Abstract
Leucine-rich repeat receptor-like kinases (LRR-RLKs) have been reported to play important roles in plant growth, development, and stress responses. However, no comprehensive analysis of this family has been performed in cotton (Gossypium spp.), which is an important economic crop that suffers various stresses in growth and development. Here we conducted a comprehensive analysis of LRR-RLK family in four Gossypium species (Gossypium arboreum, Gossypium barbadense, Gossypium hirsutum, and Gossypium raimondii). A total of 1641 LRR-RLK genes were identified in the four Gossypium species involved in our study. The maximum-likelihood phylogenetic tree revealed that all the LRR-RLK genes were divided into 21 subgroups. Exon-intron organization structure of LRR-RLK genes kept relatively conserved within subfamilies and between Arabidopsis and Gossypium genomes. Notably, subfamilies XI and XII were found dramatically expanded in Gossypium species. Tandem duplication acted as an important mechanism in expansion of the Gossypium LRR-RLK gene family. Functional analysis suggested that Gossypium LRR-RLK genes were enriched for plant hormone signaling and plant-pathogen interaction pathways. Promoter analysis revealed that Gossypium LRR-RLK genes were extensively regulated by transcription factors (TFs), phytohormonal, and various environmental stimuli. Expression profiling showed that Gossypium LRR-RLK genes were widely involved in stress defense and diverse developmental processes including cotton fiber development and provides insight into potential functional divergence within and among subfamilies. Our study provided valuable information for further functional study of Gossypium LRR-RLK genes.
1. Introduction
Receptor-like protein kinases (RLKs) represent a large number of transmembrane kinases which perceive stimulus at the cellular surface and mediate the cellular signaling transduction via autophosphorylation and subsequent downstream phosphorylation for intercellular communication or response to the extracellular environment [1,2]. In land plants, RLKs form a large family and expand extensively [3,4,5]. Commonly, RLKs have an N-terminal extracellular domain (ECD) that varies in structure, a transmembrane domain (TM), and a relatively conserved cytoplasmic protein kinase catalytic domain (KD) [6]. The ECD region, which is thought to act as a ligand-binding site, has a variety of structural features, allowing it to interact with proteins, polysaccharides, lipids, and other ligands [4,7]. The leucine-rich repeat RLKs (LRR-RLKs) comprise the largest group of plant RLKs [7,8], which contain a varying number of leucine-rich repeat (LRR) kinases in the ECD region. The LRR is a 20–30 amino acid residue sequence motif, and appears to provide the structural framework for recognition of ligands [9]. The number and arrangement pattern of LRRs may vary among different LRR-RLKs, partly contributing to the diversity of LRR-RLKs. A comprehensive LRR-RLK analysis among a diversity of plants lineages classified plant LRR-RLKs into 19 subfamilies, and phylogenetic analysis demonstrated much of the diversity of plant LRR-RLKs was established in early land plants [10].
In plants, LRR-RLK genes play various important roles in plant development and response to biotic and abiotic stresses [10]. In terms of plant growth and development, the best characterized LRR-RLK gene member is CLAVATA1 (CLV1) in Arabidopsis, which is involved in the development of shoot and flower apical meristem. By combining with receptor-like protein CLV2, the LRR-RLK CLV1 is dimerized and recognizes small secreted dodecapeptide CLV3 as the ligand to regulate expression of the downstream transcription factor WUSCHEL (WUS), which in return up-regulates the expression of CLV3, resulting in a feedback mechanism to adjust the meristem size [11,12,13]. The heterodimeric LRR-RLK complex BAK1/BRI1 initiates the brassinosteroid signaling cascade [14,15]. The one LRR-RLK gene HAESA (HAE) in Arabidopsis is involved in floral organ abscission [16], and RPK1 and TOAD2 encode LRR-RLKs required for proper embryo morphogenesis [17]. In addition, some LRR-RLKs are defense related. For instance, Arabidopsis FLS2 and EFR perceive bacterial antigens and mediate the defense against pathogens [18,19]. The rice LRR-RLK gene Xa21 is an effective rice bacterial blight resistance gene [20]. As the metabolic pathway intertwines, some LRR-RLKs function on several aspects. For instance, somatic embryogenesis receptor-like kinases (SERKs) participate in the process of microsporogenesis and embryogenesis, and enhance acquisition of embryogenic competence in culture regeneration [21,22,23]. Meanwhile, recent research demonstrates that SERKs are indispensable in brassinosteroid signaling [24,25,26], including the rice OsSERK1 (encoded by the first identified SERK gene in rice) host defense response against fungal infection by mediating defense signaling transduction [23].
As LRR-RLKs have many functional roles, genome-wide identification and analysis of LRR-RLK genes has been carried out extensively. Based on KD sequence phylogeny and gene structure, at least 213 identified LRR-RLK genes in Arabidopsis thaliana were classified into 15 groups [3,8], 309 identified in Oryza sativa were classified into 5 groups [27], 379 identified in Populus trichocarpa were classified into 14 groups [28], 303 identified in Brassica rapa were classified into 15 groups [29], 467 identified in Glycine max were classified into 14 groups, and 234 identified in Solanum lycopersicum were classified into 10 groups [30]. To date, LRR-RLK genes have been identified and phylogenetically analyzed in more than 31 plant species [10,31]. However, no such analysis has been conducted on polyploid cotton (Gossypium spp.), except diploid species Gossypium raimondii, in which more than 300 LRR-RLK genes were identified [31,32].
Cotton, which comprises several Gossypium genus species, is an important economic crop, producing large amounts of natural fibers for the textile industry. Given the significant resource value of Gossypium genus, genome sequencing has been extensively promoted in Gossypium. To date, genomes of four Gossypium species including two widely cultivated allotetraploid Gossypium species (Gossypium barbadense and Gossypium hirsutum) and two diploid progenitor relatives species (Gossypium arboreum and Gossypium raimondii) have been sequenced and well-assembled [33,34,35,36,37,38], which laid a solid foundation for cotton research at the genomic level. For a long time, cotton suffered a variety of biotic and abiotic stresses during planting, and many efforts have been taken on the development of cotton fiber to improve the quality and yield. Considering the multifunction of LRR-RLK genes in plant defense response and development processes, in this study we conducted genome-wide identification and phylogenetic analysis of LRR-RLK genes on four genome-sequenced Gossypium genus species. In addition, the function and expression profiles of Gossypium LRR-RLK genes in several important developmental and stress response processes were analyzed. Our investigations provide insights into the evolution of Gossypium LRR-RLK genes and the roles of the LRR-RLK gene family in development and stress defense.
