Kinetics and Thermodynamics of DNA Processing by Wild Type DNA-Glycosylase Endo III and Its Catalytically Inactive Mutant Forms


Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of the pET28c-Endo III Expression Vector and Mutagenesis
2.2. Protein Expression and Purification
2.3. Oligodeoxynucleotides
2.4. Stopped-Flow Experiments
2.5. Product Analysis
2.6. Kinetic Data Analysis
2.7. Thermodynamic Analysis
3. Results
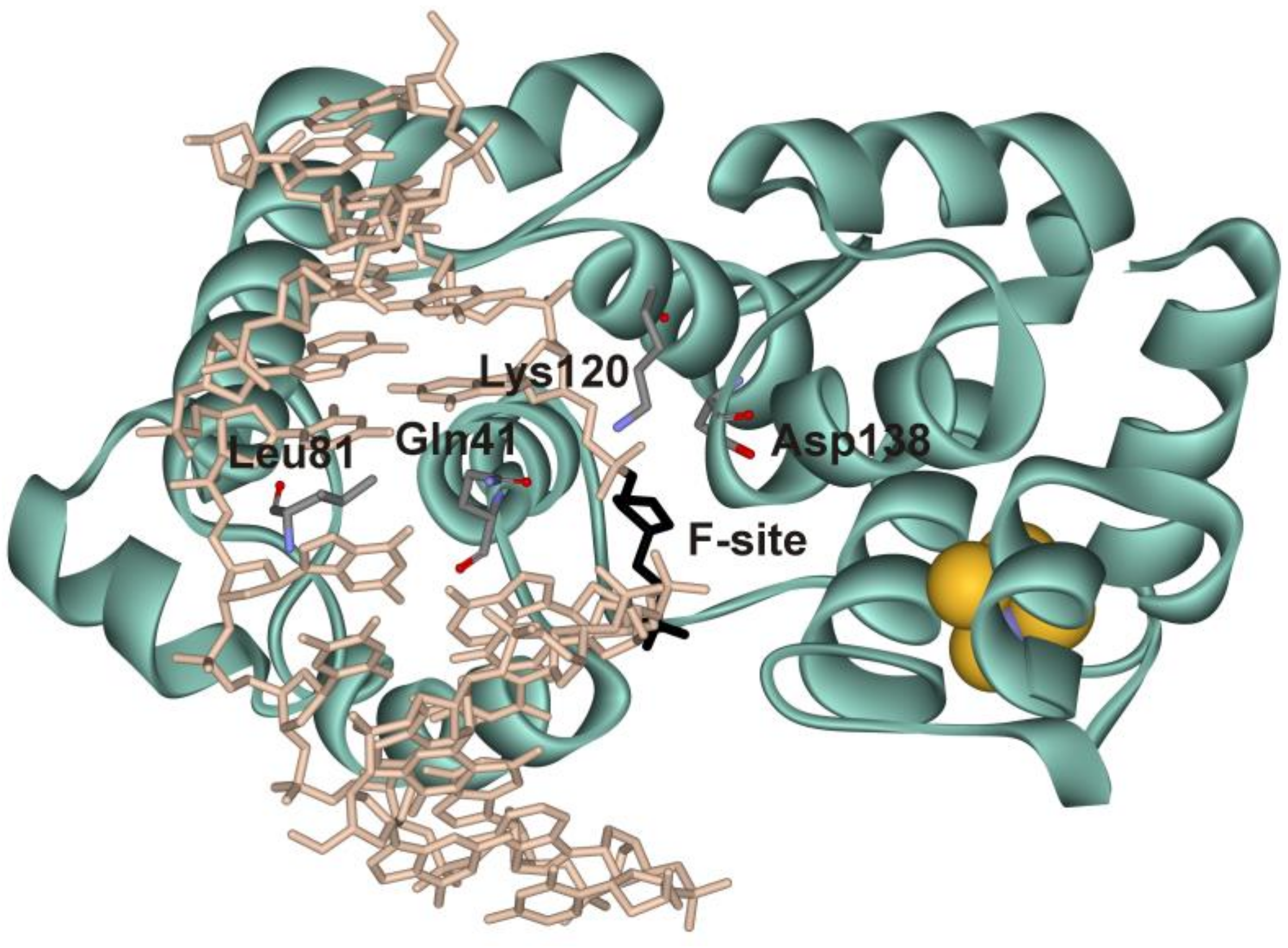
3.1. Rationale
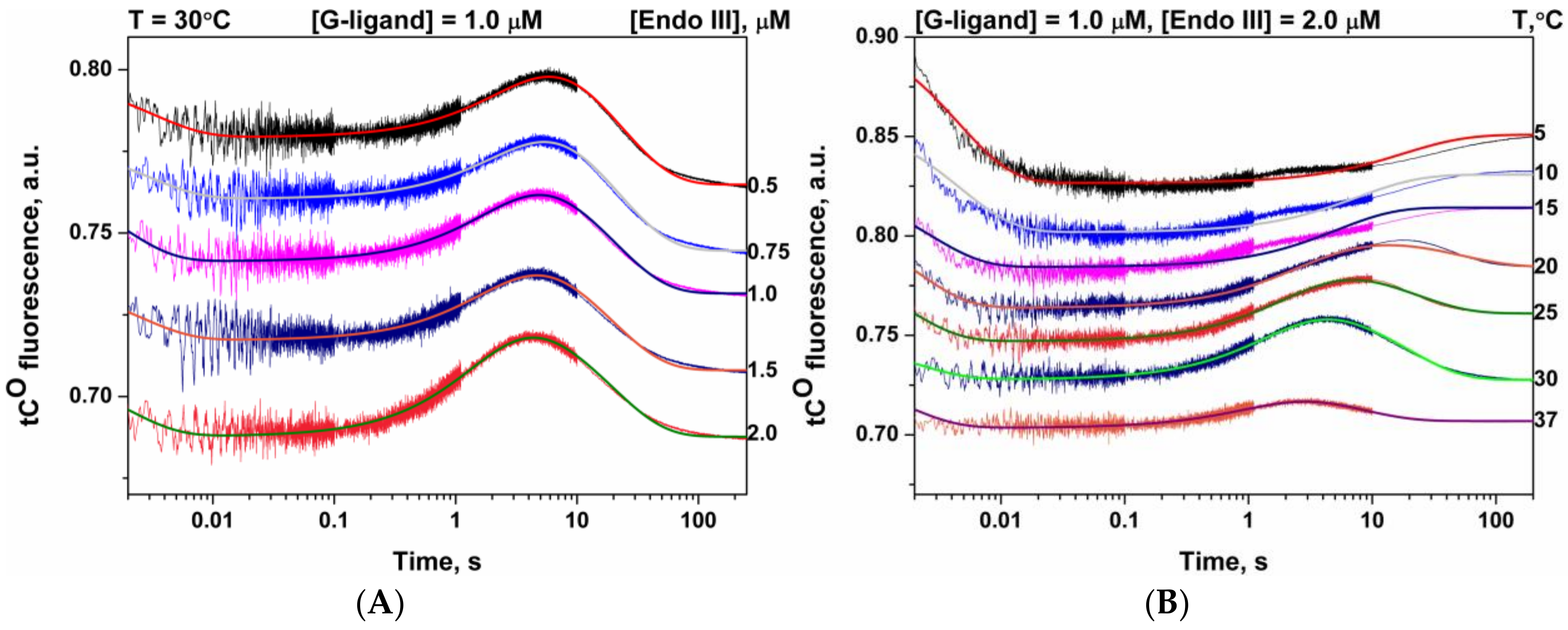
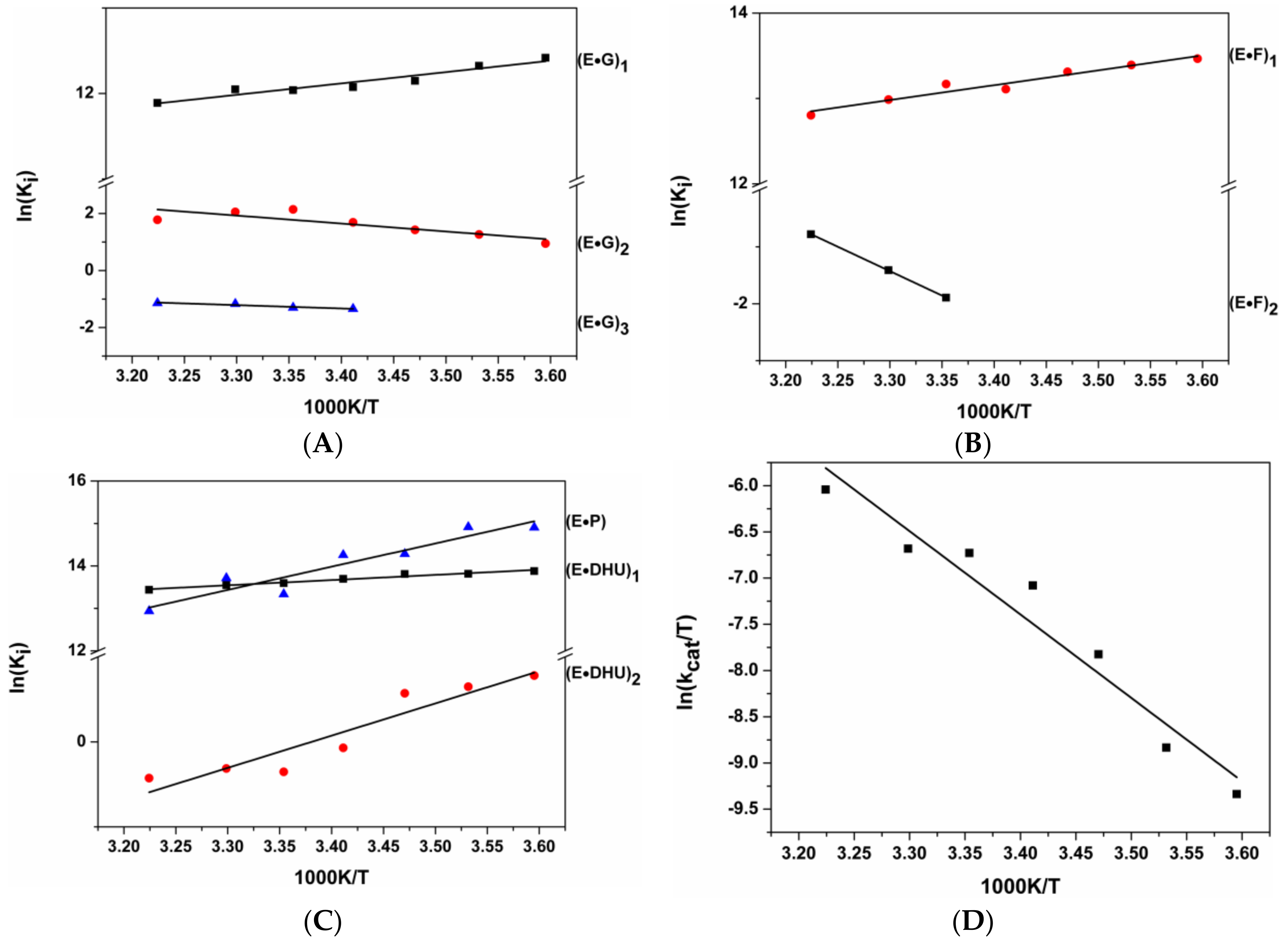
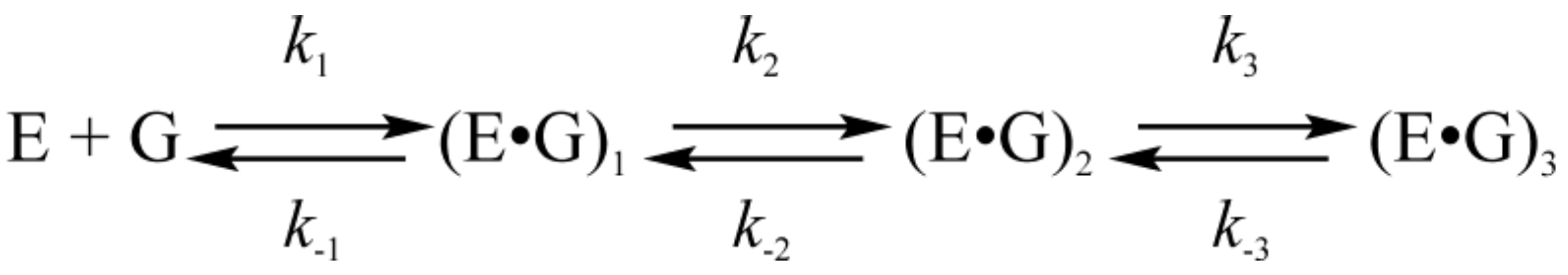
3.2. Interactions of Endonuclease III with G-Ligand
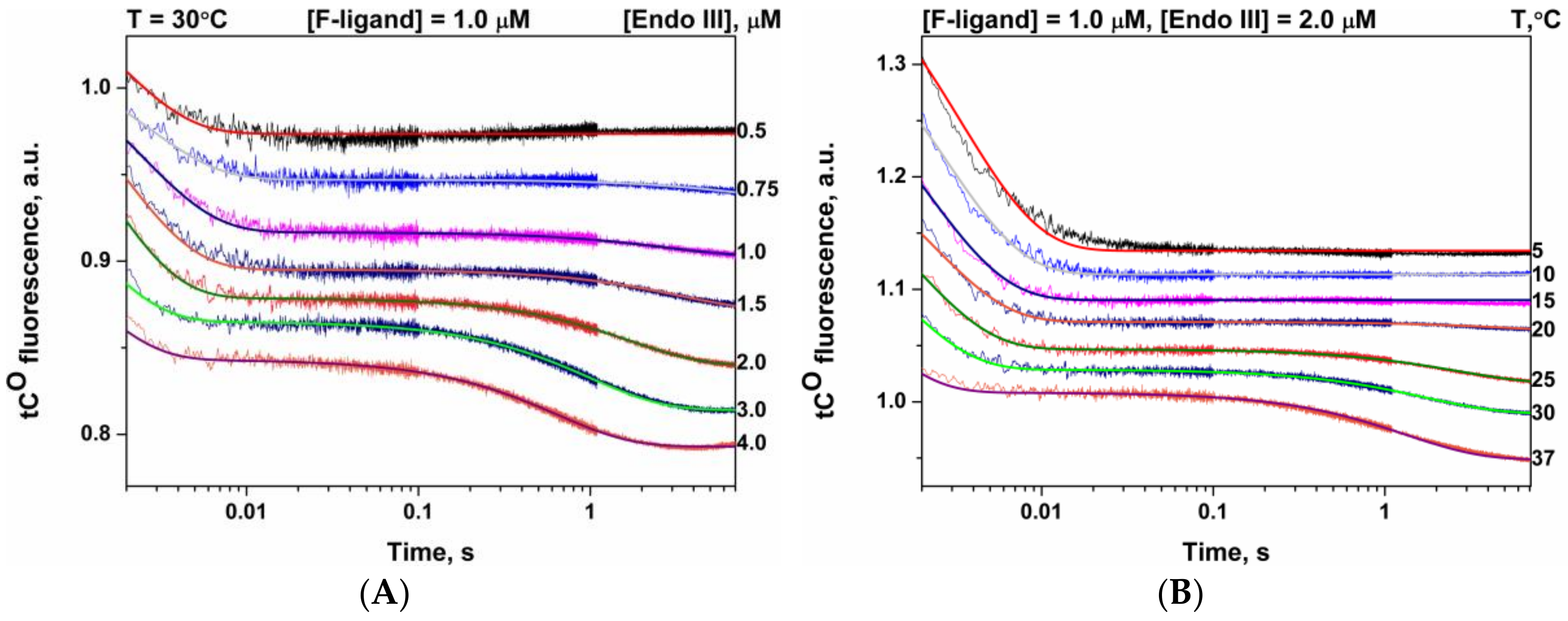
3.3. Interactions of Endonuclease III with F-Ligand
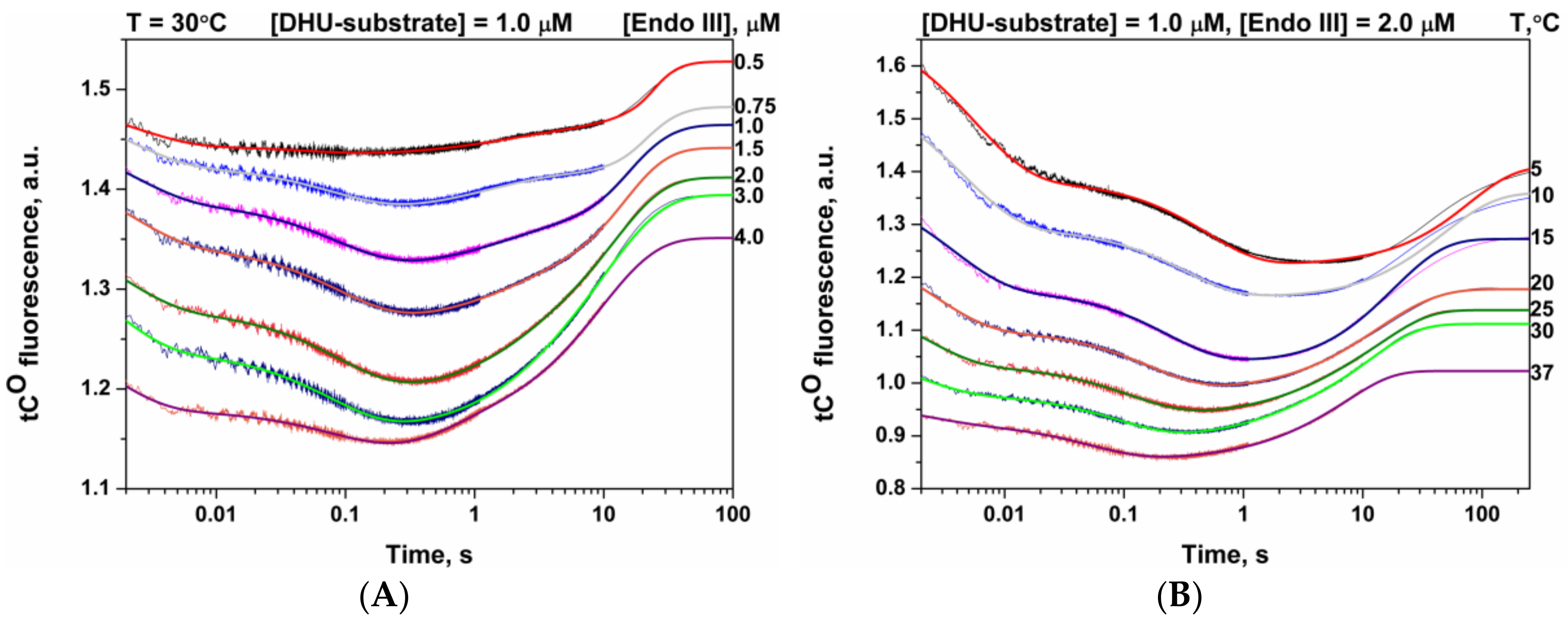
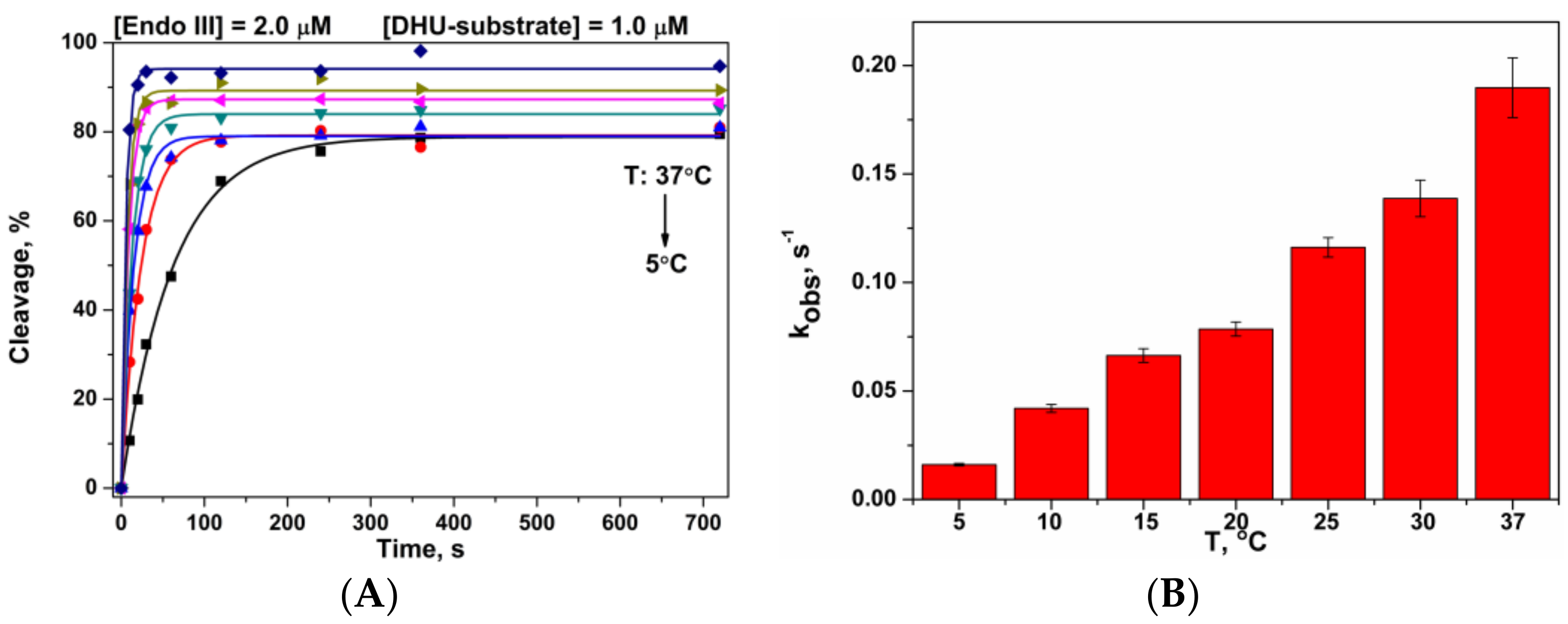
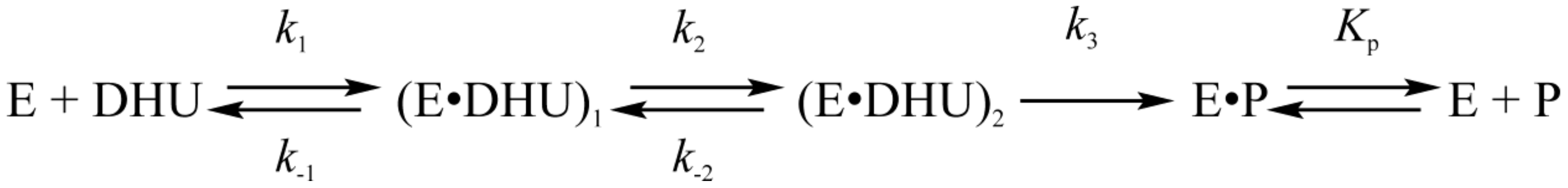
3.4. Interactions of Endonuclease III with DHU-Substrate
3.5. Thermodynamic Analysis
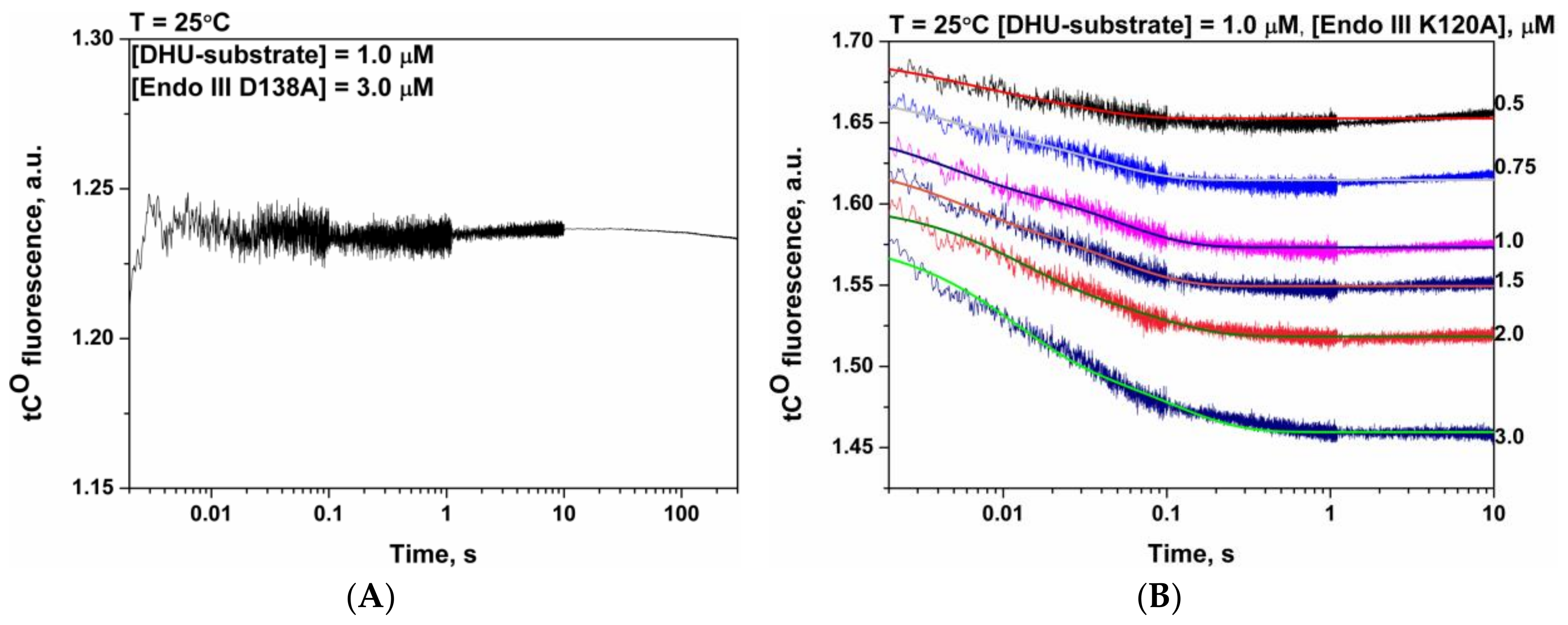
3.6. Mutational Analysis
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wallace, S.S. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002, 33, 1–14. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P.; Birincioglu, M.; Rodriguez, H. Free radical-induced damage to DNA: Mechanisms and measurement. Free Radic. Biol. Med. 2002, 32, 1102–1115. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, Z.; Kow, Y.W.; Purmal, A.A.; Cunningham, R.P.; Wallace, S.S. New substrates for old enzymes. 5-hydroxy-2′-deoxycytidine and 5-hydroxy-2′-deoxyuridine are substrates for Escherichia coli endonuclease iii and formamidopyrimidine DNA N-glycosylase, while 5-hydroxy-2′-deoxyuridine is a substrate for uracil DNA N-glycosylase. J. Biol. Chem. 1994, 269, 18814–18820. [Google Scholar] [PubMed]
- Dizdaroglu, M.; Bauche, C.; Rodriguez, H.; Laval, J. Novel substrates of Escherichia coli Nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry 2000, 39, 5586–5592. [Google Scholar] [CrossRef] [PubMed]
