Resistance to 6-Methylpurine is Conferred by Defective Adenine Phosphoribosyltransferase in Tetrahymena

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Culture Conditions, and Cell Counting
2.2. Genomic Exclusion
2.3. Cloning and Sequencing of APRT1
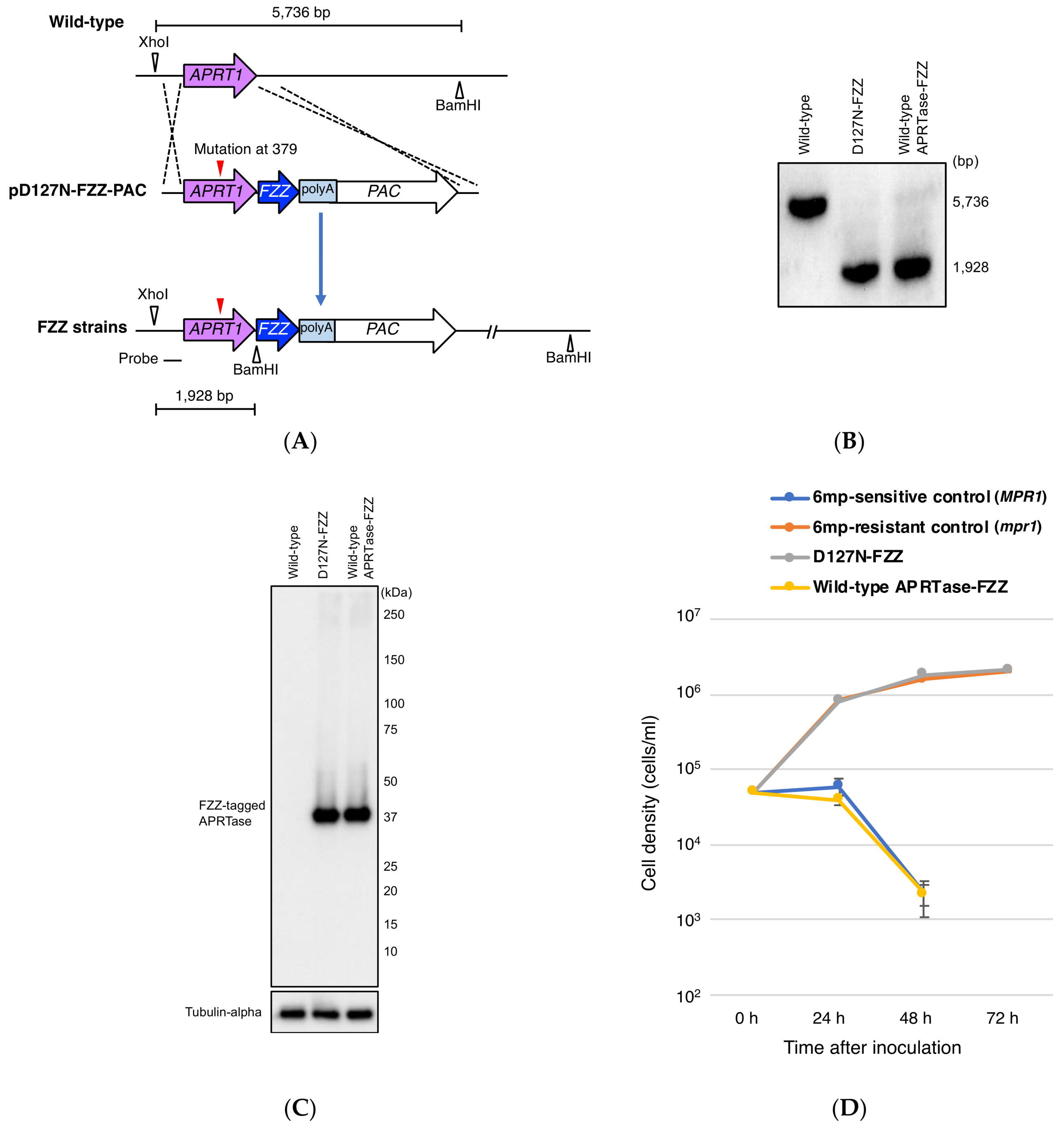
2.4. Construction of C-Terminal Epitope-Tagged Vectors
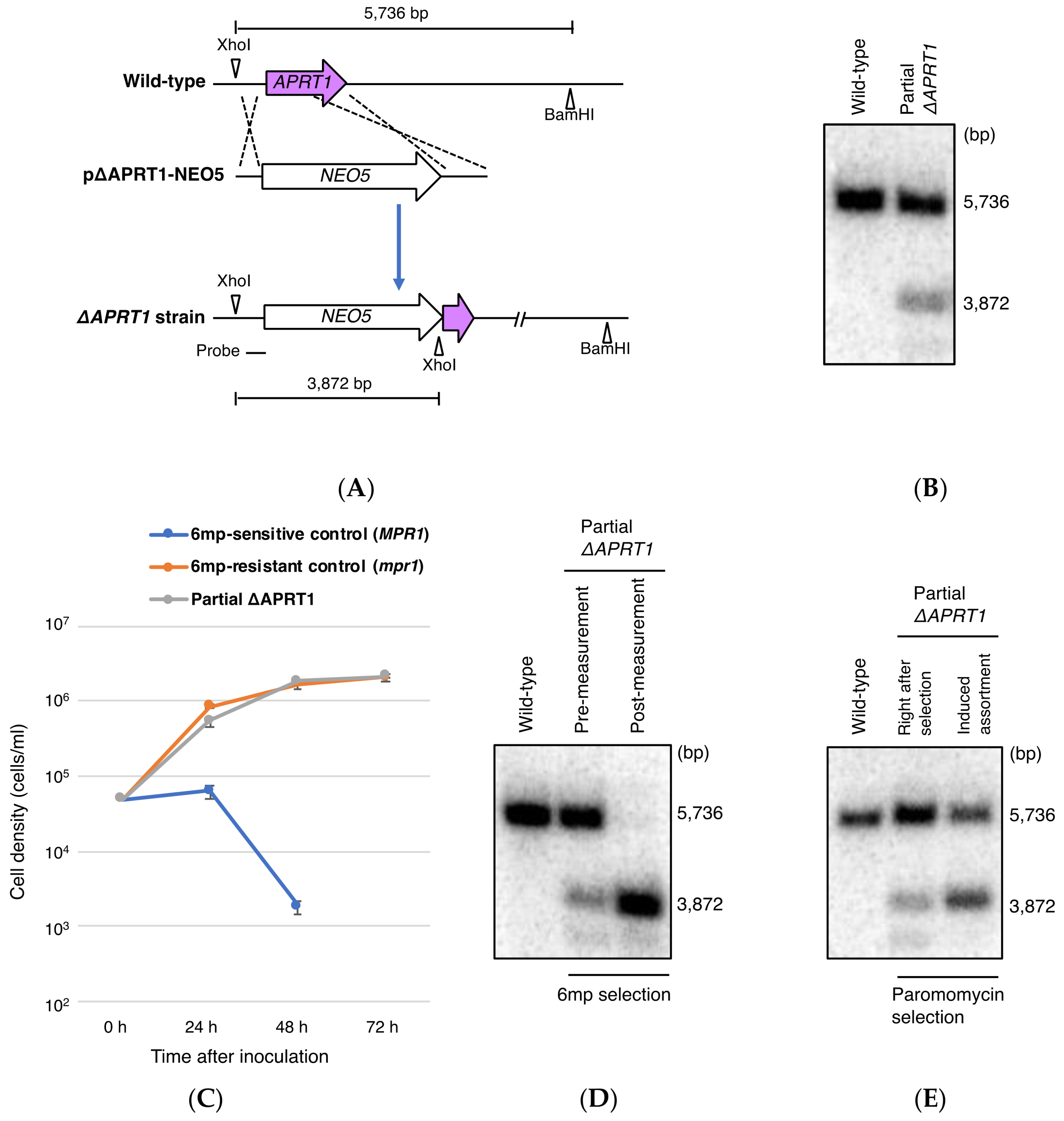
2.5. Construction of Macronucleus (MAC) APRT1 Disruption Vector
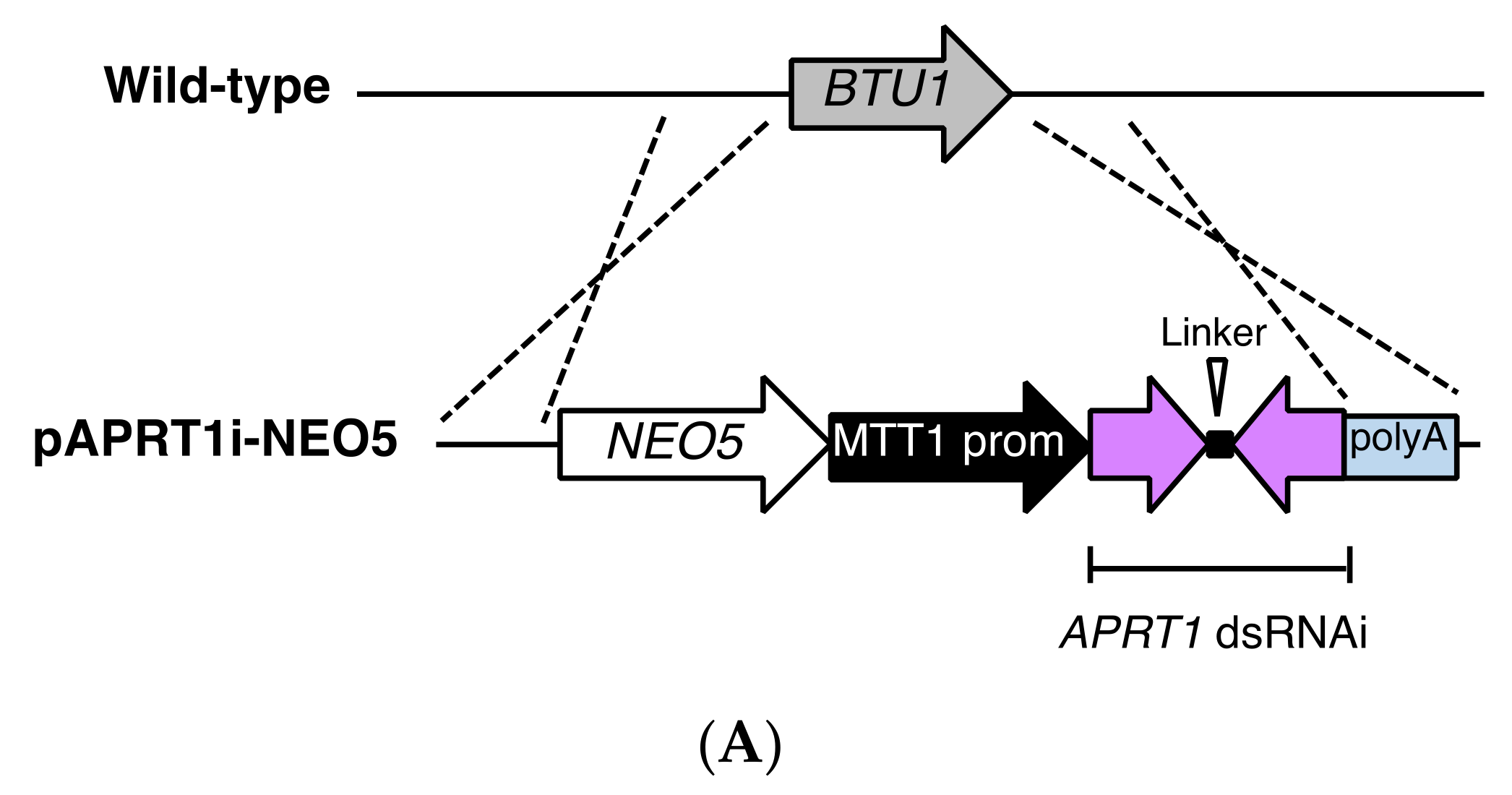
2.6. APRT1 RNA Interference (RNAi) Vector Construction and Gene Knockdown
2.7. Southern Blotting
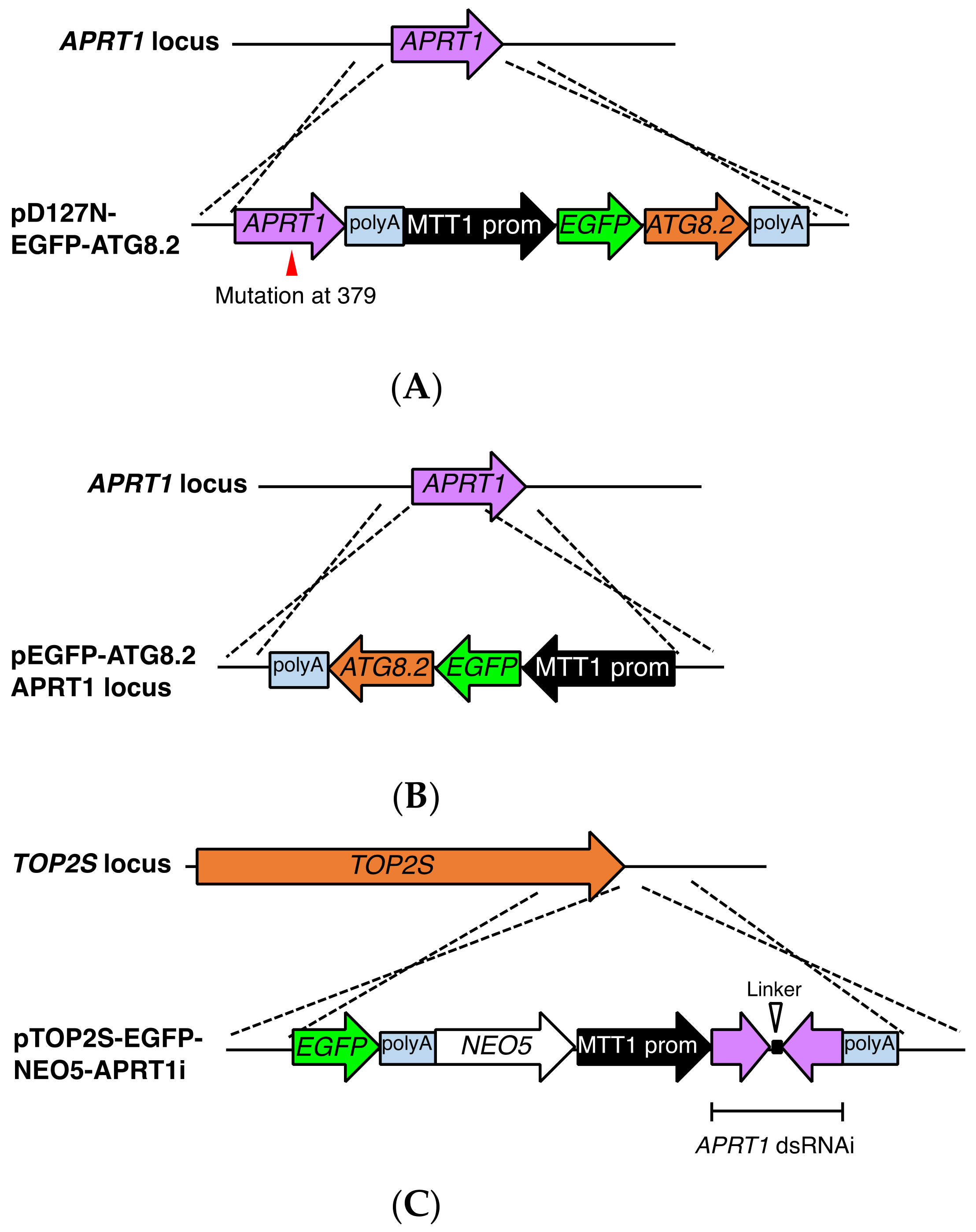
2.8. Construction of EGFP-Tagged ATG8.2 Expression Vectors Based on pD127-FZZ-PAC
2.9. Construction of EGFP-Tagged ATG8.2 Expression Vectors Based on pΔAPRT1-NEO5
2.10. Construction of EGFP-Tagged TOP2S Expression Vectors Based on pAPRT1i-NEO5
3. Results and Discussion
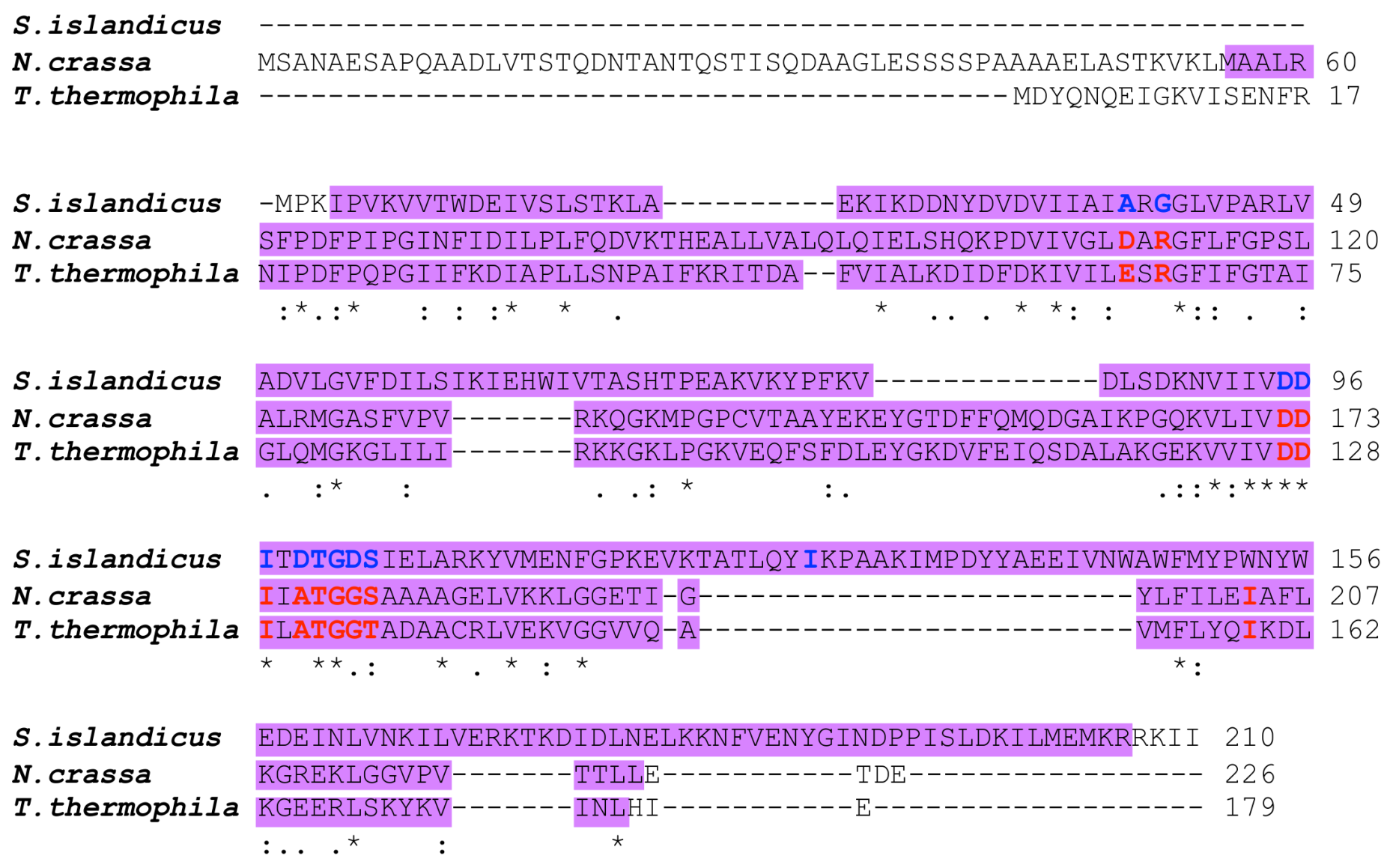
3.1. Mutation in APRT1 of 6mpr Strains
3.2. Expression of D127N from APRT1 Genomic Locus Confers 6mp Resistance
3.3. Partial Loss of MAC APRT1 Copies is Sufficient to Confer 6mp Resistance
3.4. RNAi Against APRT1 Enables Survival in the Presence of 6mp
3.5. Potential Utility of APRT1 as a New Selection Marker