2. Materials and Methods
2.1. Identification of LRR-RLK Genes in Four Gossypium Genus Species
Up to now, four Gossypium species—G. arboreum, G. barbadense, G. hirsutum, and G. raimondii—have finished genome sequencing and assembly [34,35,36,37,38,39]. G. hirsutum and G. barbadense are the two most widely cultivated cotton species, both of which are allotetraploids and formed by inter-genomic hybridization of the A genome ancestral diploid and D genome ancestral diploid. G. arboreum (AA) and G. raimondii (DD) were recognized as species of progenitor relatives, whose progenitors were the putative A genome ancestor and D genome ancestor of G. hirsutum (AD1) and G. barbadense (AD2) [40,41]. Proteomes data of G. arboreum, G. barbadense, G. hirsutum, and G. raimondii were download from public databases (ftp://bioinfo.ayit.edu.cn/downloads; http://database.chgc.sh.cn/cotton/index.html; https://phytozome.jgi.doe.gov/ [42]), and the corresponding whole-genome gene annotations were downloaded as well. As LRR-RLKs are featured with KD domain, LRR domain, and TM domain, the corresponding Hidden Markov Model (HMM) models of KD including Pkinase (PF00069) and Pkinase_Tyr (PF07714), and HMM models of LRR including LRR_1 (PF00560), LRR_2 (PF07723), LRR_3 (PF07725), LRR_4 (PF12799), LRR_5 (PF13306), LRR_6 (PF13516), LRR_8 (PF13855), LRR_9 (PF14580)and LRV (PF01816), were downloaded from Pfam database (http://pfam.xfam.org/) [43] and provided as queries to conduct homologues search (E-value < 1 × 10−10) against the protein database of the four Gossypium species respectively by using HMMER 3.1b2 software [44]. The resulting hits that were obtained in both KD domain search results and LRR domain search results were collected for further filtering. To make sure that we get as close as possible to the whole LRR-RLKs, the amino acid sequences of Arabidopsis LRR-RLKs members reported by Shiu et al. [7] were retrieved from The Arabidopsis Information Resource (TAIR) database v10.0 (http://www.arabidopsis.org/) [45] and served as the query to perform a similarity search (E-value < 1 × 10−5, identity > 50%) against the protein database of the four Gossypium species using BLAST+ v.2.6.0 [46]. The sum total items of the HMMER search result and BLAST+ result was used for subsequent validation analysis. InterProScan v.5.24-63.0 [47] was used to confirm the presence of KD domain and LRR domain and other characteristic domains. TMHMM server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/) [48] and Phobius (http://phobius.binf.ku.dk/) [49] were used for TM domain prediction. When either TMHMM server or Phobius server indicated a TM domain, we decided it contained TM domain. Proteins that contained both KD domain, LRR domain, and TM domain were considered as LRR-RLKs.
2.2. Phylogenetic Analysis of Gossypium LRR-RLKs
LRR-RLK genes identified in four Gossypium species and previously reported in Arabidopsis [8] were involved in phylogenetic analysis. Complete amino acid sequences of identified LRR-RLKs were used to perform multiple sequence alignment by MUSCLE [50]. The maximum likelihood (ML) tree was constructed by FastTree 2 [51] with default arguments. Neighbor-joining (NJ) tree was constructed by MEGA 7 [52] with 1000 bootstrap.
2.3. Gene Structure Analysis
Exon-intron structure information of identified LRR-RLK genes was retrieved from the whole-genome gene annotations. LRR, TM, and KD domain coordinates were derived from InterProScan annotation results and TM domain prediction results. TBtools [53] was used to display the gene exon-intron structure and domain coding regions within the default parameters.
2.4. Genomic Distribution of LRR-RLKs and Tandem Duplication Identification
The genomic coordinates of genes were extracted from genome annotation release data, and then used to map LRR-RLK genes on chromosome by TBtools [53]. Tandem duplication of LRR-RLK genes were identified when genes belonged to the same LRR-RLK subfamily and separated by ten or less genes in a 200 kb distance.
2.5. Gene Ontology and Pathway Analysis of Gossypium LRR-RLKs
Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway annotation information for G. barbadense, G. hirsutum, and G. raimondii were download from CottonFGD [54]. According to the gene functional annotation method used in CottonFGD, GO annotation of G. arboreum LRR-RLKs were conducted by InterProScan. KEGG Orthology was assigned to G. arboreum LRR-RLKs using KEGG Automatic Annotation Server (KAAS) [55] and then mapped to KEGG pathways. GO and KEGG pathway enrichment analysis was conducted by R package clusterProfiler v3.6.0 [56].
2.6. Promoter and Regulatory Analysis of Gossypium LRR-RLK Genes
The upstream 1.5 kb sequence of gene start codon was recognized and extracted as a promoter region. Promoter sequences of all Gossypium LRR-RLK genes were submitted to PlantCARE database [57] to predict potential cis-acting regulatory elements. Transcription factor (TF) binding sites were predicted by Binding site prediction tools on PlantTFDB 4.0 [58].
2.7. Gene Expression Profile Analysis of Gossypium LRR-RLK Genes
To investigate Gossypium LRR-RLK gene expression pattern during development and stress responses the following was conducted: RNA sequencing data of TM-1 ovule samples from −3, −1, 0, 1, 3, 5, 10, 20, and 25 days post anthesis (DPA), fiber samples from 5, 10, 20, and 25 DPA, and true leaves of the seedlings at different timepoints (1, 3, 6, and 12 h after treatment), after being treated with cold, heat, polyethylene glycol (PEG), and salt, were available at NCBI Sequence Read Archive (SRA) database (BioProject: PRJNA248163) [35]. Transcription level data were downloaded from CottonFGD [52] and analyzed. Transcriptomes of G. barbadense and Gossypium hirsutum in resistance response to Verticillium dahlia have been studied [59]. Two true leaf seedlings were inoculated with two different V. dahlia strains (highly aggressive strain V991 and intermediately aggressive strain D07038) by watering injured roots with V. dahlia spore suspension, while roots of control seedlings were watering with distilled water. For each treatment and control, plant samples from 24, 48, and 96 h after inoculation were mixed for sequencing. RNA sequencing data of response to infection were accessed from NCBI SRA (BioProject: PRJNA89721). The fragments per kilobase million (FPKM) value for each gene were computed to represent gene expression levels. An expression heatmap was drawn by R software package ComplexHeatmap (for k-means clustering) [60] and pheatmap (for hierarchy clustering) [61] based on log10-transformed FPKM values.
3. Results and Discussion
3.1. Identified LRR-RLK Genes of Four Gossypium Genus Species
A total of 298, 511, 515, and 317 LRR-RLK genes were identified in G. arboreum, G. barbadense, G. hirsutum, and G. raimondii, respectively. All of these identified LRR-RLK genes contained LRR domain, KD domain, and TM domain simultaneously, and the conserved protein domain arranged in the order of LRR-TM-KD from N-terminal to C-terminal (Figure S1). The number of Gossypium LRR-RLK genes accounted for 0.73%, 0.66%, 0.73%, and 0.85% of whole genome protein coding genes in each species, respectively. The proportions of LRR-RLK genes largely fit with the result of Liu et al. [10], which demonstrated 0.67–1.39% proportions in angiosperm species.
3.2. Phylogenetic Analysis and Gene Structure of Gossypium LRR-RLK Genes
The amino acid sequences of 1641 LRR-RLK genes identified in this present study and previously reported 213 A. thaliana LRR-RLK genes were aligned (Supplementary data 1) for phylogenetic tree construction. Referring to the classification of A. thaliana LRR-RLK genes, the ML tree showed LRR-RLK genes from Gossypium were classified into 21 distinct clades (Figure 1, Supplementary data 2). To further validate the phylogenetic relationship of LRR-RLK genes in the ML tree, another tree based on the NJ method was constructed (Figure S2). Results showed that the topologies of both trees were somewhat different, while gene member assignment among different clades remained relatively stable. Therefore, the subfamilies classification in the ML tree was reliable and could be used for further analysis. Clades were named to correspond with subfamilies according to the nomenclature of A. thaliana LRR-RLK genes [8]. Most of LRR-RLK subfamilies of Gossypium were consistent with A. thaliana, while subfamilies VI, VII, VIII, and XI were further divided into VI-1 and VI-2, VII-1 and VII-2, VIII-1 and VIII-2, XI-1, XI-2, and XI-3, respectively. The detailed classification of Gossypium and A. thaliana LRR-RLK genes was described in Table S1. Overall, subfamilies III, XI, and XII showed the highest number of LRR-RLKs. Meanwhile, in terms of A. thaliana, the majority of LRR-RLKs were distributed in subfamilies I, III, XI and (Table 1, Figure 1).
