Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria



Abstract
1. Introduction
2. Experimental Procedures
2.1. Growth Conditions
2.2. Isolation of DNA
2.3. Genome Sequencing and Assembly
2.4. Genome Annotation and Genome Comparison
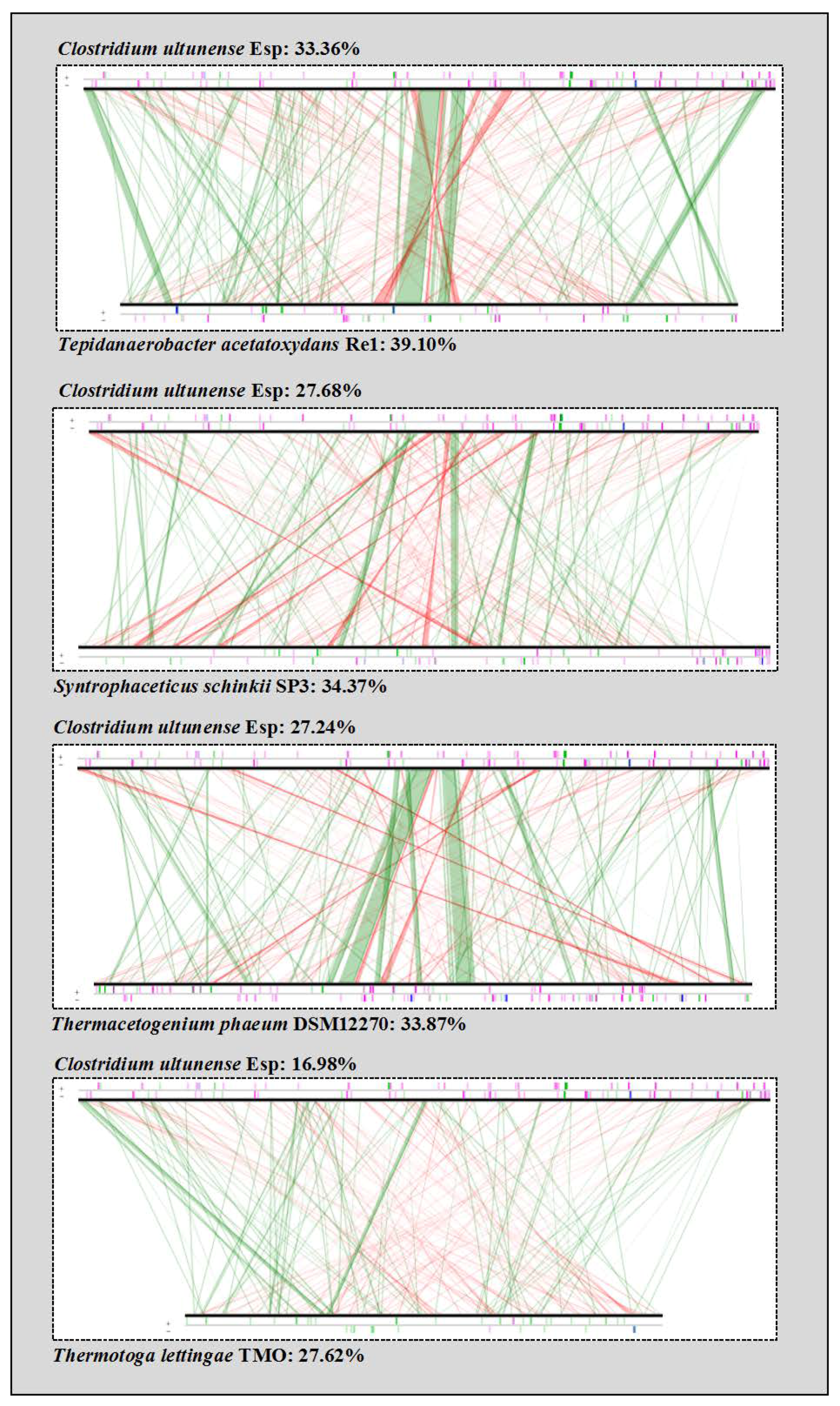
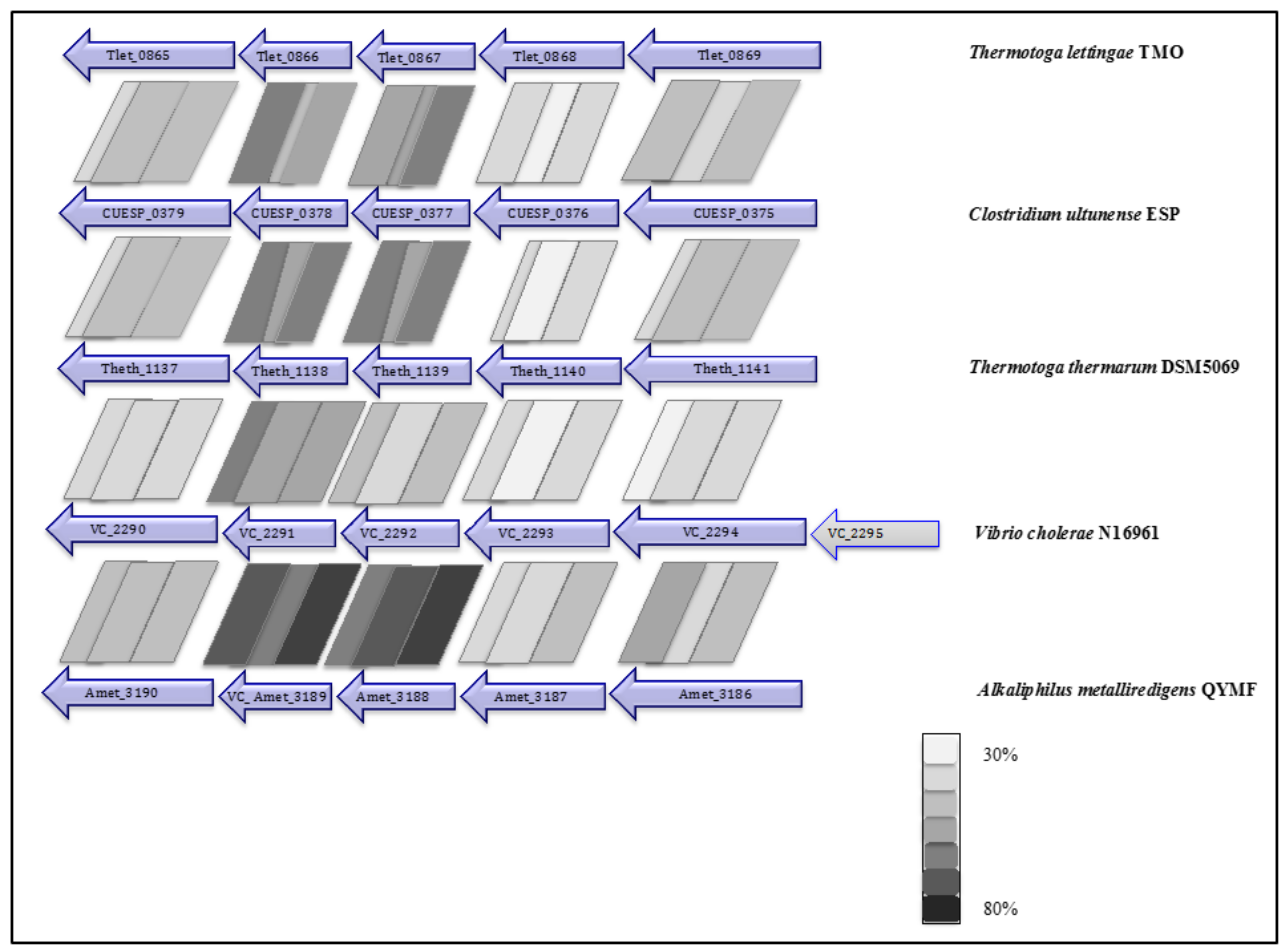
2.5. Phylogenetic Placement and Synteny
3. Results
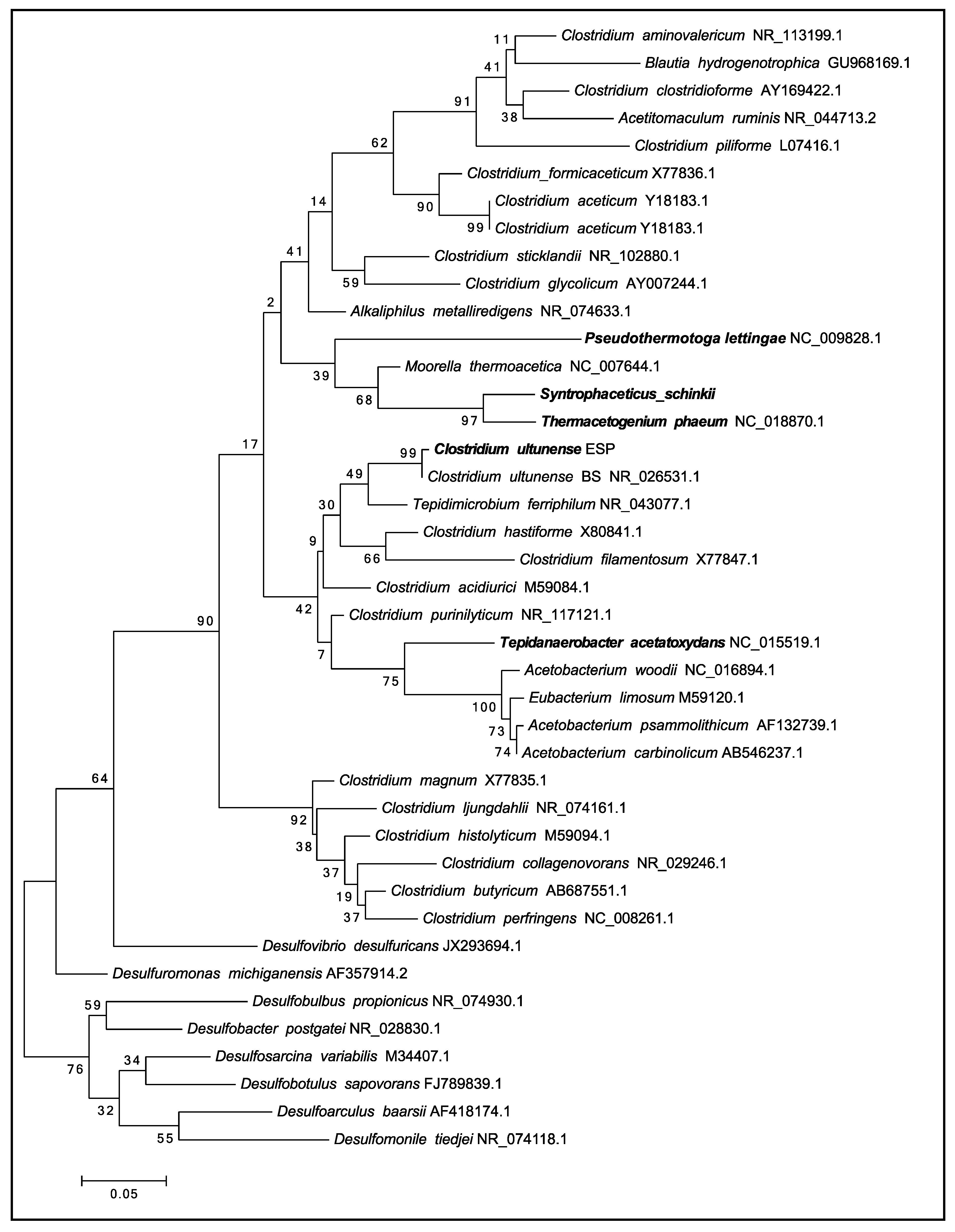
3.1. Phylogenetic Placement
3.2. Genome Properties
3.3. Morphological and Physiological Traits
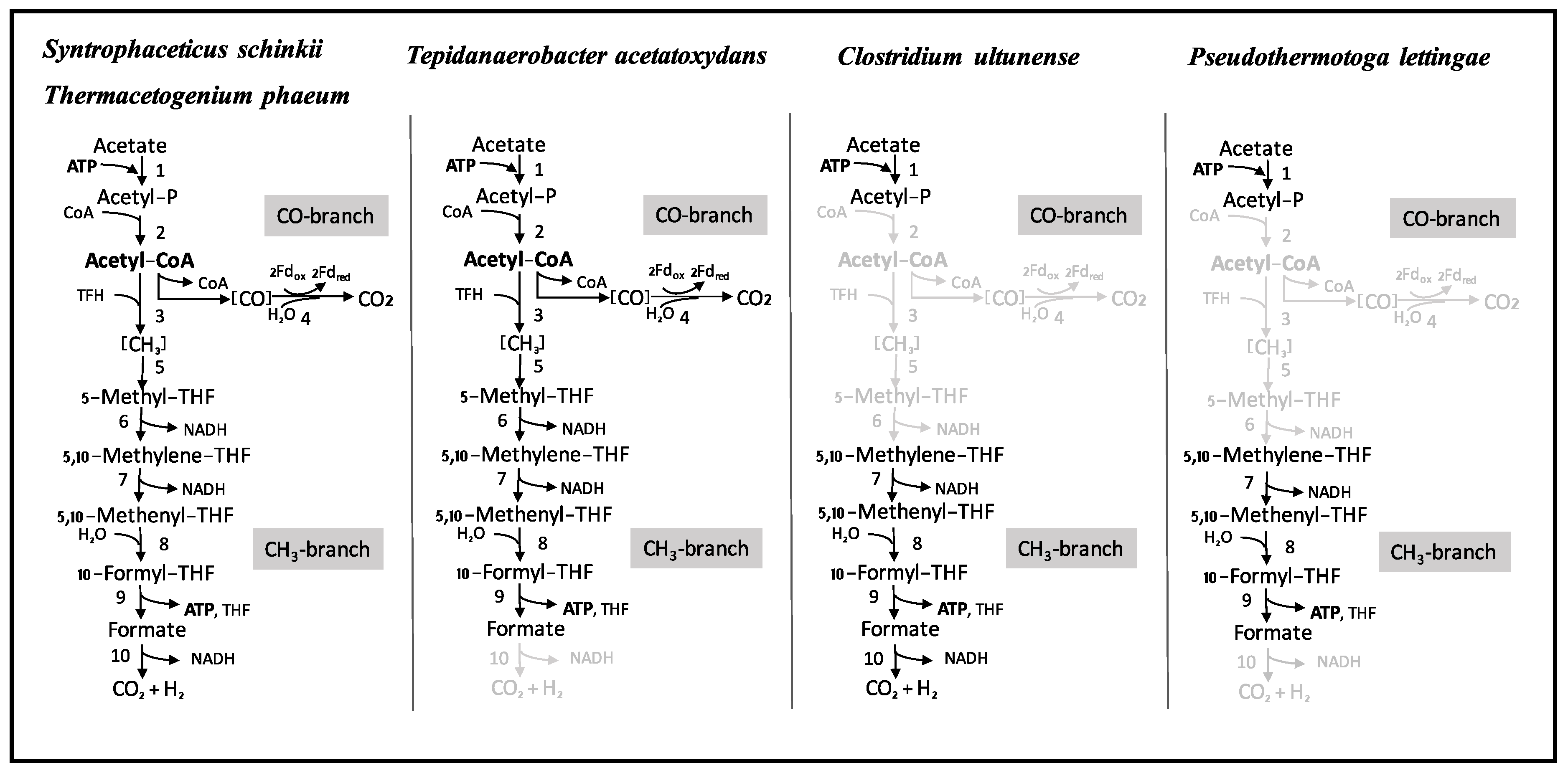
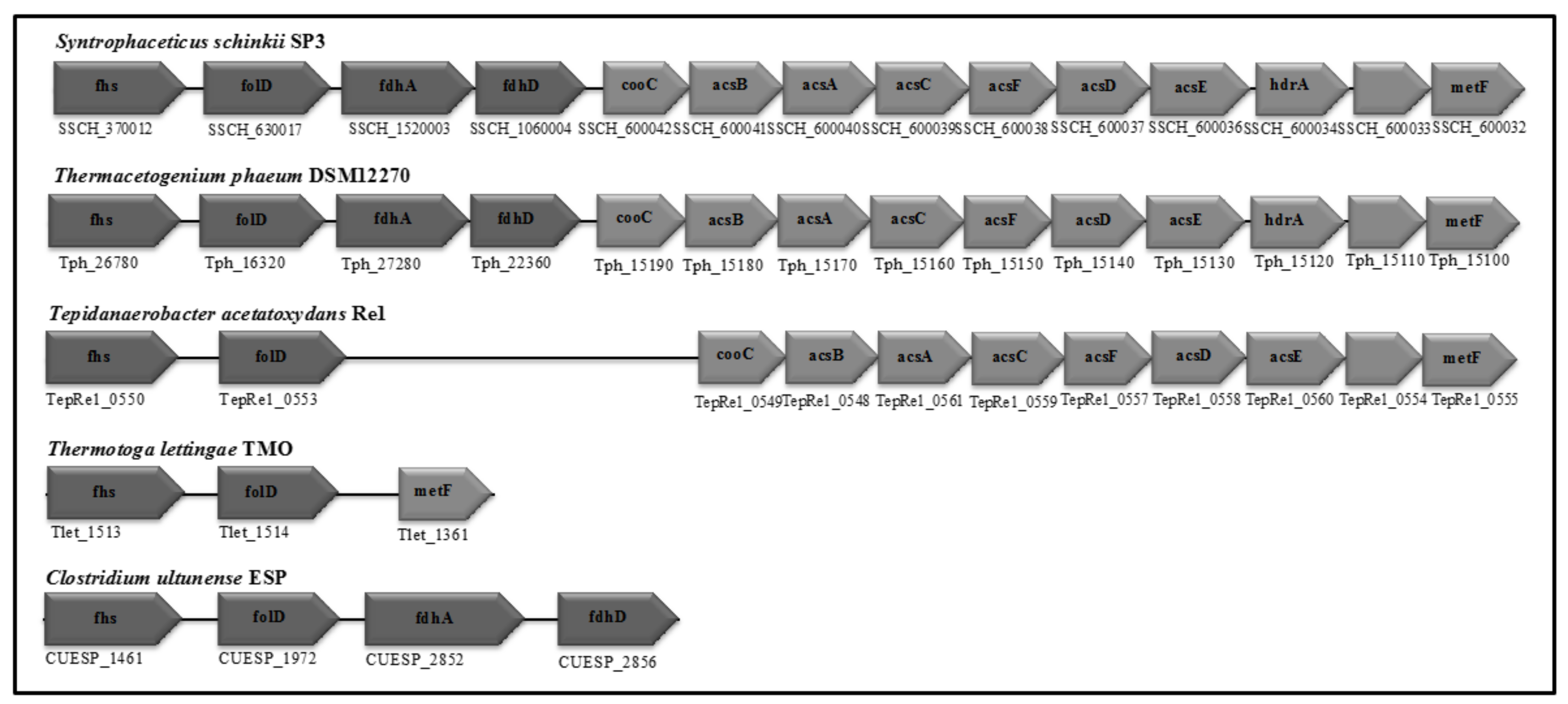
3.4. Wood-Ljungdahl Pathway and Acetate Activation
3.5. Respiratory Complexes
3.6. Soluble Electron Transfer Proteins
3.7. Hydrogenases
3.8. Adenosine Triphosphate (ATP) Synthase and Pyrophosphatase (PPase)
4. Discussion
4.1. Attributes Linked to Anaerobic Digestion Environments
4.2. Acetate Oxidation in Mesophilic and Thermophilic Syntrophic Acetate-Oxidising Bacteria
4.3. Energy Conservation Associated with Syntrophic Acetate Oxidation
5. Summary
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Y.; Cheng, J.J.; Creamer, K.S. Inhibition of anaerobic digestion process: A review. Bioresour. Technol. 2008, 99, 4044–4064. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Massé, D.I.; Singh, G. A critical review on inhibition of anaerobic digestion process by excess ammonia. Bioresour. Technol. 2013, 143, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Moestedt, J.; Schnürer, A. Biogas production through syntrophic acetate oxidation and deliberate operating strategies for improved digester performance. Appl. Energy 2016, 179, 124–135. [Google Scholar] [CrossRef]
- Sowers, K.R. Methanogenesis. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 265–286. ISBN 978-0-12-373944-5. [Google Scholar]
