (Festuca arundinacea Schreb.) to salinity stress. Eur. J. Hortic. Sci. 2014, 79, 123–128. [Google Scholar]
- Yin, L.; Ren, M.; Wei, M.; Wu, L.; Zhou, Y.; Li, X.; Gao, Y. Neotyphodium coenophialum-infected tall fescue and its potential application in the phytoremediation of saline soils. Int. J. Phytoremediation 2014, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Nakazawa, R. Effects of the timing of calcium application on the alleviation of salt stress in the maize, tall fescue, and reed canarygrass seedlings. Biol. Plant. 2008, 52, 153–156. [Google Scholar] [CrossRef]
- Mian, M.R.; Zhang, Y.; Wang, Z.; Zhang, J.Y.; Cheng, X.; Chen, L.; Chekhovskiy, K.; Dai, X.; Mao, C.; Cheung, F.; et al. Analysis of tall fescue ESTs representing different abiotic stresses, tissue types and developmental stages. BMC Plant Biol. 2008, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.C.; Glover-Cutter, K.; Baldwin, J.C.; Dombrowski, J.E. Identification and characterization of a salt stress-inducible zinc finger protein from Festuca arundinacea. BMC Res. Notes 2012, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, D.; Xue, Z.; Liu, H.; Xia, G. Enhanced salt tolerance of transgenic progeny of tall fescue (Festuca arundinacea) expressing a vacuolar Na+/H+ antiporter gene from Arabidopsis. J. Plant Physiol. 2007, 164, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.M.; Xu, W.R.; Li, H.W.; Jin, F.X.; Guo, L.N.; Wang, J.; Da, H.J.; Xu, X. Co-expression of the Arabidopsis SOS genes enhances salt tolerance in transgenic tall fescue (Festuca arundinacea Schreb.). Protoplasma 2014, 251, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, J.; Zhang, Y.; Fan, F.; Li, W.; Wang, F.; Zhong, W.; Wang, C.; Yang, J. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of Indica rice at seedling stage. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Mohanty, B.; Wijaya, E.; Lee, D.Y.; Lim, T.M.; Lin, Q.; Xu, J.; Loh, C.S.; Kumar, P.P. Transcriptomics analysis of salt stress tolerance in the roots of the mangrove Avicennia officinalis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, Q.; Lin, Q.; Tao, C.; Zhu, C.; Xing, S.; Fan, Y.; Liu, W.; Yan, Y.; Li, J.; et al. Transcriptomic characterization of candidate genes responsive to salt tolerance of Miscanthus energy crops. Bioenergy 2017, 9, 1222–1237. [Google Scholar] [CrossRef]
- Upadhyaya, A.; Gaonkara, T.; Upadhyay, A.K.; Jogaiaha, S.; Shinde, M.P.; Kadoo, N.Y.; Gupta, N.S. Global transcriptome analysis of grapevine (Vitis vinifera L.) leaves under salt stress reveals differential response at early and late stages of stress in table grape cv. Thompson Seedless. Plant Physiol. Biochem. 2018, 129, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xu, X.; Zhu, H.; Liu, A.; Liu, L.; Li, J.; Hua, H. Comparative transcriptomic profiling of a salt-tolerant wild tomato species and a salt-sensitive tomato cultivar. Plant Cell Physiol. 2010, 51, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Choi, J.; An, G.; Kim, S.R. Ectopic expression of OsSta2 enhances salt stress tolerance in rice. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Lee, W.K.; Ali, A.; Kumar, M.; Yang, T.J.; Song, K. Genome-wide identification and classification of the AP2/EREBP gene family in the Cucurbitaceae species. Plant Breed. Biotechnol. 2017, 5, 123–133. [Google Scholar] [CrossRef]
- Lee, S.C.; Lee, W.K.; Ali, A.; Kumar, M.; Yang, T.J.; Kihwan, S. Genome-wide identification of the dehydrin genes in the Cucurbitaceae species. Plant Breed. Biotechnol. 2017, 5, 282–292. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, X.; Wang, W.; Pan, Y.; Huang, L.; Liu, X.; Zong, Y.; Zhu, L.; Yang, D.; Fu, B. Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla, P.I.; Maiale, S.J.; Ruiz, O.A.; Escaray, F.J. Transcriptome response mediated by cold stress in Lotus japonicas. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lee, S.C.; Kim, J.Y.; Kim, S.J.; Aye, S.S.; Kim, S.R. Over-expression of dehydrin gene, OsDhn1, improves drought and salt stress tolerance through scavenging of reactive oxygen species in rice (Oryza sativa L.). J. Plant Biol. 2014, 57, 383–393. [Google Scholar] [CrossRef]