- Katafuchi, A.; Nakano, T.; Masaoka, A.; Terato, H.; Iwai, S.; Hanaoka, F.; Ide, H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J. Biol. Chem. 2004, 279, 14464–14471. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Karahalil, B.; Senturker, S.; Buckley, T.J.; Roldan-Arjona, T. Excision of products of oxidative DNA base damage by human Nth1 protein. Biochemistry 1999, 38, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Eide, L.; Luna, L.; Gustad, E.C.; Henderson, P.T.; Essigmann, J.M.; Demple, B.; Seeberg, E. Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry 2001, 40, 6653–6659. [Google Scholar] [CrossRef] [PubMed]
- Asagoshi, K.; Odawara, H.; Nakano, H.; Miyano, T.; Terato, H.; Ohyama, Y.; Seki, S.; Ide, H. Comparison of substrate specificities of Escherichia coli endonuclease III and its mouse homologue (mNTH1) using defined oligonucleotide substrates. Biochemistry 2000, 39, 11389–11398. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Laval, J.; Boiteux, S. Substrate specificity of the Escherichia coli endonuclease III: Excision of thymine- and cytosine-derived lesions in DNA produced by radiation-generated free radicals. Biochemistry 1993, 32, 12105–12111. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.L.; Michaels, M.L.; Lloyd, R.S. Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem. 1994, 269, 32709–32712. [Google Scholar] [PubMed]
- Nash, H.M.; Lu, R.; Lane, W.S.; Verdine, G.L. The critical active-site amine of the human 8-oxoguanine DNA glycosylase, hOGG1: Direct identification, ablation and chemical reconstitution. Chem. Biol. 1997, 4, 693–702. [Google Scholar] [CrossRef]
- Denver, D.R.; Swenson, S.L.; Lynch, M. An evolutionary analysis of the helix-hairpin-helix superfamily of DNA repair glycosylases. Mol. Biol. Evol. 2003, 20, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Thayer, M.M.; Ahern, H.; Xing, D.; Cunningham, R.P.; Tainer, J.A. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995, 14, 4108–4120. [Google Scholar] [PubMed]
- Fromme, J.C.; Verdine, G.L. Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 2003, 22, 3461–3471. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Kladova, O.A.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.K.; Zharkov, D.O.; Fedorova, O.S. Conformational dynamics of DNA repair by Escherichia coli endonuclease III. J. Biol. Chem. 2015, 290, 14338–14349. [Google Scholar] [CrossRef] [PubMed]