- Angenent, L.T.; Sung, S.; Raskin, L. Methanogenic population dynamics during startup of a full-scale anaerobic sequencing batch reactor treating swine waste. Water Res. 2002, 36, 4648–4654. [Google Scholar] [CrossRef]
- Fotidis, I.A.; Karakashev, D.; Angelidaki, I. The dominant acetate degradation pathway/methanogenic composition in full-scale anaerobic digesters operating under different ammonia levels. Int. J. Environ. Sci. Technol. 2014, 11, 2087–2094. [Google Scholar] [CrossRef]
- Schnürer, A.; Nordberg, A. Ammonia, a selective agent for methane production by syntrophic acetate oxidation at mesophilic temperature. Water Sci. Technol. 2008, 57, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Schnürer, A.; Zellner, G.; Svensson, B.H. Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol. Ecol. 1999, 29, 249–261. [Google Scholar] [CrossRef]
- Sun, L.; Muller, B.; Westerholm, M.; Schnurer, A. Syntrophic acetate oxidation in industrial CSTR biogas digesters. J. Biotechnol. 2014, 171, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Leven, L.; Schnurer, A. Bioaugmentation of syntrophic acetate-oxidizing culture in biogas reactors exposed to increasing levels of ammonia. Appl. Environ. Microbiol. 2012, 78, 7619–7625. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Morgenroth, E.; Tandukar, M.; Pavlostathis, S.G.; Smith, A.; Raskin, L.; Kilian, R.E. Syntrophic acetate oxidation in two-phase (acid-methane) anaerobic digesters. Water Sci. Technol. 2011, 64, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.; Luo, H.; Shoun, H.; Kamagata, Y. Involvement of formate as an interspecies electron carrier in a syntrophic acetate-oxidizing anaerobic microorganism in coculture with methanogens. J. Biosci. Bioeng. 2001, 91, 294–298. [Google Scholar] [CrossRef]