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ruehle, M.D.; Orias, E.; Pearson, C.G. Tetrahymena as a unicellular model eukaryote: Genetic and genomic tools. Genetics 2016, 203, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Price, J.V.; Kieft, G.L.; Kent, J.R.; Sievers, E.L.; Cech, T.R. Sequence requirements for self-splicing of the Tetrahymena thermophila pre-ribosomal RNA. Nucleic Acids Res. 1985, 13, 1871–1889. [Google Scholar] [CrossRef] [PubMed]
- Noto, T.; Mochizuki, K. Whats, hows and whys of programmed DNA elimination in Tetrahymena. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Lukaszewicz, A.; Howard-Till, R.A.; Loidl, J. Mus81 nuclease and Sgs1 helicase are essential for meiotic recombination in a protist lacking a synaptonemal complex. Nucleic Acids Res. 2013, 41, 9296–9309. [Google Scholar] [CrossRef] [PubMed]
- Akematsu, T.; Kobayashi, T.; Osada, E.; Fukuda, Y.; Endoh, H.; Pearlman, R.E. Programmed nuclear death and its relation to apoptosis and autophagy during sexual reproduction in Tetrahymena thermophila. Jpn. J. Protozool. 2012, 45, 1–12. [Google Scholar]
- Gaertig, J.; Gu, L.; Hai, B.; Gorovsky, M.A. High frequency vector-mediated transformation and gene replacement in Tetrahymena. Nucleic Acids Res. 1994, 22, 5391–5398. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K. High efficiency transformation of Tetrahymena using a codon-optimized neomycin resistance gene. Gene 2008, 425, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Marsala, C.; Kosoy, R.; Gaertig, J. Kinesin-II is preferentially targeted to assembling cilia and is required for ciliogenesis and normal cytokinesis in Tetrahymena. Mol. Biol. Cell 1999, 10, 3081–3096. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Morgan, G.; Winey, M.; Sperling, L.; Turkewitz, A.P. Elucidation of clathrin-mediated endocytosis in Tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genet. 2005, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Mori, C.; Hiraoka, Y.; Haraguchi, T. Puromycin resistance gene as an effective selection marker for ciliate Tetrahymena. Gene 2014, 534, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Xiong, J.; Zhang, C.; Berquist, B.R.; Yang, R.; Zhao, M.; Molascon, A.J.; Kwiatkowski, S.Y.; Yuan, D.; Qin, Z.; et al. Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev. 2013, 27, 1662–1679. [Google Scholar] [CrossRef] [PubMed]
- Howard-Till, R.A.; Lukaszewicz, A.; Novatchkova, M.; Loidl, J. A single cohesin complex performs mitotic and meiotic functions in the protist Tetrahymena. PLoS Genet. 2013, 9, e1003418. [Google Scholar] [CrossRef] [PubMed]