- Lukina, M.V.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. The kinetic analysis of recognition of the damaged nucleotides by mutant forms of the 8-oxoguanine DNA glycosylase hOGG1. Russ. J. Bioorg. Chem. 2017, 43, 1–12. [Google Scholar] [CrossRef]
- Tyugashev, T.E.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Interaction features of adenine DNA glycosylase Muty from E. coli with DNA substrates. Russ. J. Bioorg. Chem. 2017, 43, 13–22. [Google Scholar] [CrossRef]
- Yakovlev, D.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Search for modified DNA sites with the human methyl-CpG-binding enzyme MBD4. Acta Nat. 2017, 9, 88–98. [Google Scholar]
- Nelson, S.R.; Dunn, A.R.; Kathe, S.D.; Warshaw, D.M.; Wallace, S.S. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl. Acad. Sci. USA 2014, 111, E2091–E2099. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Step-by-step mechanism of DNA damage recognition by human 8-oxoguanine DNA glycosylase. Biochim. Biophys. Acta 2014, 1840, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Bergonzo, C.; Campbell, A.J.; Li, H.; Mechetin, G.V.; de los Santos, C.; Grollman, A.P.; Fedorova, O.S.; Zharkov, D.O.; Simmerling, C. Active destabilization of base pairs by a DNA glycosylase wedge initiates damage recognition. Nucleic Acids Res. 2015, 43, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Wallace, S.S. Visualizing the search for radiation-damaged DNA bases in real time. Radiat. Phys. Chem. 2016, 128, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Iakovlev, D.A.; Misovets, I.V.; Ishchenko, A.A.; Saparbaev, M.K.; Kuznetsov, N.A.; Fedorova, O.S. Pre-steady-state kinetic analysis of damage recognition by human single-strand selective monofunctional uracil-DNA glycosylase SMUG1. Mol. Biosyst. 2017, 13, 2638–2649. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Wallace, S.S. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Kladova, O.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and kinetic analysis of lesion recognition by Escherichia coli endonuclease VIII. Genes 2017, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Krasnoperov, L.N.; Fedorova, O.S. Thermodynamics of the DNA damage repair steps of human 8-oxoguanine DNA glycosylase. PLoS ONE 2014, 9, e98495. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Vorobjev, Y.N.; Krasnoperov, L.N.; Fedorova, O.S. Thermodynamics of the multi-stage DNA lesion recognition and repair by formamidopyrimidine-DNA glycosylase using pyrrolocytosine fluorescence—Stopped-flow pre-steady-state kinetics. Nucleic Acids Res. 2012, 40, 7384–7392. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of damaged DNA binding and catalysis by human AP endonuclease 1. Acta Nat. 2016, 8, 103–110. [Google Scholar]
- Kuznetsov, N.A.; Fedorova, O.S. Thermodynamic analysis of fast stages of specific lesion recognition by DNA repair enzymes. Biochemistry 2016, 81, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.M.; Yu, J.; Cillo, J.; Lu, A.-L. The active site of the Escherichia coli muty DNA adenine glycosylase. J. Biol. Chem. 1999, 274, 29011–29018. [Google Scholar] [CrossRef] [PubMed]
- Manuel, R.C.; Hitomi, K.; Arvai, A.S.; House, P.G.; Kurtz, A.J.; Dodson, M.L.; McCullough, A.K.; Tainer, J.A.; Lloyd, R.S. Reaction intermediates in the catalytic mechanism of Escherichia coli Muty DNA glycosylase. J. Biol. Chem. 2004, 279, 46930–46939. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Grollman, A.P. Muty DNA glycosylase: Base release and intermediate complex formation. Biochemistry 1998, 37, 12384–12394. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.D.; David, S.S. A single engineered point mutation in the adenine glycosylase Muty confers bifunctional glycosylase/AP lyase activity. Biochemistry 2000, 39, 10098–10109. [Google Scholar] [CrossRef] [PubMed]
- Dalhus, B.; Forsbring, M.; Helle, I.H.; Vik, E.S.; Forstrom, R.J.; Backe, P.H.; Alseth, I.; Bjoras, M. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 2011, 19, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Stengel, G.; Purse, B.W.; Wilhelmsson, L.M.; Urban, M.; Kuchta, R.D. Ambivalent incorporation of the fluorescent cytosine analogues TC and TCO by human DNA polymerase alpha and Klenow fragment. Biochemistry 2009, 48, 7547–7555. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H.; Wistort, P.M.; Bank, J.F.; Bakerian, R.H.; Cunningham, R.P. Purification and characterization of Escherichia coli endonuclease III from the cloned nth gene. Biochemistry 1989, 28, 4444–4449. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Vorobiev, Y.N.