Transcription Factor Binding Site Enrichment Analysis in Co-Expression Modules in Celiac Disease

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Results and Bioinformatics Analyses
2.2. Transcription Factor Binding Site and Gene Ontology Enrichment Analysis
2.3. Selection of Candidates
2.4. Patients and Biopsies
2.5. Cell Culture and Stimulation
2.6. Reverse Transcription and Quantitative PCR
2.7. Western Blot Analyses
2.8. Statistical Analyses
3. Results
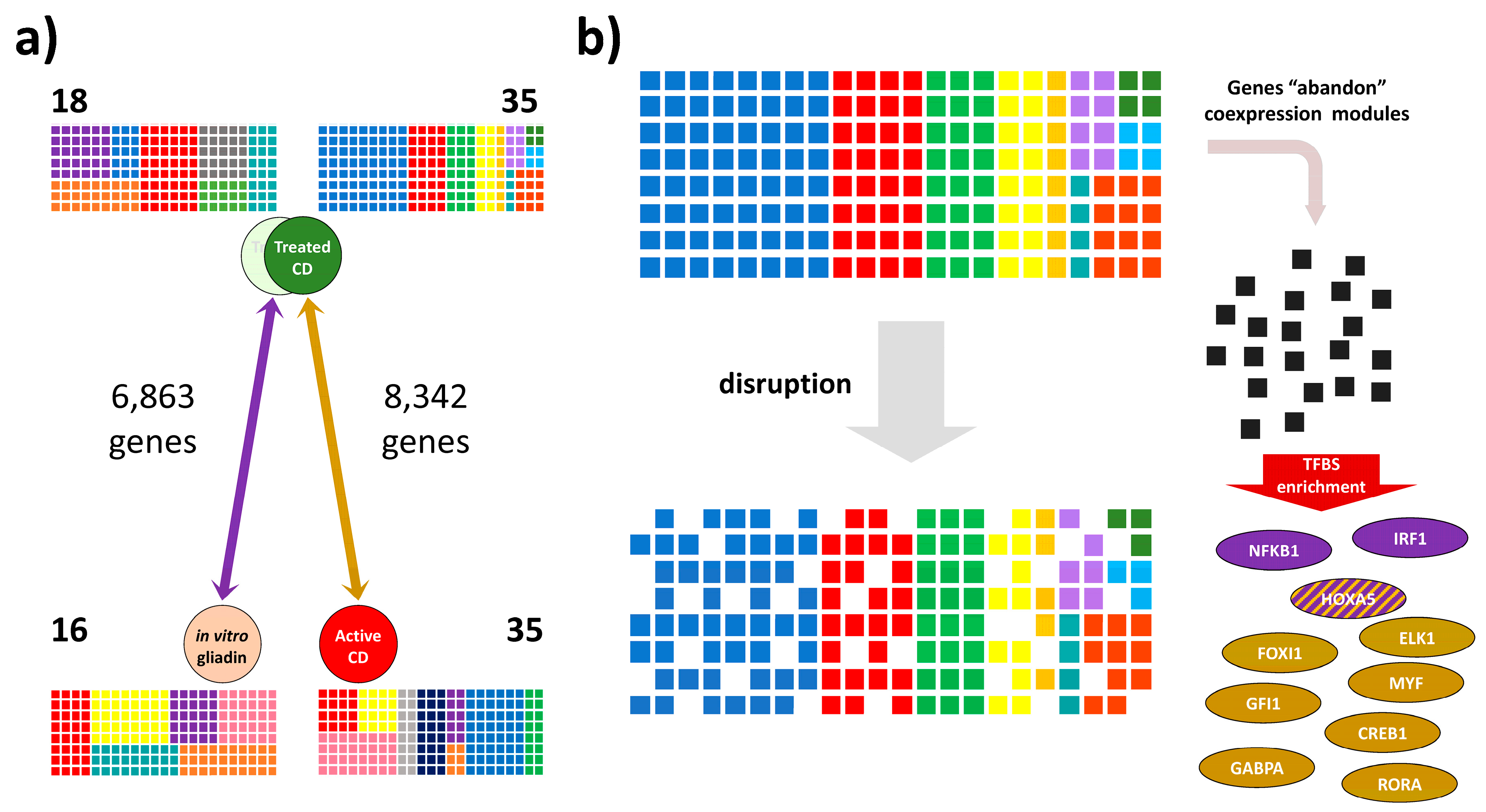
3.1. Identification of Coexpressed Gene Modules and Differentially Coexpressed Genes in Celiac Disease
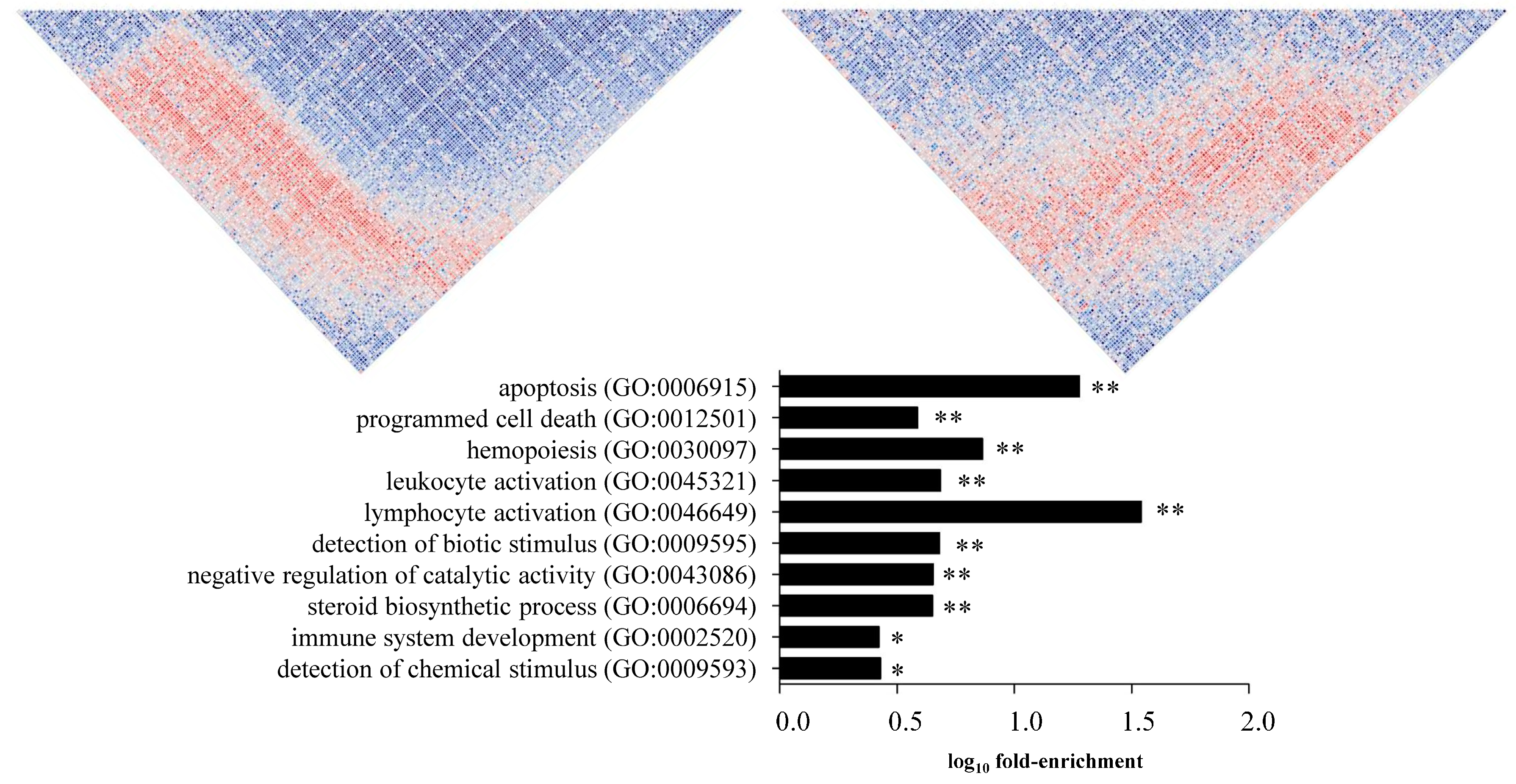
3.2. Transcription Factor Binding Site and Gene Ontology Enrichment Analyses in Differentially Co-Expressed Genes
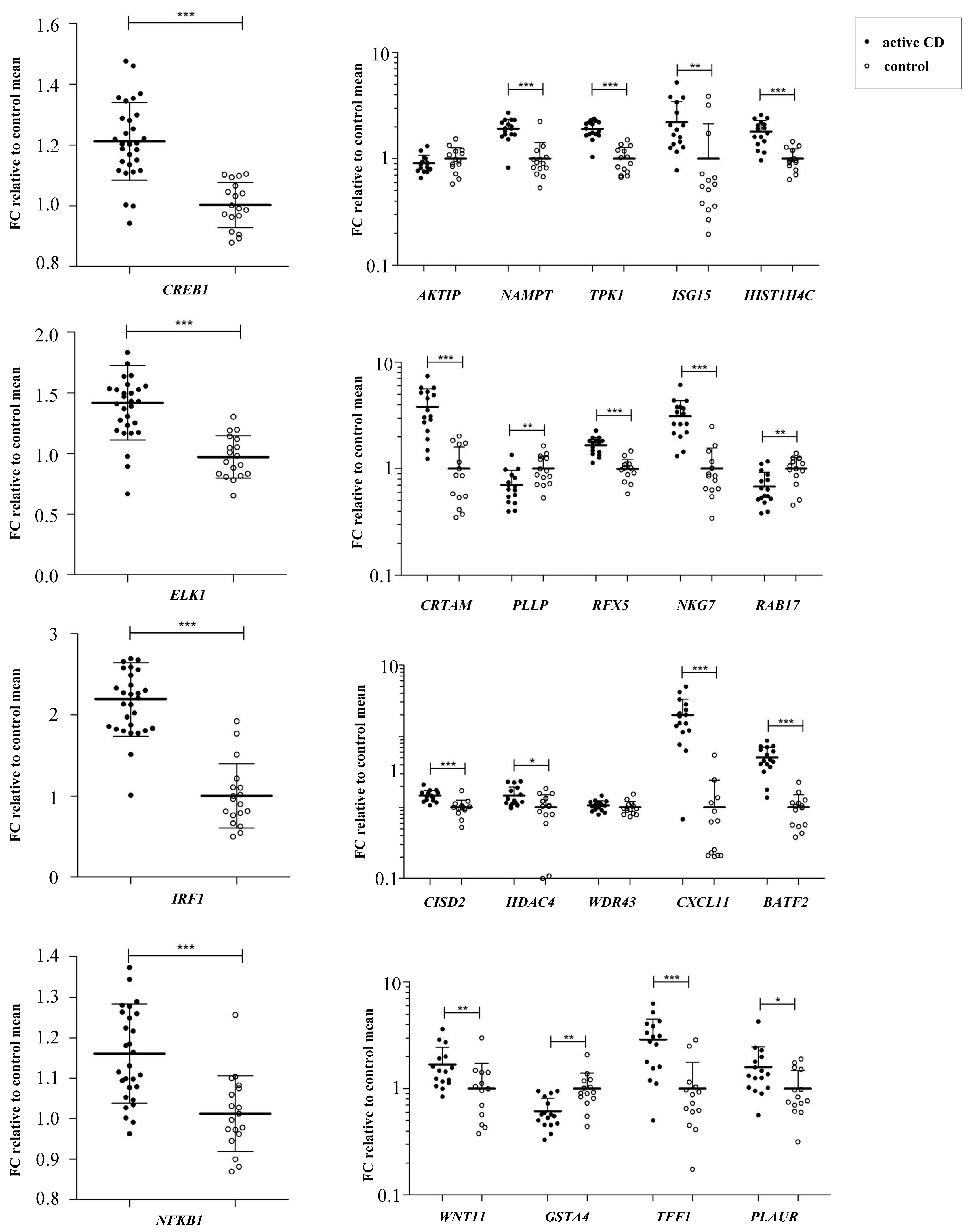
3.3. Altered Candidate Transcription Factors Expression in Celiac Disease
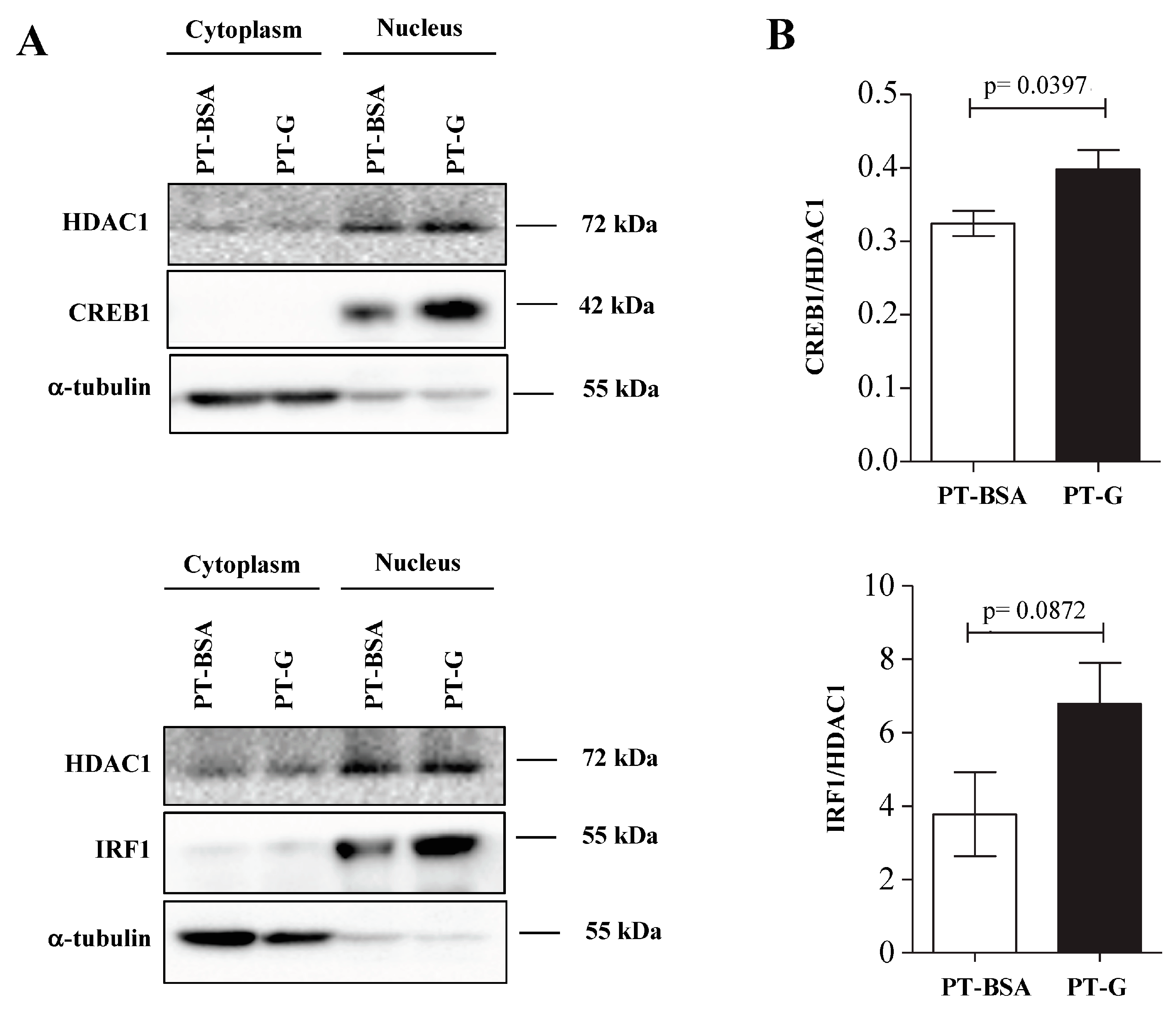
3.4. Candidate Transcription Factors in C2BBe1 Cells
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Green, P.H.R.; Cellier, C. Celiac disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Greco, L.; Romino, R.; Coto, I.; Di Cosmo, N.; Percopo, S.; Maglio, M.; Paparo, F.; Gasperi, V.; Limongelli, M.G.; Cotichini, R.; et al. The first large population based twin study of coeliac disease. Gut 2002, 50, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, D.A.; Franke, L.; Hunt, K.A.; Gwilliam, R.; Zhernakova, A.; Inouye, M.; Wapenaar, M.C.; Barnardo, M.C.; Bethel, G.; Holmes, G.K.; et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 2007, 39, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Ádány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Etxebarria, K.; Jauregi-Miguel, A.; Romero-Garmendia, I.; Plaza-Izurieta, L.; Legarda, M.; Irastorza, I.; Bilbao, J.R. Ancestry-based stratified analysis of Immunochip data identifies novel associations with celiac disease. Eur. J. Hum. Genet. 2016, 24, 1831–1834. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Izurieta, L.; Fernandez-Jimenez, N.; Irastorza, I.; Jauregi-Miguel, A.; Romero-Garmendia, I.; Vitoria, J.C.; Bilbao, J.R. Expression analysis in intestinal mucosa reveals complex relations among genes under the association peaks in celiac disease. Eur. J. Hum. Genet. 2015, 23, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Bondar, C.; Plaza-Izurieta, L.; Fernandez-Jimenez, N.; Irastorza, I.; Withoff, S.; Wijmenga, C.; Chirdo, F.; Bilbao, J.R. THEMIS and PTPRK in celiac intestinal mucosa: coexpression in disease and after in vitro gliadin challenge. Eur. J. Hum. Genet. 2014, 22, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jimenez, N.; Castellanos-Rubio, A.; Plaza-Izurieta, L.; Irastorza, I.; Elcoroaristizabal, X.; Jauregi-Miguel, A.; Lopez-Euba, T.; Tutau, C.; De Pancorbo, M.M.; Vitoria, J.C.; et al. Coregulation and modulation of NFκB-related genes in celiac disease: Uncovered aspects of gut mucosal inflammation. Hum. Mol. Genet. 2014, 23, 1298–1310. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Tovar, H.; García-Herrera, R.; Espinal-Enríquez, J.; Hernández-Lemus, E. Transcriptional master regulator analysis in breast cancer genetic networks. Comput. Biol. Chem. 2015, 59, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Sawle, A.D.; Kebschull, M.; Demmer, R.T.; Papapanou, P.N. Identification of master regulator genes in human periodontitis. J. Dent. Res. 2016, 95, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.M.; Coleman, C.; Molloy, B.; Dominguez Castro, P.; Cormican, P.; Trimble, V.; Mahmud, N.; McManus, R. Transcriptome analysis of CD4+ T cells in coeliac disease reveals imprint of BACH2 and IFNγ regulation. PLoS ONE 2015, 10, e0140049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos-Rubio, A.; Martin-Pagola, A.; Santin, I.; Hualde, I.; Aransay, A.M.; Castaño, L.; Vitoria, J.C.; Bilbao, J.R. Combined functional and positional gene information for the identification of susceptibility variants in celiac disease. Gastroenterology 2008, 134, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Santin, I.; Martin-Pagola, A.; Irastorza, I.; Castaño, L.; Vitoria, J.C.; Bilbao, J.R. Long-term and acute effects of gliadin on small intestine of patients on potentially pathogenic networks in celiac disease. Autoimmunity 2010, 43, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, H.; Liu, B.H.; Zhao, Z.; Liu, L.; Ma, L.X.; Li, Y.X.; Li, Y.Y. DCGL v2.0: An R package for unveiling differential regulation from differential co-expression. PLoS ONE 2013, 8, e79729. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahrour, F.; Díaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Medina, I.; Carbonell, J.; Pulido, L.; Madeira, S.C.; Goetz, S.; Conesa, A.; Tárraga, J.; Pascual-Montano, A.; Nogales-Cadenas, R.; Santoyo, J.; et