- Orias, E.; Cervantes, M.D.; Hamilton, E.P. Tetrahymena thermophila, a unicellular eukaryote with separate germline and somatic genomes. Res. Microbiol. 2011, 162, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Sugai, T. Amitotic division of the macronucleus in Tetrahymena thermophila: DNA distribution by genomic unit. Zoolog. Sci. 2011, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.; Sugai, T. Developmental progression of Tetrahymena through the cell cycle and conjugation. Methods Cell Biol. 2012, 109, 177–236. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.L.; Straight, S.; Allan, P.W. Use of Tetrahymena pyriformis to evaluate the effects of purine and pyrimidine analogs. J. Protozool. 1970, 17, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, L.; Wellman, A.M. Purine biosynthesis and its regulation in Neurospora crassa. BBA-Nucleic Acids Protein Synth. 1980, 607, 350–360. [Google Scholar] [CrossRef]
- Byrne, B.C.; Brussard, T.B.; Bruns, P.J. Induced resistance to 6-methylpurine and cycloheximide in Tetrahymena. I. Germ line mutants of T. thermophila. Genetics 1978, 89, 695–702. [Google Scholar] [PubMed]
- Bruns, P.J.; Katzen, A.L.; Martin, L.; Blackburn, E.H. A drug-resistant mutation in the ribosomal DNA of Tetrahymena. Proc. Natl. Acad. Sci. USA 1985, 82, 2844–2846. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.L. Cytogenetics of genomic exclusion in Tetrahymena. Genetics 1967, 55, 797–822. [Google Scholar] [PubMed]