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Kinetic basis of lesion specificity and opposite-base specificity of Escherichia coli formamidopyrimidine-DNA glycosylase. Biochemistry 2007, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Zharkov, D.O.; Koval, V.V.; Buckle, M.; Fedorova, O.S. Reversible chemical step and rate-limiting enzyme regeneration in the reaction catalyzed by formamidopyrimidine-DNA glycosylase. Biochemistry 2009, 48, 11335–11343. [Google Scholar] [CrossRef] [PubMed]
- Koval, V.V.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Real-time studies of conformational dynamics of the repair enzyme E. coli formamidopyrimidine-DNA glycosylase and its DNA complexes during catalytic cycle. Mutat. Res. 2010, 685, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kuzmic, P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Fedorova, O.S. Conformational dynamics of the interaction of Escherichia coli endonuclease VIII with DNA substrates. DNA Repair 2012, 11, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; Paula, J. Atkins' Physical Chemistry, 8th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Ragone, R.; Colonna, G.; Ambrosone, L. Reliability of the van’t Hoff plots. J. Phys. Chem. 1995, 99, 13050. [Google Scholar] [CrossRef]
- Rodgers, B.J.; Elsharif, N.A.; Vashisht, N.; Mingus, M.M.; Mulvahill, M.A.; Stengel, G.; Kuchta, R.D.; Purse, B.W. Functionalized tricyclic cytosine analogues provide nucleoside fluorophores with improved photophysical properties and a range of solvent sensitivities. Chemistry 2014, 20, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.F.; McRee, D.E.; Fisher, C.L.; O’Handley, S.F.; Cunningham, R.P.; Tainer, J.A. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science 1992, 258, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Sandin, P.; Borjesson, K.; Li, H.; Martensson, J.; Brown, T.; Wilhelmsson, L.M.; Albinsson, B. Characterization and use of an unprecedentedly bright and structurally non-perturbing fluorescent DNA base analogue. Nucleic Acids Res. 2008, 36, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Dragan, A.I.; Crane-Robinson, C.; Breslauer, K.J.; Remeta, D.P.; Minetti, C.A. What drives proteins into the major or minor grooves of DNA? J. Mol. Biol. 2007, 365, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Crenshaw, C.M.; Nam, K.; Oo, K.; Kutchukian, P.S.; Bowman, B.R.; Karplus, M.; Verdine, G.L. Enforced presentation of an extrahelical guanine to the lesion recognition pocket of human 8-oxoguanine glycosylase, hOGG1. J. Biol. Chem. 2012, 287, 24916–24928. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Santos, W.L.; Verdine, G.L. Structure of a DNA glycosylase searching for lesions. Science 2006, 311, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Spong, M.C.; Nam, K.; Banerjee, A.; Jiralerspong, S.; Karplus, M.; Verdine, G.L. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature 2009, 462, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Nam, K.; Spong, M.C.; Banerjee, A.; Sung, R.J.; Zhang, M.; Karplus, M.; Verdine, G.L. Strandwise translocation of a DNA glycosylase on undamaged DNA. Proc. Natl. Acad. Sci. USA 2012, 109, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Fromme, J.C.; Verdine, G.L. DNA lesion recognition by the bacterial repair enzyme Mutm. J. Biol. Chem. 2003, 278, 51543–51548. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; van Oijen, A.M.; Banerjee, A.; Verdine, G.L.; Xie, X.S. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 5752–5757. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; Luo, G.; Kou, S.C.; Mangel, W.F.; Verdine, G.L.; Bagchi, B.; Xie, X.S. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009, 16, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; Kad, N.M.; Nelson, S.R.; Warshaw, D.M.; Wallace, S.S. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011, 39, 7487–7498. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
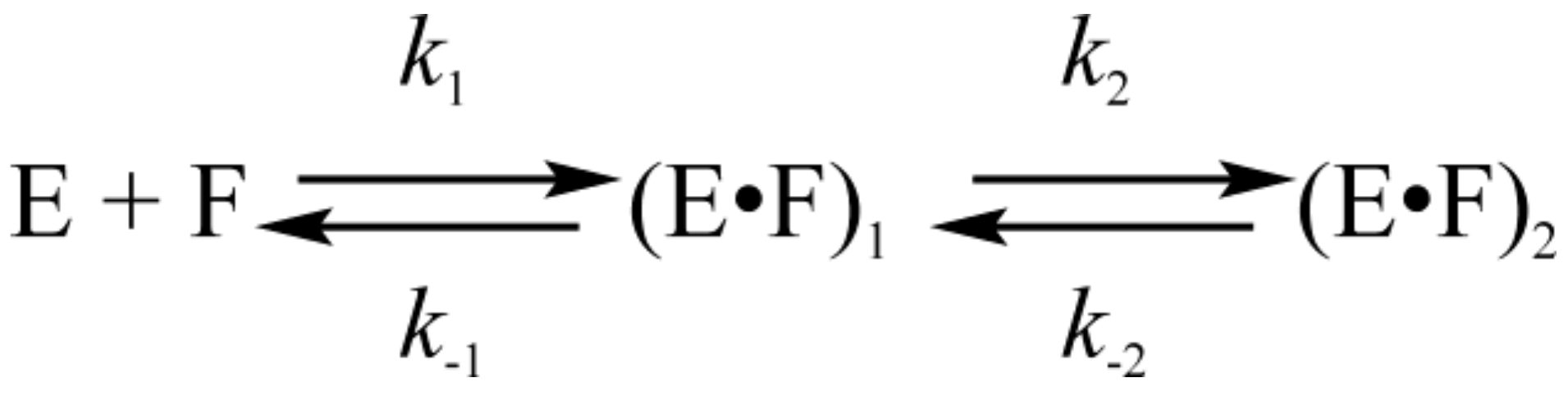{kind=link}
{kind=link}
| Shorthand | Sequence |
|---|---|
| DHU-substrate, X = DHU F-ligand, X = F-site G-ligand, X = G | 5′-TCTCTCTC X CCTTCCTT-3′ 3′-AGAGAGAG(tCO)GGAAGGAA-5′ |

| T (°C) Constants | 5 | 10 | 15 | 20 | 25 | 30 | 37 |
|---|---|---|---|---|---|---|---|
| k1, M−1 s−1 | (27 ± 2)×106 | (30 ± 4)×106 | (45 ± 5)×106 | (52 ± 5)×106 | (61 ± 4)×106 | (65 ± 11)×106 | (72 ± 3)×106 |
| k−1, s−1 | 110 ± 3 | 131 ± 11 | 239 ± 11 | 299 ± 20 | 360 ± 14 | 379 ± 36 | 494 ± 24 |