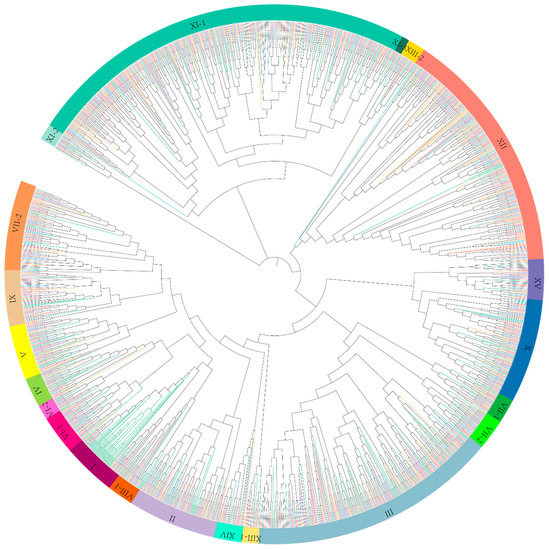
Figure 1.
Phylogenetic tree of leucine-rich repeat receptor-like kinases (LRR-RLK) genes from four Gossypium species and Arabidospsis thaliana. The phylogenetic tree was constructed by maximum likelihood (ML) method based on kinase domain amino acid sequences of LRR-RLKs. All LRR-RLK genes were divided into 21 distinct clades, marked by bold curves with different colors. LRR-RLKs from A. thaliana, Gossypium arboreum, Gossypium barbadense, Gossypium hirsutum, and Gossypium raimondii were represented by branches colored within green, red, yellow, purple, and blue, respectively.

Table 1.
Statistics of A. thaliana and Gossypium LRR-RLK gene distribution among different subfamilies. For both A. thaliana and four Gossypium species, the number of LRR-RLK genes belonging to each subfamily were counted respectively. The corresponding percentage in all LRR-RLK genes from specific species was computed and indicated in brackets.
Gene exon-intron structures and characteristic domain organizations of Gossypium LRR-RLK genes were investigated (Figure S1). For each subfamily, gene structures of representative genes from each species were displayed and compared. The number of introns in the ECD region varied both within and between subfamilies, while the number of introns in the KD domain were relatively constant (Figure 2D). Variable ECD regions might help LRR-RLKs to perceive diverse environmental stimuli. As the KD domain is more conserved than the LRR domain in LRR-RLK family genes [10,31], we classified the Gossypium LRR-RLK genes into three groups based on exon-intron organization of KD domain (Figure 2A–C). LRR-RLK genes in Group A, which contained subfamilies VII-1, VII-2, and XV, had KD domains located on an integral exon (Figure 2A), while KD domains of LRR-RLK genes in Group B, containing subfamilies III, IV, IX, X, XI-1, XI-2, XI-3, and XII, were separated by one intron (Figure 2B). In Group C, containing subfamilies I, II, V, VI-1, VI-2, VIII-1, VIII-2, XIII-1, XIII-2, and XIV, KD domains of LRR-RLK gene members were separated into 3–6 exons by introns. Meanwhile, the ECD regions of Group C genes (except for subfamilies XIV and VI-1 members) showed highly discrete distributed exons (Figure 2C). As a result, the LRR domains of Group C gene members were distributed in many different exons. Conversely, the majority of genes’ LRR domains in Group A and B were located in an integral exon. Referring to subfamily division based on the ML tree, exon-intron structures of LRR-RLK genes were variable between different subfamilies, while LRR-RLK genes in the same subfamily showed comparable gene structures. This indicated that gene structures were relatively conserved within each subfamily, implying that evolutionary relationships of these genes coincided with the ML tree. For each subfamily, gene structures of LRR-RLK genes from A. thaliana and four Gossypium species were almost identical, suggesting that exon-intron structures of LRR-RLK genes were relatively conserved between A. thaliana and Gossypium. However, there were some exceptions. In subfamily IX, there was a subclade containing five gene members (GOBAR_DD21842, evm.model.Ga14G2553, Gorai.010G147700, GOBAR_AA01355, and evm.model.Ga06G1442) which showed different exon-intron structures, in which the KD coding regions were divided into five exons (except for GOBAR_AA01355, which has a truncated KD domain), which differed from the representative two-exon KD (kinase catalytic domain) domain of subfamily IX (Figure S1). The phylogenic tree showed that this subclade was distinctly separated from other subfamily IX members with strong bootstrap support (0.98) (Supplementary data 2), further implying the consistency between the phylogenic tree and gene structure. Some LRR-RLK genes lost all introns compared to their close homologues, including 8 subfamily XI-1 gene members (Gh_D11G2499, Gh_A07G0162, Gh_D09G1175, GOBAR_AA08625, GOBAR_AA07945, Gh_A06G0294, Gh_D09G0173, and Gh_D09G0176), 6 subfamily III gene members (GOBAR_DD29294, GOBAR_AA04771, Gh_D05G0350, GOBAR_AA35240, GOBAR_DD18442, and Gh_A05G0258) and 3 subfamily XII gene members (GOBAR_DD24038, Gh_D05G3552, Gh_D10G2231). These genes could be the retrotransposed genes. Retrotransposition is an important mechanism for genome expansion and is ubiquitous in plants [62]. For the LRR-RLK family in Gossypium, all these retrotransposed genes were found in the three largest subfamilies, suggesting that retrotransposition may contribute to the high gene numbers of these subfamilies to some degree.
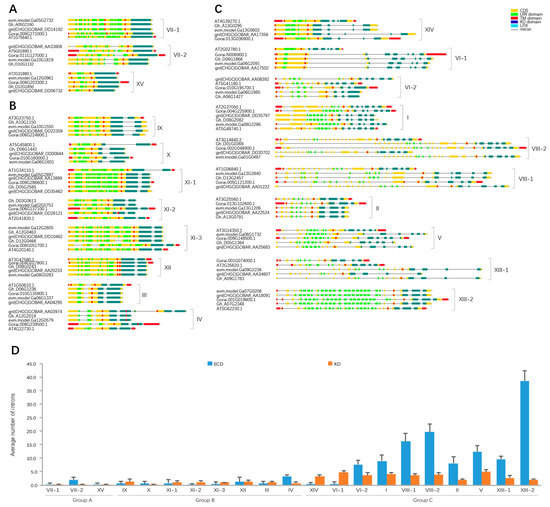
Figure 2.
Exon-intron structures of representative LRR-RLK genes of each subfamily from four Gossypium species and A. thaliana. LRR (leucine-rich repeat), KD (kinase catalytic domain), and TM (transmembrane) domain coding regions were marked on exons by different colored rectangles. Based on the exon-intron structures of KD domain, Gossypium LRR-RLK genes were classified into Group A (A), Group B (B), and Group C (C). The number of introns in ECD (extracellular domain) and KD domain were counted and then average values were computed for each subfamily (D). The error bar on the column represented standard deviation of the subfamily.
3.3. Dramatic Expansion of LRR-RLK Subfamilies XI and XII in Gossypium Species
Statistics of LRR-RLK gene distribution among different subfamilies was conducted in both Gossypium species and A. thaliana. As shown in the phylogenetic tree (Figure 1), some subfamilies exhibit linage specific expansion. For example, in subfamily XII clade, the overwhelming majority of members belong to Gossypium, with only 7 of the total 282 members belonging to A. thaliana. Another example is the subfamily I clade, which is mainly (41 out of 61) composed of A. thaliana LRR-RLKs (Table 1, Figure 1). An uncoordinated proportion of LRR-RLK members between Gossypium and A. thaliana in these subfamilies suggested different expansion patterns of LRR-RLK genes between them.