- Pendyala, L.; Smyth, J.; Wellman, A.M. Nature of 6-methylpurine inhibition and characterization of two 6-methylpurine-resistant mutants of Neurospora crassa. J. Bacteriol. 1979, 137, 248–255. [Google Scholar] [PubMed]
- Wang, C.C. Parasite enzymes as potential targets for antiparasitic chemotherapy. J. Med. Chem. 1984, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; She, Q.; Bi, H.; Whitaker, R.J. The apt/6-methylpurine counterselection system and Its applications in genetic studies of the hyperthermophilic archaeon Sulfolobus islandicus. Appl. Environ. Microbiol. 2016, 82, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Schoeberl, U.E.; Mochizuki, K. Modules for C-terminal epitope tagging of Tetrahymena genes. J. Microbiol. Methods 2010, 82, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Akematsu, T.; Fukuda, Y.; Garg, J.; Fillingham, J.S.; Pearlman, R.E.; Loidl, J. Post-meiotic DNA double-strand breaks occur in Tetrahymena, and require Topoisomerase II and Spo11. eLife 2017, 6, e26176. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, F.; Santovito, G.; Formigari, A.; Bisharyan, Y.; Cassidy-Hanley, D.; Clark, T.G.; Piccinni, E. MTT2, a copper-inducible metallothionein gene from Tetrahymena thermophila. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2008, 147, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Hanley, D.; Bowen, J.; Lee, J.H.; Cole, E.; VerPlank, L.A.; Gaertig, J.; Gorovsky, M.A.; Bruns, P.J. Germline and somatic transformation of mating Tetrahymena thermophila by particle bombardment. Genetics 1997, 146, 135–147. [Google Scholar] [PubMed]
- Shang, Y.; Song, X.; Bowen, J.; Corstanje, R.; Gao, Y.; Gaertig, J.; Gorovsky, M.A. A robust inducible-repressible promoter greatly facilitates gene knockouts, conditional expression, and overexpression of homologous and heterologous genes in Tetrahymena thermophila. Proc. Natl. Acad. Sci. USA 2002, 99, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Birien, T.; Thiel, A.; Henneke, G.; Flament, D.; Moalic, Y.; Jebbar, M. Development of an effective 6-methylpurine counterselection marker for genetic manipulation in Thermococcus barophilus. Genes 2018, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Hanley, D.M. Tetrahymena in the laboratory: strain resources, methods for culture, maintenance, and storage. Methods Cell Biol. 2012, 109, 237–276. [Google Scholar] [CrossRef] [PubMed]
- Bleyman, L.K.; Bruns, P.J. Genetics of cycloheximide resistance in Tetrahymena. Genetics 1977, 87, 275–284. [Google Scholar] [PubMed]