| K1, M−1 | (0.25 ± 0.01)×106 | (0.23 ± 0.04)×106 | (0.19 ± 0.02)×106 | (0.18 ± 0.02)×106 | (0.17 ± 0.01)×106 | (0.17 ± 0.03)×106 | (0.15 ± 0.01)×106 |
| k2, s−1 | 0.034 ± 0.006 | 0.15 ± 0.05 | 0.33 ± 0.01 | 0.47 ± 0.14 | 0.58 ± 0.14 | 0.9 ± 0.3 | 1.2 ± 0.1 |
| k−2, s−1 | 0.013 ± 0.003 | 0.04 ± 0.01 | 0.078 ± 0.004 | 0.090 ± 0.030 | 0.070 ± 0.030 | 0.120 ± 0.060 | 0.200 ± 0.050 |
| K2 | 2.6 ± 0.8 | 3.5 ± 1.8 | 4.2 ± 0.2 | 5.42 ± 2.55 | 8.5 ± 4.2 | 7.8 ± 4.5 | 5.9 ± 1.5 |
| k3, s−-1 | N/A | N/A | N/A | 0.004 ± 0.002 | 0.014 ± 0.004 | 0.017 ± 0.003 | 0.029 ± 0.006 |
| k−3, s−1 | N/A | N/A | N/A | 0.015 ± 0.003 | 0.050 ± 0.004 | 0.054 ± 0.016 | 0.09 ± 0.04 |
| K3 | N/A | N/A | N/A | 0.26 ± 0.12 | 0.27 ± 0.09 | 0.3 ± 0.1 | 0.32 ± 0.15 |
| Kass, M–1 | (0.9 ± 0.01)×106 | (1.04 ± 0.04)×106 | (0.99 ± 0.02)×106 | (1.4 ± 0.02)×106 | (2.0 ± 0.01)×106 | (1.9 ± 0.03)×106 | (1.3 ± 0.01)×106 |
| T (°C) Constants | 5 | 10 | 15 | 20 | 25 | 30 | 37 |
|---|---|---|---|---|---|---|---|
| k1, M−1 s−1 | (86 ± 15)×106 | (98 ± 17)×106 | (102 ± 20)×106 | (106 ± 10)×106 | (156 ± 15)×106 | (172 ± 6)×106 | (192 ± 27)×106 |
| k−1, s−1 | 122 ± 12 | 150 ± 18 | 169 ± 15 | 214 ± 15 | 298 ± 50 | 395 ± 6 | 528 ± 34 |
| K1, M−1 | (0.71 ± 0.14)×106 | (0.65 ± 0.14)×106 | (0.60 ± 0.13)×106 | (0.49 ± 0.06)×106 | (0.52 ± 0.1)×106 | (0.44 ± 0.017)×106 | (0.36 ± 0.06)×106 |
| k2, s−1 | N/A | N/A | N/A | 0.005 ± 0.001 | 0.06 ± 0.01 | 0.19 ± 0.01 | 0.23 ± 0.06 |
| k−2, s−1 | N/A | N/A | N/A | 0.17 ± 0.04 | 0.37 ± 0.02 | 0.804 ± 0.003 | 0.50 ± 0.02 |
| K2 | N/A | N/A | N/A | 0.03 ± 0.01 | 0.15 ± 0.04 | 0.24 ± 0.01 | 0.46 ± 0.1 |
| Kass, M−1 | (0.71 ± 0.14)×106 | (0.65 ± 0.14)×106 | (0.60 ± 0.13)×106 | (0.50 ± 0.06)×106 | (0.41 ± 0.10)×106 | (0.54 ± 0.17)×106 | (0.52 ± 0.06)×106 |
| T (°C) Constants | 5 | 10 | 15 | 20 | 25 | 30 | 37 |
|---|---|---|---|---|---|---|---|
| k1, M−1 s−1 | (50 ± 7)×106 | (63 ± 11)×106 | (88 ± 8)×106 | (98 ± 16)×106 | (116 ± 5)×106 | (127 ± 30)×106 | (144 ± 7)×106 |
| k−1, s−1 | 47 ± 6 | 63 ± 11 | 88 ± 15 | 111 ± 17 | 145 ± 11 | 166 ± 16 | 210 ± 35 |
| K1, M−1 | (1.1 ± 0.2)×106 | (1.0 ± 0.3)×106 | (1.0 ± 0.2)×106 | (0.9 ± 0.2)×106 | (0.8 ± 0.1)×106 | (0.8 ± 0.2)×106 | (0.7 ± 0.1)×106 |
| k2, s−1 | 2.1 ± 0.6 | 2.6 ± 0.4 | 3.5 ± 0.3 | 3.8 ± 0.4 | 3.8 ± 0.4 | 5.3 ± 1.2 | 9.5 ± 1.5 |