To further investigate the expansion pattern of different linages, we compared the distribution of LRR-RLK genes among different subfamilies between A. thaliana and Gossypium. In consideration of the genome size difference among different species, the proportion of each subfamily was compared instead of member number of each subfamily. In general, LRR-RLK genes showed similar proportion in most subfamilies between Gossypium and A. thaliana, while Gossypium LRR-RLK genes had a significantly smaller proportion of subfamily I distribution, and a larger proportion of subfamily XI and XII than A. thaliana (Figure 3). There were 41 LRR-RLK I members in A. thaliana, accounting for 19.2% of all A. thaliana LRR-RLK genes, while the counterpart percentage was 0.9–1.4% in Gossypium species. A. thaliana had about 13 times more LRR-RLK I members compared to Gossypium species. On the contrary, Gossypium species showed 26.5–27.4% and 11.8–20.0% of LRR-RLKs distributed in subfamily XI and XII, respectively. The proportions were about two to four times more than in A. thaliana, in which the corresponding percentage were only 15.0% and 3.3%.
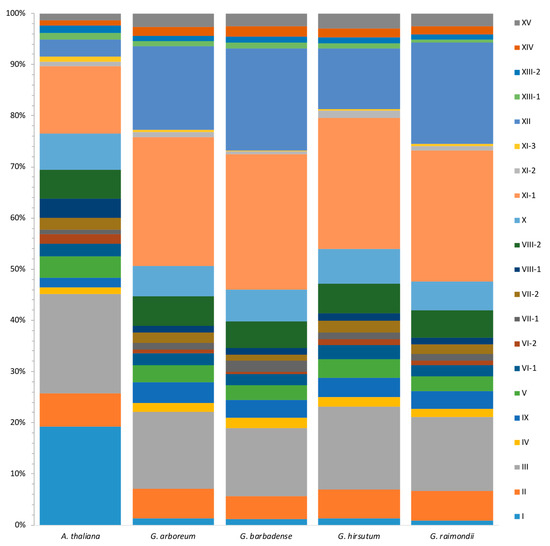
Figure 3.
Comparison of LRR-RLK gene distribution among different subfamilies between A. thaliana and Gossypium. For each species, all the LRR-RLKs were divided into 21 subfamilies, represented by rectangles within different colors. The area of each rectangle within a specific color represented the proportion of the corresponding subfamily.
As the last common ancestor of angiosperms (LCAA) was estimated to contain about seven LRR-RLK subfamily I members, A. thaliana LRR-RLK I subfamily shows dramatic expansion due to whole-genome duplication (WGD) of Brassicaceae [31]. The significant expansion of subfamily I in A. thaliana made it to be the largest subfamily, while a slight reduction of this subfamily was found in diploid Gossypium, which perhaps suggested gene loss during evolution. With respect to LRR-RLK subfamily XI and XII, compared with LCAA, all four Gossypium species exhibited significant expansion in these two subfamilies. As described in a previous study [31], the LRR-RLK XI subfamily almost kept stable member numbers in most angiosperms, as verified by A. thaliana in this present study, while in Gossypium, subfamily XI expands dramatically (predominantly in XI-1), making it to be the largest LRR-RLK subfamily (accounting for about 25% of the total LRR-RLK genes). Current knowledge suggests that many of the A. thaliana LRR-RLK genes that fall into XI were involved in plant organ and tissue development. For example, RGFR genes are involved in root growth and development. CLV1 and BAM are involved in shoot and floral meristem development and function. PXY is involved in vascular-tissue development. HAE and HSL control floral organ abscission. IKU and GSO are involved in embryo development. Besides some other A. thaliana XI gene members, such as PEPR1 and PEPR2 which are defense and stress response-related, expanded subfamily XI might help Gossypium to defend against stresses. Interestingly, according to the phylogenic tree, the one large clade of subfamily XI-1 containing AT4G08850 (MIK2) and AT1G35710 was almost entirely comprised of Gossypium LRR-RLK members (Figure 1), suggesting that the orthologs of these two A. thaliana XI genes were largely expanded in Gossypium, which largely accounted for the expansion of subfamily XI-1 in Gossypium. According to previous studies [28,31,63], the dramatic expansion of homologues of AT4G08850 (MIK2) and AT1G35710 was also detected in many other plant species, such as P. trichocarpa, G. max, Malus × domestica, and Prunus persica, though the corresponding clade was sometimes assigned a different name. It has been reported that MIK2 acts as a key component of the male receptor heteromer in the pollen tube cell perceiving female attractants in plants [64]. Other studies showed that MIK2 is stress defense related. MIK2 up-regulates expression in the root under salt exposure, and enhances rosette growth maintenance under salt stress conditions [65,66]. In cell wall damage response-processes, MIK2 acts as an important sensor and regulator, involved in response to abiotic and biotic stresses [67]. Little information about the function of AT1G35710 gene was known, however, a transcriptome study found that the AT1G35710 gene was repressed by linolenic acid, which is a precursor of the phytohormone jasmonic acid (JA) and launched a set of defense responses to pathogen attacks [68], suggesting that AT1G35710 may be involved in defense response. As homologues within the same cluster may have similar functions, we supposed that the expansion of subfamily XI-1 in Gossypium may enhance defense of cotton against diverse environmental stimuli and stresses. Likewise, subfamily XII expands greatly in Gossypium. It is already known that the expansion of LRR-RLK XII is extensive in many different species [31]. Most genes in subfamily XII are involved in biotic and abiotic stress response, so we supposed that the expansion of LRR-RLK XII helps in perceiving and adapting to diverse environments. All of these results were consistent with previous findings that lineage-specific expanded LRR-RLK genes predominantly belong to subgroups involved in environmental interactions [31].
As allotetraploid species G. hirsutum and G. barbadense are known to have derived from hybridization of diploid ancestors of G. arboreum and G. raimondii [33,34,41], the number of LRR-RLK members in each subfamily between the two allotetraploid species were compared. In general, G. hirsutum and G. barbadense have similar proportions in terms of most of the subfamilies. However, when regarding subfamily XII, G. barbadense showed a significantly higher proportion than G. hirsutum (Figure 3). In further detail, G. barbadense had 102 LRR-RLK genes assigned into subfamily XII, as there were only 61 LRR-RLK subfamily XII members in G. hirsutum. As most subfamily XII members are defense-related, we supposed that more subfamily XII LRR-RLK members may confer better resistance in G. barbadense than G. hirsutum to some extent. We supposed that G. barbadense is more likely to retain copies of LRR-RLK subfamily XII gene members from diploid ancestors, while G. hirsutum tends to lose some copies. Different retain/loss models of duplicates after polyploidization between G. hirsutum and G. barbadense may be due to adaptation under different environments and different selection pressures.
3.4. Genomic Distribution and Gene Duplication of LRR-RLK Genes in Gossypium
The genomic distribution of identified LRR-RLK genes form four Gossypium species was displayed respectively (Figure S3). Most of the LRR-RLK genes were mapped on chromosomes, with only 11(3.7%), 19(3.7%), 40(7.8%), and 4(1.3%) LRR-RLK genes from G. arboreum, G. barbadense, G. hirsutum, G. raimondii being located on scaffolds, respectively. The LRR-RLK genes were distributed in all chromosomes but unevenly across different chromosomes (Table S2). The distribution across different chromosomes was comparative in two polyploid species, chromosome A5, A10, D5, and D10 containing the highest proportion of LRR-RLK genes in both G. barbadense and G. hirsutum. The comparable genomic distribution pattern of this large gene family between G. barbadense and G. hirsutum suggested the close phylogenic relationship of these two allotetraploid species. However, the distribution across different chromosomes was quite different between two diploid species G. arboreum and G. raimondii (Figure S3).