al. Babelomics: An integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010, 38, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Rauhavirta, T.; Qiao, S.W.; Jiang, Z.; Myrsky, E.; Loponen, J.; Korponay-Szabó, I.R.; Salovaara, H.; Garcia-Horsman, J.A.; Venäläinen, J.; Männistö, P.T.; et al. Epithelial transport and deamidation of gliadin peptides: A role for coeliac disease patient immunoglobulin A. Clin. Exp. Immunol. 2011, 164, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Martin-Pagola, A.; Ortiz, L.; De Nanclares, G.P.; Vitoria, J.C.; Castaño, L.; Bilbao, J.R. Analysis of the expression of MICA in small intestinal mucosa of patients with celiac disease. J. Clin. Immunol. 2003, 23, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Mao, X.; Ji, Q.; Lang, M.; Li, S.; Peng, Y.; Zhou, W.; Xiong, B.; Zeng, Q. Inhibition of IFN regulatory factor-1 down-regulate Th1 cell function in patients with acute coronary syndrome. J. Clin. Immunol. 2010, 30, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-α modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am. J. Pathol. 2013, 183, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, L.; Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Pettoello-Mantovani, M.; Guido, S.; Cexus, O.N.; Peake, N.; Londei, M.; et al. Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARγ downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010, 59, 311–319. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Lambert, M.; David-Cordonnier, M.H. Targeting transcription factor binding to DNA by competing with DNA binders as an approach for controlling gene expression. Curr. Top. Med. Chem. 2015, 15, 1323–1358. [Google Scholar] [CrossRef] [PubMed]