- Singh, D.; Kumar, C.K.; Taunk, J.; Tomar, R.S.S.; Chaturvedi, A.K.; Gaikwad, K.; Pal, M. Transcriptome analysis of lentil (Lensculinaris medikus) in response to seedling drought stress. BMC Genom. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shukla, V.; Merewitz, E.B. Transcriptome analysis of creeping bentgrass exposed to drought stress and polyamine treatment. PLoS ONE 2017, 12, e0175848. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Kim, H.C.; Shin, S.; Kim, K.H.; Moon, J.C.; Kim, J.Y.; Lee, B.M. Transcriptome analysis of flowering time genes under drought stress in maize leaves. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.N.; Zhang, L.; Yang, L.; Rabiul, M.D.; Liu, Y.; Luo, H.; Yang, P.F.; Wang, Q.F.; Chan, Z.L. Transcriptomic profiling of tall fescue in response to heat stress and improved thermotolerance by melatonin and 24-epibrassinolide. BMC Genom. 2018, 19, 224. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, S.S.; Guimarães, F.C.; Carvalho, J.F.; Stolf-Moreira, R.; Oliveira, M.C.; Rolla, A.A.; Farias, J.R.; Neumaier, N.; Nepomuceno, A.L. Transcription factors expressed in soybean roots under drought stress. Genet. Mol. Res. 2011, 10, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.J.; García-Gómez, M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization, and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The universal protein resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 9, D1114–D1117. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucl. Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 2, 357–360. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenki, A.; Cibulskis, K.; Kerutsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Robin, A.H.; Yi, G.E.; Laila, R.; Yang, K.; Park, J.I.; Kim, H.R.; Nou, I.S. Expression profiling of glucosinolate biosynthetic genes in Brassica oleracea L. var. capitate inbred lines reveals their association with glucosinolate content. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Peng, Z.; He, S.; Gong, W.; Sun, J.; Pan, Z.; Xu, F.; Lu, Y.; Du, X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genom. 2014, 15, 760. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Frugis, G.; Colgan, D.F.; Chua, N. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Mieulet, D.; Hubberten, H.M.; Obata, T.; Hoefgen, R.; Fernie, A.R.; Fisahn, J.; San Segundo, B.; Guiderdoni, E.; Schippers, J.H.; et al. Salt-responsive ERF1 regulates reactive oxygen species-dependent signaling during the initial response to salt stress in rice. Plant Cell 2013, 6, 2115–2131. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Cai, H.; Zhai, H.; Luo, Z.; Wang, Z.; Cui, L.; Bai, X. Overexpression of Glycine soja WRKY20 enhances both drought and salt tolerance in transgenic alfalfa (Medicago sativa L.). Plant Cell Tissue Organ Cult. 2014, 118, 77–86. [Google Scholar] [CrossRef]
- Ding, Z.J.; Yan, J.Y.; Li, C.X.; Li, G.X.; Wu, Y.R.; Zheng, S.J. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 2015, 84, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Guo, A.; Jin, X.; Yang, Q.; Wang, D.; Sun, Y.; Huang, Q.; Wang, L.; Peng, C.; Wang, X. The β subunit of glyceraldehyde 3-phosphate dehydrogenase is an important factor for maintaining photosynthesis and plant development under salt stress—based on an integrative analysis of the structural, physiological and proteomic changes in chloroplasts in Thellungiella halophila. Plant Sci. 2015, 236, 223–238. [Google Scholar] [PubMed]