Mapping LRR-RLK genes on chromosomes allows us to detect gene duplication. In our analysis, LRR-RLK genes which fell into the same subfamily and were separated by 10 or less genes in a 200 kb chromosomal distance were recognized as a tandem duplication set [5,28]. There were 26, 47, 33, and 25 tandem duplication sets involving 79, 146, 92, and 96 tandem duplicates found in G. arboreum, G. barbadense, G. hirsutum, and G. raimondii, respectively. Gene numbers contained in each tandem duplication set ranged from 2 to 11. Furthermore, tandem duplication occurred unevenly among different subfamilies (Table 2). The dramatically expanded subfamilies XI and XII contained the majority of all tandem duplication sets (73.1%, 78.7%, 69.7%, and 72.0% for G. arboreum, G. barbadense, G. hirsutum, and G. raimondii, respectively) and tandem duplicated genes (78.5%, 84.2%, 75.0%, and 81.3% for G. arboreum, G. barbadense, G. hirsutum, and G. raimondii, respectively). Subfamily VIII-2 contained about 14.5% of tandem duplication sets and 11.7% of tandem duplicated genes, corresponding to somewhat of an expansion of subfamily VIII-2. Other subfamilies (II, III, VII-1, and IX) contained a few sets of tandem duplication. No tandem duplication was found in subfamilies I, IV, V, VI-1, VI-2, VII-2, VIII-1, X, XI-1, XI-3, XIII, XIV, and XV) (Table 2 and Table S3). When concerning the two dramatically expanded subfamilies XI and XII, we found that about 40% of LRR-RLK subfamilies XI members and about 60% of LRR-RLK XII members were involved in tandem duplication (Table S3), implying that tandem duplication played an important role in vast expansion of these subfamilies. Furthermore, just as in subfamilies XI and XII, about half of the LRR-RLK subfamily VIII-2 members were derived from tandem duplication (Table S3), suggesting the same important role of tandem duplication in expansion of subfamily VIII-2. We deduced that tandem duplication existed extensively and acted as an important expansion mechanism in expanded LRR-RLK subfamilies.
Table 2.
Number of identified tandem duplicated LRR-RLK genes among different subfamilies in four Gossypium species. Number of tandem duplicated gene sets in each subfamily were indicated in brackets.
3.5. Functional and Pathway Annotation Analysis of Gossypium LRR-RLK Genes
Gene ontology annotation information was available from public Gossypium databases. We conducted GO enrichment on all LRR-RLK genes from G. arboreum, G. barbadense, G. hirsutum, and G. raimondii. Results showed that the molecular function of “protein kinase activity”, “protein binding”, “ATP binding”, and biological processes of “protein phosphorylation” were significantly enriched in Gossypium LRR-RLK genes (Figure S4). The GO enrichment results were confirmed with the basic kinase attributes of LRR-RLKs. KEGG pathway enrichment analysis showed that “plant pathogen interaction” and “plant hormone signal transduction” were significantly enriched in all four Gossypium species (Figure 4). Furthermore, “toll-like receptor signaling” and “NOD-like receptor signaling pathway” were found to be enriched in G. arboreum and G. hirsutum. These results suggested that the functional roles of LRR-RLK genes in plant development and defense may be largely mediated by related signaling.
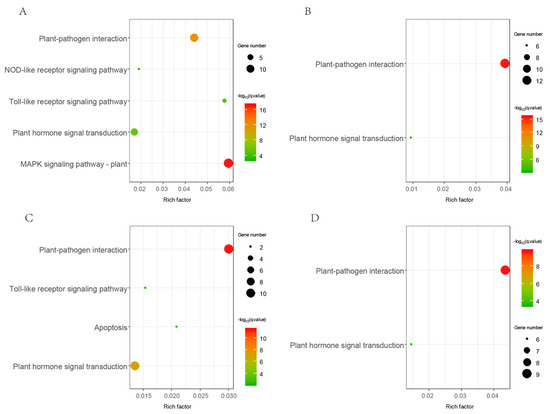
Figure 4.
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment result of Gossypium LRR-RLK genes. Results of G. arboreum, G. barbadense, G. hirsutum, and G. raimondii were shown by (A–D) respectively.
3.6. A cis-Acting Regulatory Analysis of Gossypium LRR-RLK Genes’ Promoters
We found that cis-acting regulatory elements in the promoter regions (1.5 kb sequence upstream of start codon) of Gossypium LRR-RLK genes were detected by searching PlantCARE database. Phytohormone, stresses defense, and cell cycle related cis-acting regulatory elements were widespread in the promoter regions of Gossypium LRR-RLK genes (Table 3 and Table S4). More than 90% of Gossypium LRR-RLK genes had water stress, drought stress, and light response cis-acting regulatory elements in their promoters. Anoxic stresses response, wounding, and pathogen response cis-acting regulatory elements were found in promoter regions of about 80% of Gossypium LRR-RLK genes. Additionally, more than half of Gossypium LRR-RLK genes had heat, osmotic stress, low pH, nutrient starvation, ethylene (ETH), and abscisic acid (ABA) response cis-acting regulatory elements in their promoter regions. Jasmonate response elements existed in many Gossypium LRR-RLK genes’ promoters (50.7%, 42.5%, 44.7%, and 48.3% of G. arboreum, G. barbadense, G. hirsutum, and G. raimondii LRR-RLK genes, respectively). Salicylic acid (SA) and gibberellin response elements were also found in about 40% of Gossypium LRR-RLK genes’ promoters. Auxin response elements were found in about 30% of Gossypium LRR-RLK genes’ promoters. TC-rich repeats (defense and stress response) and LTR (low temperature response) elements were found in promoters of about 30% of Gossypium LRR-RLK genes. More than 45% of Gossypium LRR-RLK genes had cell cycle and cell proliferation related elements in promoter regions. Some (about 5%) Gossypium LRR-RLK genes had heavy metal ions response-related cis-acting regulatory elements. The cis-acting regulatory elements analysis revealed that the expression of Gossypium LRR-RLK genes was extensively regulated by phytohormone and other diverse abiotic and biotic environmental signals, implying the important roles of Gossypium LRR-RLK genes in stresses defense and development. Compared with A. thaliana, there were more heat, osmotic stress, low pH, nutrient starvation, and ETH response cis-acting elements but less ABA, JA, and auxin response cis-acting elements in promoter regions of Gossypium LRR-RLK genes. We further conducted enrichment analysis on each subfamily to investigate the probable over-represented cis-acting regulatory elements. As a result, circadian response element and Cd (cadmium) response element were overrepresented in subfamily XV and subfamily VIII-2 of G. barbadense, respectively. Cold response element was overrepresented in subfamily IX of G. hirsutum.
Table 3.
Statistics of cis-acting regulatory elements detected in promoter regions of Gossypium LRR-RLK genes. (*cis-acting regulatory elements that have no functional description were not shown, see Table S3 for details.).
Transcription factors (TFs) play key roles in many cellular and biological processes by regulating expression of corresponding target genes. To investigate the possible regulation relationship between TFs and Gossypium LRR-RLK genes, TF binding sites were predicted by an online tool—binding site prediction on PlantTFDB [58]. Results showed that Gossypium LRR-RLK genes could be regulated by 39 TF families (Figure 5, Table S5). Dof, MIKC_MADS, MYB, AP2, C2H2, and ERF were the most widely functioning TF families and could regulate the majority of Gossypium LRR-RLK genes. Most of the top TF families were implicated in various aspects of plant development, hormonal signal transduction, plant defense, and stresses response, suggesting that Gossypium LRR-RLKs might participate in diverse plant development and stress defense processes by TF mediated regulation.