- Schnürer, A.; Svensson, B.H.; Schink, B. Enzyme activities in and energetics of acetate metabolism by the mesophilic syntrophically acetate-oxidizing anaerobe Clostridium ultunense. FEMS Microbiol. Lett. 1997, 154, 331–336. [Google Scholar] [CrossRef]
- Schnürer, A.; Houwen, F.; Svensson, B. Mesophilic syntrophic acetate oxidation during methane formation by a triculture at high ammonium concentration. Arch. Microbiol. 1994, 162, 70–74. [Google Scholar] [CrossRef]
- Chauhan, A.; Ogram, A. Phylogeny of acetate-utilizing microorganisms in soils along a nutrient gradient in the florida everglades. Appl. Environ. Microbiol. 2006, 72, 6837–6840. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.D.; Sherry, A.; Grant, R.J.; Rowan, A.K.; Hubert, C.R.J.; Callbeck, C.M.; Aitken, C.M.; Jones, D.M.; Adams, J.J.; Larter, S.R.; et al. The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ. Microbiol. 2011, 13, 2957–2975. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Matthies, C.; Küsel, K.; Schramm, A.; Drake, H.L. Hydrogenotrophic methanogenesis by moderately acid-tolerant methanogens of a methane-emitting acidic peat. Appl. Environ. Microbiol. 2003, 69, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Head, I.M.; Gray, N.D.; Adams, J.J.; Rowan, A.K.; Aitken, C.M.; Bennett, B.; Huang, H.; Brown, A.; Bowler, B.F.J.; et al. Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 2008, 451, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Conrad, R. Thermoanaerobacteriaceae oxidize acetate in methanogenic rice field soil at 50 °C. Environ. Microbiol. 2010, 12, 2341–2354. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, D.; Dolfing, J.; Sakata, S.; Maeda, H.; Miyagawa, Y.; Ikarashi, M.; Tamaki, H.; Takeuchi, M.; Nakatsu, C.H.; Kamagata, Y. Carbon dioxide concentration dictates alternative methanogenic pathways in oil reservoirs. Nat. Commun. 