- Zhao, F.; Zhang, H. Salt and paraquat stress tolerance results from co-expression of the Suaeda salsa glutathione S-transferase and catalase in transgenic rice. Plant Cell Tissue Organ Cult. 2006, 86, 349–358. [Google Scholar] [CrossRef]
- Sofa, A.; Scopa, A.; Nuzzaci, M.; Vitti, A. Ascorbate Peroxidase and Catalase Activities and Their Genetic Regulation in Plants Subjected to Drought and Salinity Stresses. Int. J. Mol. Sci. 2015, 16, 13561–13578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustroph, A.; Stock, J.; Hess, N.; Aldous, S.; Dreilich, A.; Grimm, B. Characterization of the phosphofructokinase gene family in rice and its expression under oxygen deficiency stress. Front. Plant Sci. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Naested, H.; Holm, A.; Jenkins, T.; Nielsen, H.B.; Harris, C.A.; Beale, M.H.; Andersen, M.; Mant, A.; Scheller, H.; Camara, B.; et al. Arabidopsis VARIEGATED3 encodes a chloroplast-targeted, zinc-finger protein required for chloroplast and palisade cell development. J. Cell Sci. 2004, 117, 4807–4818. [Google Scholar] [CrossRef] [PubMed]
- Lamppa, G.K.; Morelli, G.; Chua, N.H. Structure and developmental regulation of a wheat gene encoding the major chlorophyll a/b-binding polypeptide. Mol. Cell. Biol. 1985, 5, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Kaufman, L.S. Blue-light regulation of the Arabidopsis thaliana Cab1 Gene. Plant Physiol. 1994, 104, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Ramanathan, V.; Nallathambi, J.; Duraialagaraja, S.; Muthurajan, R. Over-expression of a NAC 67 transcription factor from finger millet (Eleusine coracana L.) confers tolerance against salinity and drought stress in rice. BMC Biotechnol. 2016, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.C.; Cramer, G.R.; Devitt, D.A. Effect of salinity and nitrogen status on nitrogen uptake by tall fescue turf. J. Plant Nutr. 2006, 29, 1481–1490. [Google Scholar] [CrossRef]
- Talukder, S.K.; Azhaguvel, P.; Mukherjee, S.; Young, C.; Tang, Y.; Krom, N.; Saha, M.C. De novo assembly and characterization of tall fescue transcriptome under water stress. Plant Genome 2015, 2, 1–13. [Google Scholar] [CrossRef]
- Hu, T.; Sun, X.; Zhang, X.; Nevo, A.; Fu, J. An RNA sequencing transcriptome analysis of the high-temperature stressed tall fescue reveals novel insights into plant thermotolerance. BMC Genom. 2014, 15, 1147. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, T.; Amombo, E.; Fu, J. Transcriptome profiling of two tall fescue (Festuca arundinacea) cultivars in response to lead (Pb) stress. BMC Genom. 2017, 18, 145. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Munawar, J.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, J.; Chen, H.; Wang, Z.; Gu, X.; Wei, C.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X. Exogenous melatonin confers salt stress tolerance to watermelon by improving photosynthesis and redox homeostasis. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Cavaliere, C.; Guarino, C.; Gubbiotti, R.; Foglia, P.; Laganà, A. Identification of changes in Triticum durum L. leaf proteome in response to salt stress by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Fatehi, F.; Hosseinzadeh, A.; Alizadeh, H.; Brimavandi, T.; Struik, P.C. The proteome response of salt-resistant and salt-sensitive barley genotypes to long-term salinity stress. Mol. Biol. Rep. 2012, 39, 6387–6397. [Google Scholar] [CrossRef] [PubMed]
- Rasoulnia, A.; Bihamta, M.R.; Peyghambari, S.A.; Alizadeh, H.; Rahnama, A. Proteomic response of barley leaves to salinity. Mol. Biol. Rep. 2011, 38, 5055–5063. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Rakwal, R.; Agrawal, G.K.; Jung, Y.H.; Shibato, J.; Jwa, N.S.; Iwahashi, Y.; Iwahashi, H.; Kim, D.H.; Shim, I.S.; et al. A hydroponic rice seedling culture model system for investigating proteome of salt stress in rice leaf. Electrophoresis 2005, 26, 4521–4539. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (Mb) | Total Clean Reads (Mb) | Total Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|