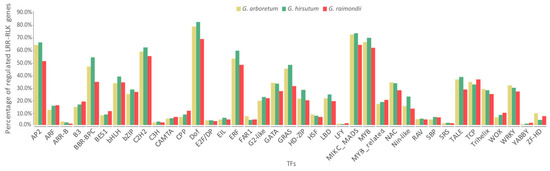
Figure 5.
Statistics of Gossypium LRR-RLK genes regulated by different families of transcription factors (TFs) (genes with TF binding sites were considered to be regulated by TFs).
3.7. Gene Expression of Gossypium LRR-RLKs during Developmental and Stress Defense Processes
As the functional analysis showed the important roles of Gossypium LRR-RLKs in diverse developmental and defense processes, the expression profilers of Gossypium LRR-RLKs in several important developmental (fiber development, ovule development) and biotic (Verticillium wilt) stress defense and abiotic (cold, hot, drought, and salt) stress defense processes were investigated. Fiber development is an important process in cotton biology, based on the transcription dynamics of LRR-RLK genes during fiber development. The k-means clustering result showed that G. hirsutum LRR-RLK genes were clustered into two groups. The majority of Group 1 gene members showed relatively lower or moderate expression throughout the fiber development, while gene members in Group 2 were obviously actively expressed. Genes in Group 2 were highly expressed at 5 and 10 dpa (day post anthesis) stages, followed by down-regulation during 20 and 25 dpa (Figure 6A). In cotton fiber development, fiber cell initials start at 0 dpa and elongate subsequently from 0–18 dpa. Additionally, 20 dpa is commonly considered as the key stage of transition to secondary cell wall growth, followed by dehydration and maturation after 30 dpa [69,70,71]. Expression patterns of Group 2 genes showed high correlation with the elongation phase of fiber development, suggesting that these genes were extensively involved in the rapid elongation of fiber. There were eight LRR-RLK genes assigned into Group 2 (three subfamily IX members: Gh_D09G1268, Gh_A13G0257, and Gh_D13G0274; two subfamily V members: Gh_A10G0460 and Gh_D10G0477; two subfamily III members: Gh_A11G1546 and Gh_D11G3486; and another subfamily I member gene Gh_A07G1471). Except for Gh_A07G1471, the other Group 2 members belong to 3 homologous pairs. All three subfamily IX members in Group 2 have orthologous gene in A. thaliana known as TMK3, which is reported to play an essential role in plant growth mediated by regulation of cell expansion and auxin signaling [72]. Therefore, we suggested that these three LRR-RLK genes would likely have contributed to the elongation of cotton fiber, given that the elongation of fiber is almost a longitudinal expansion of singular fiber cells. The two homologous subfamily III members in Group 2 have the orthologs of RLK1 in A. thaliana, as two homologous subfamily V members in Group 2 are orthologs of A. thaliana SRF6. Further investigation of expression profiles in different tissues showed that all eight Group 2 genes were predominantly expressed in early-period fiber, especially the developmental stage of 5–10 dpa [73], further implying the role of these genes in fiber elongation. Cloning and functional identification of these genes in fiber development would prove a worthy finding.
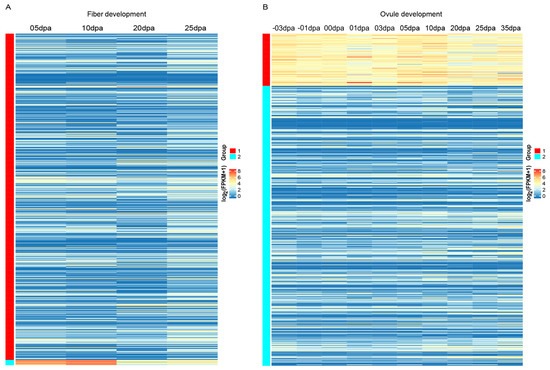
Figure 6.
Expression patterns of G. hirsutum LRR-RLK genes in fiber and ovule development. Genes were clustered by the k-means method.
In the process of ovule development, all G. hirsutum LRR-RLK genes were divided into two groups according to k-means clustering. Group 1 contained 81 LRR-RLK genes that showed high expression at almost all stages of ovule development (Figure 6B), implying their important role in ovule development. Most of the Group 1 genes belonged to subfamily III, II, and XI-1. Protein orthologs of Group 1 genes contained several members involved in embryo and gamete development (such as BAM, BAK1, EMS1, RPK2, TOAD2, and ERECTA) and members involved in hormone signaling (such as RGI3 and BRI1). Moreover, the 6 LRR-RLK genes (Gh_A10G0460, Gh_D10G0477, Gh_A11G1546, Gh_D11G3486, Gh_A13G0257 and Gh_D13G0274) that participated in fiber development as described above were also assigned to Group 1 in the ovule development k-means clustering. This implied the versatility of single G. hirsutum LRR-RLK genes, which might participate in multiple developmental processes.
Cotton is inevitably threatened by diverse abiotic stresses during its growth and development. Therefore, expression profiles of G. hirsutum LRR-RLK gene responses to cold, hot, drought, and salt stresses were analyzed. The G. hirsutum LRR-RLK genes were clustered in four groups based on their expression profile responses to different abiotic stresses (Figure 7). Different gene sets responded to different abiotic stimulates by changing expressions in a temporal manner. About half of G. hirsutum LRR-RLK genes depicted low expression during the stress treatments, as shown in Group blue which contains LRR-RLK genes belonging to almost all subfamilies except for subfamily VIII-1. The majority of LRR-RLK genes in Group green, which consists of LRR-RLK genes from almost all subfamilies except for subfamily XI-3, showed relatively lower expression levels in control. Most were down-regulated in response to the four abiotic stresses compared to control, while some members subsequently up-regulated at later response stages. Genes in Group orange and Group red showed relatively higher expression level in control. Most of Group orange’s genes down-regulated under all stresses, with different genes down-regulating at different stages of stress responses, suggesting diverse mechanisms of LRR-RLK gene response to abiotic stresses. Group orange consisted of LRR-RLK genes from almost all subfamilies except for subfamilies I, XI-3, and XIV. Genes in Group red stayed highly expressed during all stress response processes. Most of them up-regulated at early stages of cold and hot exposure but at later stages of PEG and salt stress treatments, implying that genes in Group red might act as positive regulators in abiotic stress responses. There were 29 LRR-RLK genes belonging to 9 different subfamilies (IX, V, VII-2, VIII-2, X, XI-1, XIII-1, XIV, and XV) in Group red, with subfamily XI-1 accounting for the largest proportion (12 genes). These results implied that Gossypium LRR-RLK genes were multi-functional, play important roles in multiple abiotic stress responses, and might help cotton to adapt to diverse abiotic environments.
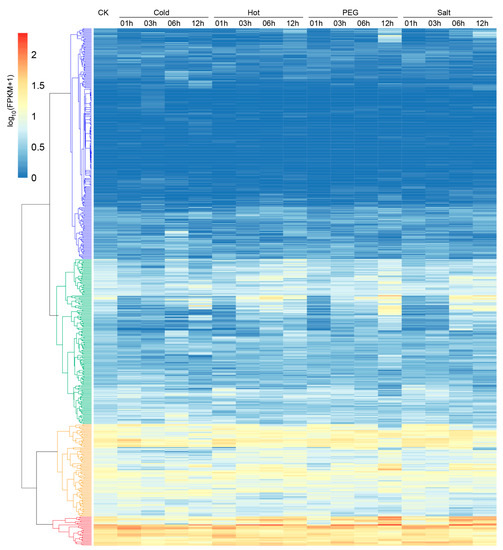
Figure 7.
Expression patterns of G. hirsutum LRR-RLK genes in response to diverse abiotic stresses (cold, hot, drought simulated by polyethylene glycol (PEG), and salt). Hierarchical clustering analysis classified genes into four distinct groups (colored by blue, green, orange, and red, respectively).