2013, 4, 1998. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Qiu, Q.; Lu, Y. Syntrophic acetate oxidation under thermophilic methanogenic condition in Chinese paddy field soil. FEMS Microbiol. Ecol. 2011, 77, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Balk, M.; Weijma, J.; Stams, A.J.M. Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int. J. Syst. Evol. Microbiol. 2002, 52, 1361–1368. [Google Scholar] [PubMed]
- Hattori, S.; Kamagata, Y.; Hanada, S.; Shoun, H. Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 2000, 50, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Schnurer, A.; Schink, B.; Svensson, B.H. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int. J. Syst. Bacteriol. 1996, 46, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Roos, S.; Schnürer, A. Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol. Lett. 2010, 309, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Roos, S.; Schnürer, A. Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst. Appl. Microbiol. 2011, 34, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.; Galushko, A.S.; Kamagata, Y.; Schink, B. Operation of the co dehydrogenase/acetyl coenzyme a pathway in both acetate oxidation and acetate formation by the syntrophically acetate-oxidizing bacterium Thermacetogenium Phaeum. J. Bacteriol. 2005, 187, 3471–3476. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Bongcam-Rudloff, E.; Schnurer, A.; Muller, B. Genome-guided analysis and whole transcriptome profiling of the mesophilic syntrophic acetate oxidising bacterium Syntrophaceticus schinkii. PLoS ONE 2016, 11, e0166520. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Sun, L.; Schnürer, A. First insights into the syntrophic acetate-oxidizing bacteria—A genetic study. MicrobiologyOpen 2013, 2, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Manzoor, S.; Niazi, A.; Bongcam-Rudloff, E.; Schnürer, A. Genome-guided analysis of physiological capacities of Tepidanaerobacter acetatoxydans provides insights into environmental adaptations and syntrophic acetate oxidation. PLoS ONE 2015, 10, e0121237. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Müller, B.; Niazi, A.; Schnürer, A.; Bongcam-Rudloff, E. Working draft genome sequence of the mesophilic acetate oxidizing bacterium Syntrophaceticus schinkii strain Sp3. Stand. Genom. Sci. 2015, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Oehler, D.; Poehlein, A.; Leimbach, A.; Müller, N.; Daniel, R.; Gottschalk, G.; Schink, B. Genome-guided analysis of physiological and morphological traits of the fermentative acetate oxidizer Thermacetogenium phaeum. BMC Genom. 2012, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, H.; Zhang, L.; Zhang, J.; Wang, Y.; Wang, S.; Zhou, Z.; Yan, X. Draft genome sequence of Clostridium ultunense strain BS (DSMZ 10521), recovered from a mixed culture. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Müller, B.; Niazi, A.; Bongcam-Rudloff, E.; Schnürer, A. Draft genome sequence of Clostridium ultunense strain Esp, a syntrophic acetate-oxidizing bacterium. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, A.J.; Huser, B.A.; Brock, T.D.; Wuhrmann, K. Characterization of an acetate-decarboxylating, non-hydrogen-oxidizing methane bacterium. Arch. Microbiol. 1980, 124, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scilifelab. Available online: https://www.scilifelab.se/platforms/ngi/ (accessed on 26 October 2016).