| CK | 87.16 | 66.65 | 6.67 | 96.48 | 89.35 | 76.47 |
| Salt-24h | 85.915 | 65.965 | 6.595 | 96.41 | 89.175 | 76.8 |
| Salt-48h | 88.405 | 65.825 | 6.585 | 96.315 | 89.01 | 74.47 |
| Sample | Total Number | Total Length | Mean Length | N50 | N70 | N90 | GC (%) |
|---|---|---|---|---|---|---|---|
| CK (a) | 150,967 | 98,232,067 | 650 | 1034 | 598 | 261 | 50.23 |
| Salt-24h (a) | 152,598.5 | 98,743,637 | 646 | 1024 | 592.5 | 261 | 50.395 |
| Salt-48h (a) | 152,316.5 | 99,403,369 | 651.5 | 1040 | 603 | 261.5 | 50.035 |
| CK (b) | 86,828 | 63,981,634 | 736 | 1147 | 679 | 301 | 49.69 |
| Salt-24 (b) | 88,639.5 | 65,201,778 | 734.5 | 1137.5 | 677 | 303 | 49.875 |
| Salt-48 (b) | 87,520.5 | 64,836,088 | 740 | 1152 | 689 | 303 | 49.5 |
| All-Unigene (b) | 144,339 | 1.3 × 108 | 898 | 1443 | 912 | 367 | 49.49 |
| Values | Total | Nr | Nt | SwissProt | KEGG | KOG | Interpro | GO | Intersection | Overall |
|---|---|---|---|---|---|---|---|---|---|---|
| Number | 144,339 | 74,388 | 62,387 | 41,836 | 48,083 | 47,776 | 45,601 | 40,017 | 17,928 | 83,213 |
| Percentage | 100% | 51.54% | 43.22% | 28.98% | 33.31% | 33.10% | 31.59% | 27.72% | 12.42% | 57.65% |
| Sample | A–G | C–T | Transition | A–C | A–T | C–G | G–T | Transversion | Total |
|---|---|---|---|---|---|---|---|---|---|
| CK | 105,203 | 104,656 | 209,859 | 28,471 | 20,647 | 39,544 | 28,720 | 117,382 | 327,241 |
| Salt-24h | 93,988 | 93,312.5 | 187,300.5 | 25,432 | 18,548.5 | 35,876 | 25,632 | 105,488.5 | 292,789 |
| Salt-48h | 95,195 | 94,725.5 | 189,920.5 | 25,844.5 | 19,188.5 | 35,741.5 | 26,083.5 | 106,858 | 296,778.5 |
| Sample | Total Bases | Total Reads | Total Mapped Reads | Unique Mapped Reads |
|---|---|---|---|---|
| CK | 6,665,213,000 | 66,652,130 | 50,987,376 | 21,680,100 |
| Salt-24h | 6,596,346,200 | 65,963,462 | 51,527,965 | 21,538,592 |
| Salt-48h | 6,582,573,600 | 65,825,736 | 50,782,868 | 21,848,553 |
| Unigene | log2 Fold Change (Salt-48h/Salt-24h) | Gene name | Function | Reference |
|---|---|---|---|---|
| CL19340.Contig2_All | 1.048 | NAC021 | Salt tolerance | [53,54] |
| CL914.Contig6_All | 8.247 | ERF1 | ROS signaling | [55] |
| CL5384.Contig1_All | 2.098 | WRKY20 | Salt tolerance | [56] |
| CL12389.Contig1_All | 2.228 | WRKY46 | Osmotic stress response | [57] |
| CL8335.Contig1_All | 1.3 | GAPC | Photosystem repair and salt tolerance | [58] |
| CL342.Contig3_All | 1.1 | CAT1 | Response to oxidative stress | [59] |
| CL16806.Contig2_All | 1.7 | APX2 | Response to oxidative stress | [60] |
| Unigene27613_All | 1.5 | PFK6 | Fructose 6-phosphate metabolic process; | [61] |
| Unigene12326_All | 2.3 | VAR3 | Chloroplast development | [62] |
| CL1733.Contig12_All | 1.4 | WHAB1.6 | Photosynthesis, light harvesting in photosystem I | [63] |
| CL1733.Contig11_All | 1.3 | CAB1 | Photosynthesis, light harvesting in photosystem I | [64] |
| Unigene16967_All | 2.362 | NAC67 | Salt tolerance | [65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amombo, E.; Li, X.; Wang, G.; An, S.; Wang, W.; Fu, J. Comprehensive Transcriptome Profiling and Identification of Potential Genes Responsible for Salt Tolerance in Tall Fescue Leaves under Salinity Stress. Genes 2018, 9, 466. https://doi.org/10.3390/genes9100466
Amombo E, Li X, Wang G, An S, Wang W, Fu J. Comprehensive Transcriptome Profiling and Identification of Potential Genes Responsible for Salt Tolerance in Tall Fescue Leaves under Salinity Stress. Genes. 2018; 9(10):466. https://doi.org/10.3390/genes9100466
Chicago/Turabian StyleAmombo, Erick, Xiaoning Li, Guangyang Wang, Shao An, Wei Wang, and Jinmin Fu. 2018. "Comprehensive Transcriptome Profiling and Identification of Potential Genes Responsible for Salt Tolerance in Tall Fescue Leaves under Salinity Stress" Genes 9, no. 10: 466. https://doi.org/10.3390/genes9100466