- Shalimar, D.M.; Das, P.; Sreenivas, V.; Gupta, S.D.; Panda, S.K.; Makharia, G.K. Mechanism of villous atrophy in celiac disease: Role of apoptosis and epithelial regeneration. Arch. Pathol. Lab. Med. 2013, 137, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Sari, C.; Bayram, N.A.; Doğan, F.E.A.; Baştuğ, S.; Bolat, A.D.; Sarı, S.Ö.; Ersoy, O.; Bozkurt, E. The evaluation of endothelial functions in patients with celiac disease. Echocardiography 2012, 29, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Jauregi-Miguel, A.; Fernandez-Jimenez, N.; Irastorza, I.; Plaza-Izurieta, L.; Vitoria, J.C.; Bilbao, J.R. Alteration of tight junction gene expression in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Lee, S.Y.; Peng, C.; Lim, D.Y.; Yamamoto, H.; Ryu, J.; Lim, T.G.; Chen, H.; Jin, G.; Zhao, Z.; et al. RSK2 is required for TRAF6 phosphorylation-mediated colon inflammation. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Lahdenperä, A.; Ludvigsson, J.; Fälth-Magnusson, K.; Högberg, L.; Vaarala, O. The effect of gluten-free diet on Th1-Th2-Th3-associated intestinal immune responses in celiac disease. Scand. J. Gastroenterol. 2011, 46, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Pietz, G.; De, R.; Hedberg, M.; Sjöberg, V.; Sandström, O.; Hernell, O.; Hammarström, S.; Hammarström, M.L. Immunopathology of childhood celiac disease—Key role of intestinal epithelial cells. PLoS ONE 2017, 12, e0185025. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lin, A.; Chen, J.; Zhang, X.; Liu, H.; Li, H.; Hu, Y.; Zhang, X.; Zhang, J.; Qiu, L.; et al. CREB1 directly activates the transcription of ribonucleotide reductase small subunit M2 and promotes the aggressiveness of human colorectal cancer. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Rodón, L.; Gonzàlez-Juncà, A.; Del Mar Inda, M.; Sala-Hojman, A.; Martínez-Sáez, E.; Seoane, J. Active CREB1 promotes a malignant TGFβ2 autocrine loop in glioblastoma. Cancer Discov. 2014, 4, 1230–1241. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Garmendia, I.; Garcia-Etxebarria, K.; Hernandez-Vargas, H.; Santin, I.; Jauregi-Miguel, A.; Plaza-Izurieta, L.; Cros, M.-P.; Legarda, M.; Irastorza, I.; Herceg, Z.; et al. Transcription Factor Binding Site Enrichment Analysis in Co-Expression Modules in Celiac Disease. Genes 2018, 9, 245. https://doi.org/10.3390/genes9050245
Romero-Garmendia I, Garcia-Etxebarria K, Hernandez-Vargas H, Santin I, Jauregi-Miguel A, Plaza-Izurieta L, Cros M-P, Legarda M, Irastorza I, Herceg Z, et al. Transcription Factor Binding Site Enrichment Analysis in Co-Expression Modules in Celiac Disease. Genes. 2018; 9(5):245. https://doi.org/10.3390/genes9050245
Chicago/Turabian StyleRomero-Garmendia, Irati, Koldo Garcia-Etxebarria, Hector Hernandez-Vargas, Izortze Santin, Amaia Jauregi-Miguel, Leticia Plaza-Izurieta, Marie-Pierre Cros, Maria Legarda, Iñaki Irastorza, Zdenko Herceg, and et al. 2018. "Transcription Factor Binding Site Enrichment Analysis in Co-Expression Modules in Celiac Disease" Genes 9, no. 5: 245. https://doi.org/10.3390/genes9050245