- Zimmerman, E.F.; Magasanik, B. Utilization and interconversion of purine bases and ribonucleosides by Salmonella typhimurium. J. Biol. Chem. 1964, 239, 293–300. [Google Scholar] [PubMed]
- Sarver, A.E.; Wang, C.C. The adenine phosphoribosyltransferase from Giardia lamblia has a unique reaction mechanism and unusual substrate binding properties. J. Biol. Chem. 2002, 277, 39973–39980. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Jayaram, H.N.; Pillwein, K.; Natsumeda, Y.; Reardon, M.A.; Zhen, Y.S. Salvage pathways as targets of chemotherapy. Adv. Enzyme Regul. 1987, 26, 335–352. [Google Scholar] [CrossRef]
- Liu, M.-L.; Yao, M.-C. Role of ATG8 and autophagy in programmed nuclear degradation in Tetrahymena thermophila. Eukaryot. Cell 2012, 11, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Sledz, C.A.; Williams, B.R.G. RNA interference in biology and disease. Blood 2005, 106, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Chalker, D.L. Transformation and strain engineering of Tetrahymena. Methods Cell Biol. 2012, 109, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Spangler, E.A.; Blackburn, E.H. The nucleotide sequence of the 17S ribosomal RNA gene of Tetrahymena thermophila and the identification of point mutations