- Andrews, S. FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 March 2016).
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. Comput. Sci. Biol. Proc. German Conf. Bioinform. 1999, 99, 45–56. [Google Scholar]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinform. 2003, 2. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Liolios, K.; Mavromatis, K.; Tavernarakis, N.; Kyrpides, N.C. The Genomes on Line Database (GOLD) in 2007: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008, 36, D475–D479. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Labarre, L.; Rouy, Z.; Barbe, V.; Bocs, S.; Cruveiller, S.; Lajus, A.; Pascal, G.; Scarpelli, C.; Médigue, C. MaGe: A microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006, 34, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Bocs, S.; Cruveiller, S.; Vallenet, D.; Nuel, G.; Medigue, C. AMIGene: Annotation of microbial genes. Nucleic Acids Res. 2003, 31, 3723–3726. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the phast phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Alauzet, C.; Marchandin, H.; Courtin, P.; Mory, F.; Lemee, L.; Pons, J.L.; Chapot-Chartier, M.P.; Lozniewski, A.; Jumas-Bilak, E. Multilocus analysis reveals diversity in the genus Tissierella: Description of Tissierella carlieri sp. nov. in the new class Tissierellia classis nov. Syst. Appl. Microbiol. 2014, 37, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, Y.; Ma, S.; Chen, L.; Zhang, H.; Deng, Y. Isolation and characterization of Keratinibaculum paraultunense gen. nov., sp. nov., a novel thermophilic, anaerobic bacterium with keratinolytic activity. FEMS Microbiol. Lett. 2013, 345, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Sabja, D.; Shen, A.; Sorg, J.A. Clostridium difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.B.; Lolkema, J.S. CcpA-dependent carbon catabolite repression in bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Bernson, V.S.M. Acetyl-CoA hydrolase; activity, regulation and physiological, significance of the enzyme in brown adipose tissue from hamster. Eur. J. Biochem. 1976, 67, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Wohlfarth, G.; Buckel, W. A sodium ion gradient as energy source for Peptostreptococcus asaccharolyticus. Arch. Microbiol. 1985, 142, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Juarez, O.; Barquera, B. Insights into the mechanism of electron transfer and sodium translocation of the Na+-pumping NADH:quinone oxidoreductase. Biochim. Biophys. Acta 2012, 1817, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Casutt, M.S.; Nedielkov, R.; Wendelspiess, S.; Vossler, S.; Gerken, U.; Murai, M.; Miyoshi, H.; Moller, H.M.; Steuber, J. Localization of ubiquinone-8 in the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 2011, 286, 40075–40082. [Google Scholar] [CrossRef] [PubMed]
- Goker, M.; Spring, S.; Scheuner, C.; Anderson, I.; Zeytun, A.; Nolan, M.; Lucas, S.; Tice, H.; Del Rio, T.G.; Cheng, J.F.; et al. Genome sequence of the Thermotoga thermarum type strain (LA3T) from an African solfataric spring. Stand. Genom. Sci. 2014, 9, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Biegel, E.; Schmidt, S.; González, J.M.; Müller, V. Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 2011, 68, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Ahring, B.K.; Schmidt, J.E.; Winther-Nielsen, M.; Macario, A.J.; de Macario, E.C. Effect of medium composition and sludge removal on the production, composition, and architecture of thermophilic (55 °C) acetate-utilizing granules from an upflow anaerobic sludge blanket reactor. Appl. Environ. Microbiol. 1993, 59, 2538–2545. [Google Scholar] [PubMed]
- Hao, L.-P.; Lü, F.; He, P.-J.; Li, L.; Shao, L.-M. Predominant contribution of syntrophic acetate oxidation to thermophilic methane formation at high acetate concentrations. Environ. Sci. Technol. 2011, 45, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, D.; Batstone, D.J.; Trably, E.; Angelidaki, I. Acetate oxidation is the dominant methanogenic pathway from acetate in the absence of Methanosaetaceae. Appl. Environ. Microbiol. 2006, 72, 5138–5141. [Google Scholar] [CrossRef] [PubMed]