Verticillium wilt is one of the most interactable diseases in cotton growth. The expression profiles of Gossypium LRR-RLK genes under Verticillium wilt infection were investigated to analyze their response to biotic stress. The main cultispecies G. hirsutum is susceptible to Verticillium wilt, while G. barbadense shows better resistance and immunity to Verticillium wilt. Transcriptomic dynamic response to Verticillium wilt was been studied between G. hirsutum and G. barbadense [59]. There were 37 significant differentially expressed (log2FC > 1 and FDR (false discovery rate) < 0.05) LRR-RLK genes detected in G. barbadense under two Verticillium wilt strain infections. The counterpart differentially expressed LRR-RLK gene numbers in G. hirsutum was 34. Among these differentially expressed LRR-RLK genes, subfamily XI, XII, and VIII-2 accounted for the largest share (Figure 8) in both G. barbadense and G. hirsutum. Comparing G. barbadense with G. hirsutum, we found that G. barbadense had more differentially expressed LRR-RLK genes belonging to subfamily XI-1 and XII than G. hirsutum (Figure 8). Expression heatmaps revealed that the majority of subfamily XI-1 and all subfamily XII LRR-RLK genes down-regulated significantly when infected by Verticillium wilt, while subfamily VIII-2 LRR-RLK genes were more likely to be up-regulated. Differentially expressed LRR-RLK gene belonging to subfamilies II, IX, V, VI, X, XI-3, XIII-2, and XIV were significantly up-regulated. The two homologous subfamily IV genes in G. hirsutum were significantly down-regulated. There was only one up-regulated subfamily III LRR-RLK gene which showed significant differential expression in G. hirsutum, while six subfamily III LRR-RLK genes, including two down-regulated and four up-regulated genes, were found to be differentially expressed in G. barbadense. The difference of LRR-RLK expression regulation between G. barbadense and G. hirsutum may be associated with a difference in disease resistance between these two species.
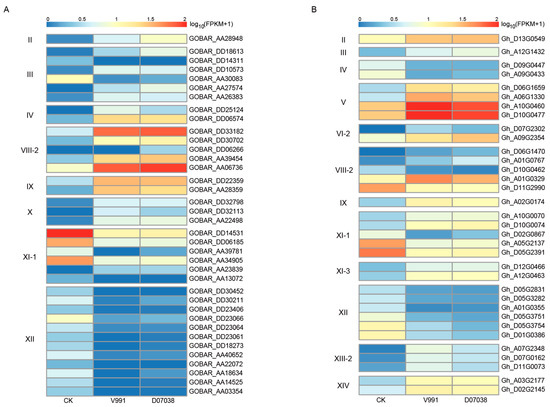
Figure 8.
Expression patterns of differentially expressed LRR-RLK genes in response to Verticillium dahlia infection. For both G. barbadense (A) and G. hirsutum (B), significant differentially expressed LRR-RLK genes simultaneously detected in both samples infected with V. dahliae strain V991 (highly toxic) and samples infected with V. dahliae strain D07038 (intermediately toxic) were used for heatmap drawings for each subfamily.
In summary, expression profiles of LRR-RLK genes varied both within and among subfamilies in Gossypium development and stress response, implying the functional divergence of LRR-RLK gene copies. It was difficult to assign distinct functional roles to different Gossypium LRR-RLK subfamilies, while the expression profile analysis in our study suggested wide involvement of Gossypium LRR-RLK genes in diverse processes of cotton development and stress response.
4. Conclusions
The present study performed a comprehensive analysis of the large LRR-RLK gene family in four Gossypium species. The Gossypium LRR-RLK genes were classified into 21 distinct subfamilies. Subfamilies XI and XII were found to be dramatically expanded in Gossypium, while tandem duplication was found to act as an important expansion mechanism in these expanded subfamilies. Functional and expression profile analysis revealed that Gossypium LRR-RLK genes were widely involved in diverse developmental processes and stress defenses. The expansion of subfamily XI and XII could be associated with more complicated development and regulation processes, and enhanced adaptability against various environments. The cis-acting regulatory elements analysis revealed that Gossypium LRR-RLK genes were extensively regulated by TFs and various abiotic and biotic stimuli. Our study provided valuable information for further functional study of Gossypium LRR-RLK genes.
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4425/9/12/592/s1. Figure S1: Domain and exon-intron organization of identified LRR-RLK family members in A. thaliana and Gossypium. Figure S2: NJ tree constructed by MEGA 7 based on amino acid sequences of LRR-RLKs from A. thaliana and Gossypium. Figure S3: Chromosomal location of LRR-RLK genes from four Gossypium species. Figure S4: GO enrichment results of Gossypium LRR-RLK genes. Table S1: Detailed assignment of A. thaliana and Gossypium LRR-RLK genes into different subfamilies. Table S2: Genomic distribution of Gossypium LRR-RLK genes among different chromosomes and scaffolds. Table S3: Number of tandem duplication gene sets and involved genes in Gossypium LRR-RLK family. Table S4: Statistics of cis-acting regulatory elements found by PlantCARE in promoter regions of Gossypium LRR-RLK genes. Table S5: Statistics of TF binding sites predicted in promoter regions of Gossypium LRR-RLK genes. Supplementary data 1: Multiple sequence alignment results of LRR-RLKs from A. thaliana and Gossypium. Supplementary data 2: Original ML tree constructed from amino acid sequences of LRR-RLKs from A. thaliana and Gossypium.
Author Contributions
R.S. conceived the research, carried out the analysis, and wrote draft manuscript. S.W. helped prepare figures. D.M., S.W., and professor C.L. supervised the research, and gave final approval of the version to be published. All the authors read and approved the final manuscript.
Funding
This research was funded by the National Science and Technology Major Project of China (2016YFD0101006) and the Central-level public welfare scientific research institutes for basic R & D special fund (161012018035; 161012018039). The APC was funded by Institute of Cotton Research, Chinese Academy of Agricultural Sciences, China.