resulting in resistance to the antibiotics paromomycin and hygromycin. J. Biol. Chem. 1985, 260, 6334–6340. [Google Scholar] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transformation Plasmids | Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 |
|---|---|---|---|---|
| pD127N-EGFP-ATG8.2 | 0/96 | 0/96 | 0/96 | 0/96 |
| pEGFP-ATG8.2 APRT1 locus | 0/96 | 0/96 | 0/96 | 0/96 |
| pTOP2S-EGFP-NEO5-APRT1i | 1/96 | 0/96 | 1/96 | 1/96 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akematsu, T.; Findlay, A.; Fukuda, Y.; Pearlman, R.E.; Loidl, J.; Orias, E.; P. Hamilton, E. Resistance to 6-Methylpurine is Conferred by Defective Adenine Phosphoribosyltransferase in Tetrahymena. Genes 2018, 9, 179. https://doi.org/10.3390/genes9040179
Akematsu T, Findlay A, Fukuda Y, Pearlman RE, Loidl J, Orias E, P. Hamilton E. Resistance to 6-Methylpurine is Conferred by Defective Adenine Phosphoribosyltransferase in Tetrahymena. Genes. 2018; 9(4):179. https://doi.org/10.3390/genes9040179
Chicago/Turabian StyleAkematsu, Takahiko, Andrew Findlay, Yasuhiro Fukuda, Ronald E. Pearlman, Josef Loidl, Eduardo Orias, and Eileen P. Hamilton. 2018. "Resistance to 6-Methylpurine is Conferred by Defective Adenine Phosphoribosyltransferase in Tetrahymena" Genes 9, no. 4: 179. https://doi.org/10.3390/genes9040179