- Moestedt, J.; Müller, B.; Westerholm, M.; Schnürer, A. Ammonia threshold for inhibition of anaerobic digestion of thin stillage and the importance of organic loading rate. Microb. Biotechnol. 2016, 9, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Sun, L.; Westerholm, M.; Schnurer, A. Bacterial community composition and fhs profiles of low- and high-ammonia biogas digesters reveal novel syntrophic acetate-oxidising bacteria. Biotechnol. Biofuels 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.P.; Ahring, B.K. Acetate oxidation in a thermophilic anaerobic sludge-digestor: The importance of non-acetoclastic methanogenesis from acetate. FEMS Microbiol. Ecol. 1991, 9, 149–157. [Google Scholar] [CrossRef][Green Version]
- Shigematsu, T.; Tang, Y.; Kobayashi, T.; Kawaguchi, H.; Morimura, S.; Kida, K. Effect of dilution rate on metabolic pathway shift between aceticlastic and nonaceticlastic methanogenesis in chemostat cultivation. Appl. Environ. Microbiol. 2004, 70, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Sprott, G.D.; Patel, G.B. Ammonia toxicity in pure cultures of methanogenic bacteria. Syst. Appl. Microbiol. 1986, 7, 358–363. [Google Scholar] [CrossRef]
- Westerholm, M.; Dolfing, J.; Sherry, A.; Gray, N.D.; Head, I.M.; Schnürer, A. Quantification of syntrophic acetate-oxidizing microbial communities in biogas processes. Environ. Microbiol. Rep. 2011, 3, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, V.; Osimani, A.; Taccari, M.; Garofalo, C.; Butta, A.; Clementi, F.; Aquilanti, L. Insight into the bacterial diversity of fermentation woad dye vats as revealed by PCR-DGGE and pyrosequencing. J. Ind. Microbiol. Biotechnol. 2017, 44, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Q.; Zhang, Q.; Wang, T.; Yue, H.; Wu, L.; Shi, J.; Qin, Z.; Zhou, J.; Zuo, J.; et al. Bacteriophage-prokaryote dynamics and interaction within anaerobic digestion processes across time and space. Microbiome 2017, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.; Palmer, N.; van Vugt, R.; Huang, W.M.; Stevenson, B.; Rosa, P.; Lathigra, R.; Sutton, G.; Peterson, J.; Dodson, R.J.; et al. A bacterial genome in flux: The twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 2000, 35, 490–516. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G.; Rassoulzadegan, F. Are viruses driving microbial diversification and diversity? Environ. Microbiol. 2004, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Smit, A.; Herndl, G.J.; Weinbauer, M.G. Impact of virioplankton on archaeal and bacterial community richness as assessed in seawater batch cultures. Appl. Environ. Microbiol. 2004, 70, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997, 61, 262–280. [Google Scholar] [PubMed]
- Sancho, J. Flavodoxins: Sequence, folding, binding, function and beyond. Cell. Mol. Life Sci. 2006, 63, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Thauer, R.K. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K.; Kaster, A.-K.; Goenrich, M.; Schick, M.; Hiromoto, T.; Shima, S. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu. Rev. Biochem. 2010, 79, 507–536. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 1873–1898. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Roh, Y.; Carroll, S.L.; Blair, B.; Zhou, J.; Zhang, C.L.; Fields, M.W. Alkaline anaerobic respiration: Isolation and characterization of a novel alkaliphilic and metal-reducing bacterium. Appl. Environ. Microbiol. 2004, 70, 5595–5602. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Saier, M.H., Jr. Phylogenetic characterization of the ubiquitous electron transfer flavoprotein families ETF-α and ETF-β. Res. Microbiol. 1995, 146, 397–404. [Google Scholar] [CrossRef]
- Baykov, A.A.; Malinen, A.M.; Luoto, H.H.; Lahti, R. Pyrophosphate-fueled Na+ and H+ transport in prokaryotes. Microbiol. Mol. Biol. Rev. 2013, 77, 267–276. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | % Age | No. of Genes |
|---|---|---|
| T. acetatoxydans Re1 | 33.36 | 1128 |
| C. ultunense BST | 98.00 | 3323 |
| S. schinkii Sp3 | 27.68 | 936 |