| k−2, s−1 | 1.0 ± 0.1 | 1.3 ± 0.1 | 2.0 ± 0.4 | 4.1 ± 0.5 | 5.5 ± 0.2 | 7.2 ± 1.3 | 14.5 ± 1.2 |
| K2 | 2.2 ± 0.7 | 1.9 ± 0.4 | 1.8 ± 0.4 | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.2 | 0.65 ± 0.12 |
| k3, s−1 | 0.027 ± 0.006 | 0.045 ± 0.007 | 0.13 ± 0.03 | 0.27 ± 0.06 | 0.39 ± 0.04 | 0.41 ± 0.09 | 0.8 ± 0.2 |
| KP, M−1 | (2.9 ± 1.7)×106 | (3.0 ± 2)×106 | (1.6 ± 1.4)×106 | (1.6 ± 1.4)×106 | (0.62 ± 0.5)×106 | (0.9 ± 0.4)×106 | (0.4 ± 0.1)×106 |
| Kass, M–1 | (3.5 ± 0.2)×106 | (2.9 ± 0.3)×106 | (2.8 ± 0.2)×106 | (1.7 ± 0.2)×106 | (1.5 ± 0.1)×106 | (1.4 ± 0.2)×106 | (1.2 ± 0.1)×106 |
| DNA | Step Number | ΔH, kcal/mol | ΔS, cal/(mol × K) | ΔG298, kcal/mol a |
|---|---|---|---|---|
| G-ligand | 1 | −2.6 ± 0.3 | 15.1 ± 1.1 | −7.1 |
| 2 | 5.5 ± 1.5 | 22 ± 5 | −1.3 | |
| 3 | 2.3 ± 0.5 | 5.3 ± 1.7 | 0.7 | |
| 5.2 ± 2.3 | 42.4 ± 7.8 | −7.7 | ||
| F-ligand | 1 | −3.5 ± 0.4 | 14 ± 1 | −7.8 |
| 2 | 17.0 ± 0.1 | 53.3 ± 0.5 | 1.1 | |
| 13.5 ± 0.5 | 67.3 ± 1.5 | −6.7 | ||
| DHU-substrate | 1 | −2.4 ± 0.2 | 19 ± 1 | −8.0 |
| 2 | −7.5 ± 1.2 | −25 ± 4 | 0.2 | |
| −9.9 ± 1.4 | −6.0 ± 5.0 | −7.8 | ||
| Transition state of catalytic step 3 b | 18 ± 2 | −1.0 ± 0.1 | 18.0 | |
| Product dissociation step 4 | −11 ± 2 | 9 ± 6 | −7.9 |
| Constants | T (25 °C) |
|---|---|
| k1, M−1 s−1 | (8 ± 2) × 106 |
| k−1, s−1 | 60 ± 15 |
| K1, M−1 | (0.13 ± 0.05) × 106 |
| k2, s−1 | 4.6 ± 1.4 |
| k−2, s−1 | 12 ± 2 |
| K2 | 0.4 ± 0.1 |
| Kass, M–1 | 0.18 × 106 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kladova, O.A.; Krasnoperov, L.N.; Kuznetsov, N.A.; Fedorova, O.S. Kinetics and Thermodynamics of DNA Processing by Wild Type DNA-Glycosylase Endo III and Its Catalytically Inactive Mutant Forms. Genes 2018, 9, 190. https://doi.org/10.3390/genes9040190
Kladova OA, Krasnoperov LN, Kuznetsov NA, Fedorova OS. Kinetics and Thermodynamics of DNA Processing by Wild Type DNA-Glycosylase Endo III and Its Catalytically Inactive Mutant Forms. Genes. 2018; 9(4):190. https://doi.org/10.3390/genes9040190
Chicago/Turabian StyleKladova, Olga A., Lev N. Krasnoperov, Nikita A. Kuznetsov, and Olga S. Fedorova. 2018. "Kinetics and Thermodynamics of DNA Processing by Wild Type DNA-Glycosylase Endo III and Its Catalytically Inactive Mutant Forms" Genes 9, no. 4: 190. https://doi.org/10.3390/genes9040190