Acknowledgments
We thank the National Science and Technology Major Project and the Central-level public welfare scientific research institutes for basic R & D special fund for its funding. We are particularly grateful to WJ Li for her support. We also thank Ghulam QANMBER for helping to revise the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Boil. 2007, 19, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 141, 1117. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol. 2003, 132, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Lehti-Shiu, M.D.; Cheng, Z.; Shiu, S.H. Origin, diversity, expansion history, and functional evolution of the plant receptor-like kinase/pelle family. Signal. Commun. Plants 2012, 13, 1–22. [Google Scholar] [CrossRef]
- Lehti-Shiu, M.D.; Zou, C.; Hanada, K.; Shiu, S.H. Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009, 150, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.C. Structure and function of the receptor-like protein kinases of higher plants. Plant Mol. Biol. 1994, 26, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Plant receptor-like kinase gene family: Diversity, function, and signaling. Sci. STKE 2001, 2001, re22. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. USA 2001, 98, 10763–10768. [Google Scholar] [CrossRef]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Boil. 2001, 11, 725. [Google Scholar] [CrossRef]
- Liu, P.-L.; Du, L.; Huang, Y.; Gao, S.-M.; Yu, M. Origin and diversification of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in plants. BMC Evol. Boil. 2017, 17. [Google Scholar] [CrossRef]
- Clark, S.E.; Williams, R.W.; Meyerowitz, E.M. The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 1997, 89, 575–585. [Google Scholar] [CrossRef]
- Somssich, M.; Je, B.I.; Simon, R.; Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 2016, 143, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.X.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef]
- Nam, K.H.; Li, J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Jinn, T.L.; Stone, J.M.; Walker, J.C. HAESA, an Arabidopsis leucine-rich repeat receptor kinase, controls floral organ abscission. Genes Dev. 2000, 14, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Nodine, M.D.; Yadegari, R.; Tax, F.E. RPK1 and TOAD2 are two receptor-like kinases redundantly required for Arabidopsis embryonic pattern formation. Dev. Cell 2007, 12, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Cyril, Z.; Gernot, K.; Delphine, C.; Anne, C.; Jones, J.D.G.; Thomas, B.; Georg, F. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 2006, 125, 749–760. [Google Scholar] [CrossRef]
- Gómez-Gómez, L.; Boller, T. FLS2: An LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 2000, 5, 1003–1011. [Google Scholar] [CrossRef]
- Song, W.Y.; Wang, G.L.; Chen, L.L.; Kim, H.S.; Pi, L.Y.; Holsten, T.; Gardner, J.; Wang, B.; Zhai, W.X.; Zhu, L.H.; et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 1995, 270, 1804–1806. [Google Scholar] [CrossRef]
- Albrecht, C.; Russinova, E.; Hecht, V.; Baaijens, E.; de Vries, S. The Arabidopsis thaliana SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASES1 and 2 control male sporogenesis. Plant Cell 2005, 17, 3337–3349. [Google Scholar] [CrossRef] [PubMed]
- Hecht, V.; Vielle-Calzada, J.P.; Hartog, M.V.; Schmidt, E.D.; Boutilier, K.; Grossniklaus, U.; de Vries, S.C. The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol. 2001, 127, 803. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xiong, L.; Yang, Y. Rice SERK1 gene positively regulates somatic embryogenesis of cultured cell and host defense response against fungal infection. Planta 2005, 222, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gou, X.; Yin, H.; He, K.; Du, J.; Yi, J.; Xu, S.; Lin, H.; Clouse, S.D.; Li, J. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinosteroid signaling. PLoS Genet. 2012, 8, e1002452. [Google Scholar] [CrossRef] [PubMed]
- Karlova, R. The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE1 protein complex includes BRASSINOSTEROID-INSENSITIVE1. Plant Cell Online 2006, 18, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Russinova, E.; Kemmerling, B.; Kwaaitaal, M.; de Vries, S.C. Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE proteins serve brassinosteroid-dependent and -independent signaling pathways. Plant Physiol. 2008, 148, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, G.L. Genome-wide identification, characterization and phylogenetic analysis of the rice LRR-kinases. PLoS ONE 2011, 6, e16079. [Google Scholar] [CrossRef]
- Zan, Y.; Ji, Y.; Zhang, Y.; Yang, S.; Song, Y.; Wang, J. Genome-wide identification, characterization and expression analysis of populus leucine-rich repeat receptor-like protein kinase genes. BMC Genom. 2013, 14, 318. [Google Scholar] [CrossRef]
- Rameneni, J.J.; Lee, Y.; Dhandapani, V.; Yu, X.; Choi, S.R.; Oh, M.H.; Lim, Y.P. Genomic and post-translational modification analysis of leucine-rich-repeat receptor-like kinases in Brassica Rapa. PLoS ONE 2015, 10, e0142255. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, J.; Yang, S.; Song, Y. Identification and expression analysis of the LRR-RLK gene family in tomato (Solanum lycopersicum) Heinz 1706. Genome 2015, 58, 121–134. [Google Scholar] [CrossRef]
- Fischer, I.; Dievart, A.; Droc, G.; Dufayard, J.F.; Chantret, N. Evolutionary dynamics of the leucine-rich repeat receptor-like kinase (LRR-RLK) subfamily in angiosperms. Plant Physiol. 2016, 170, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Dufayard, J.-F.; Bettembourg, M.; Fischer, I.; Droc, G.; Guiderdoni, E.; Périn, C.; Chantret, N.; Diévart, A. New insights on leucine-rich repeats receptor-like kinase orthologous relationships in angiosperms. Front. Plant Sci. 2017, 08. [Google Scholar] [CrossRef]
- Yuan, D.; Tang, Z.; Wang, M.; Gao, W.; Tu, L.; Jin, X.; Chen, L.; He, Y.; Zhang, L.; Zhu, L.; et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 2015, 5, 17662. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium Raimondii. Nat. Genet. 2012, 44, 1098. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Zheng, H.-J.; Hu, Y.; Lu, G.; Yang, C.-Q.; Chen, J.-D.; Chen, J.-J.; Chen, D.-Y.; Zhang, L.; et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. In Advances in Agronomy; Elsevier: Amsterdam, The Netherlands, 2003; Volume 78, pp. 139–186. ISBN 978-0-12-000796-7. [Google Scholar]
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Viklund, H.; Elofsson, A. Best α-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004, 13, 1908–1917. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Boil. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xia, R.; Chen, H.; He, Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv 2018. [Google Scholar] [CrossRef]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Boil. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Quan, S.; Jiang, H.; Zhu, X.; Wang, W.; He, X.; Shi, Y.; Yuan, Y.; Du, X.; Cai, Y. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 2013, 14, 852. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- pheatmap: Pretty Heatmaps. Available online: http://CRAN.R-project.org/package=pheatmap (accessed on 25 August 2018).
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef]
- Zhou, F.; Guo, Y.; Qiu, L.-J. Genome-wide identification and evolutionary analysis of leucine-rich repeat receptor-like protein kinase genes in soybean. BMC Plant Boil. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liang, L.; Xue, Y.; Jia, P.-F.; Chen, W.; Zhang, M.-X.; Wang, Y.-C.; Li, H.-J.; Yang, W.-C. A receptor heteromer mediates the male perception of female attractants in plants. Nature 2016, 531, 241. [Google Scholar] [CrossRef] [PubMed]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Julkowska, M.M.; Klei, K.; Fokkens, L.; Haring, M.A.; Schranz, M.E.; Testerink, C. Natural variation in rosette size under salt stress conditions corresponds to developmental differences between Arabidopsis accessions and allelic variation in the LRR-KISS gene. J. Exp. Bot. 2016, 67, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Van der Does, D.; Boutrot, F.; Engelsdorf, T.; Rhodes, J.; McKenna, J.F.; Vernhettes, S.; Koevoets, I.; Tintor, N.; Veerabagu, M.; Miedes, E.; et al. The Arabidopsis leucine-rich repeat receptor kinase MIK2/LRR-KISS connects cell wall integrity sensing, root growth and response to abiotic and biotic stresses. PLOS Genet. 2017, 13, e1006832. [Google Scholar] [CrossRef] [PubMed]
- Mata-Pérez, C.; Sánchez-Calvo, B.; Begara-Morales, J.C.; Luque, F.; Jiménez-Ruiz, J.; Padilla, M.N.; Fierro-Risco, J.; Valderrama, R.; Fernández-Ocaña, A.; Corpas, F.J.; et al. Transcriptomic profiling of linolenic acid-responsive genes in ROS signaling from RNA-seq data in Arabidopsis. Front. Plant Sci. 2015, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Song, Q.; Chen, Z.J. Polyploidy and small RNA regulation of cotton fiber development. Trends Plant Sci. 2014, 19, 516–528. [Google Scholar] [CrossRef]
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot. 2007, 100, 1391–1401. [Google Scholar] [CrossRef]
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef]
- Dai, N.; Wang, W.; Patterson, S.E.; Bleecker, A.B. The TMK subfamily of receptor-like kinases in Arabidopsis display an essential role in growth and a reduced sensitivity to auxin. PLoS ONE 2013, 8, e60990. [Google Scholar] [CrossRef]
- You, Q.; Xu, W.; Zhang, K.; Zhang, L.; Yi, X.; Yao, D.; Wang, C.; Zhang, X.; Zhao, X.; Provart, N.J.; et al. ccNET: Database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017, 45, D1090–D1099. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).