| T. phaeum DSM 12270 | 27.24 | 921 |
| T. lettingae TMO | 16.98 | 574 |
| A. metalliredigens QYMF | 43.39 | 1467 |
| M. thermoacetica ATCC 39073 | 26.44 | 894 |
| A. woodii DSM 1030 | 26.32 | 890 |
| Organism | Total COGs (% Age) | Total No. Genes | Amino Acid Transport & Metabolism | Carbohydrate Transport & Metabolism | Energy Production & Conservation | Inorganic Ion Transport & Metabolism |
|---|---|---|---|---|---|---|
| T. acetatoxydans Re1 | 81.25 | 2158 | 10.80 | 9.11 | 5.91 | 3.87 |
| C. ultunense strain Esp | 79.41 | 2623 | 9.23 | 5.26 | 5.54 | 4.60 |
| C. ultunense strain BST | 77.78 | 2594 | 9.05 | 5.18 | 5.54 | 4.58 |
| T. phaeum PB | 73.50 | 2263 | 9.09 | 3.24 | 6.78 | 4.48 |
| P. lettingae TMO | 85.39 | 1853 | 10.78 | 13.17 | 6.35 | 6.08 |
| S. schinkii Sp3 | 75.07 | 2586 | 9.84 | 4.00 | 5.92 | 5.97 |
| T. acetatoxydans Re1 (Mesophilic) | S. schinkii Sp3 (Mesophilic) | C. ultunense Strain Esp (BST) (Mesophilic) | T. phaeum PB (Thermophilic) | P. lettingae TMO (Thermophilic) | |
|---|---|---|---|---|---|
| Rnf complex (rnfCDGEAB) | TepiRe1_2026-_2031 | SSCH_420047-420053 | CUESP_2612-2617 (AZSU01_50145-50150) | - | Tlet_0286-_0291 |
| NQR complex (nqrBCDEF) | - | - | CUESP_0375-0379 (AZSU01_10374-10378) | - | Tlet_0865-0869 |
| NADH Ferredoxin-depending [Fe-Fe] hydrogenase | TepiRe1_2033-2037 TepiRe1_2699-2701 | SSCH_90017-19 SSCH_210008-10 SSCH_60009-11 SSCH_1120014-15 | CUESP_2849-2850 (AZSU_01_60160-60162) | Tph_18440-_18460 | Tlet_0952-0957 Tlet_1518-1522 |
| Ech hydrogenase (echABCDEF) | - | SSCH_170021-26 | - | Tph_21310-_21360 | - |
| Periplasmic [Ni-Fe] hydrogenase (Maturation proteins) | - | SSCH_30031-_30033 (SSCH_60028-_60030) | - | Tph_06350-_06370 (Tph_09180-_09210) | - |
| Cytoplasmic [Ni-Fe] hydrogenase | - | SSCH_370001-6 | - | Tph_26880-_26930 | - |
| Ion-translocating Ferredoxin-NADH oxidoreductase/heterodisulfide reductase complex | - | SSCH_160001-8 | - | - | - |
| Cytoplasmic formate dehydrogenase | - | SSCH_1520002-3 | CUESP_2852-2856 (AZSU01_60163-60167) | Tph_27280-_27290 Tph_18420-_18430 | - |
| Membrane bound formate dehydrogenase | - | SSCH_1490003-6 | - | Tph_15370-_15400 | - |
| Formate hydrogen lyase | - | - | - | Tph_26250-26370 | - |
| Electron transfer flavoprotein (EtfAB) | TepiRe1_2532-2533 | - | CUESP_1366-1368 (AZSU01_20759-20761) | - | Tlet_1692-1694 Tlet_1832-1833 |
| ATP synthase | - | SSCH_240003-240010 | CUESP_1228-1235 (AZSU_20626-20633) | Tph_27340-27360 | Tlet_0160-0167 |
| Membrane bound Na/H-PPase | TepiRe1_2120 | SSCH_1440001 | CUESP_0504 (AZSU01_10499) | Tph_08020 | Tlet_1947 Tlet_1318 Tlet_2017 |
| Ferredoxins | TepRe1_0333 TepRe1_0615 TepRe1_1978 | SSCH_100042 SSCH_450007 SSCH_530010 SSCH_760007 SSCH_1120013 | CUESP_0098 CUESP_0771 CUESP_1024 CUESP_1759 CUESP_2851 (AZSU01_10096 AZSU01_20179 AZSU01_30304 AZSU01_60162) | Tph_08140 Tph_08570 Tph_09550 Tph_11190 Tph_15740 Tph_17670 Tph_18150 Tph_24780 | Tlet_0408 Tlet_0921 Tlet_0956 Tlet_1345 Tlet_1467 Tlet_1761 Tlet_2059 |
| Rubredoxin | TepRe1_0396 | SSCH_180038 | CUESP_0131 (AZSU01_10127) | Thp_07990 Thp_23390 Thp_23400 | Tlet_1612 |
| Flavodoxin | - | - | CUESP_0726 CUESP_1387 CUESP_2600 CUESP_0610 (AZSU01_20134 AZSU01_20779 AZSU01_50133) | Tph_01330 Thp_21710 | Tlet_0030 Tlet_1248 Tlet_1568 Tlet_1577 |
| Cytochrome | - | - | - | - | Tlet_1388 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzoor, S.; Schnürer, A.; Bongcam-Rudloff, E.; Müller, B. Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria. Genes 2018, 9, 225. https://doi.org/10.3390/genes9040225
Manzoor S, Schnürer A, Bongcam-Rudloff E, Müller B. Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria. Genes. 2018; 9(4):225. https://doi.org/10.3390/genes9040225
Chicago/Turabian StyleManzoor, Shahid, Anna Schnürer, Erik Bongcam-Rudloff, and Bettina Müller. 2018. "Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria" Genes 9, no. 4: 225. https://doi.org/10.3390/genes9040225
APA StyleManzoor, S., Schnürer, A., Bongcam-Rudloff, E., & Müller, B. (2018). Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria. Genes, 9(4), 225. https://doi.org/10.3390/genes9040225