A High-Quality, Long-Read De Novo Genome Assembly to Aid Conservation of Hawaiiʻs Last Remaining Crow Species

 , , ,
, , ,
Abstract
:1. Introduction
Study Species and Aims
2. Materials and Methods
2.1. Library Construction and Sequencing
2.2. Genome Assembly and Quality
2.3. Repeat Composition Analysis
2.4. Candidate Gene Annotation and Analysis
2.5. MHC Functional Supertypes
2.6. Runs of Homozygosity
3. Results
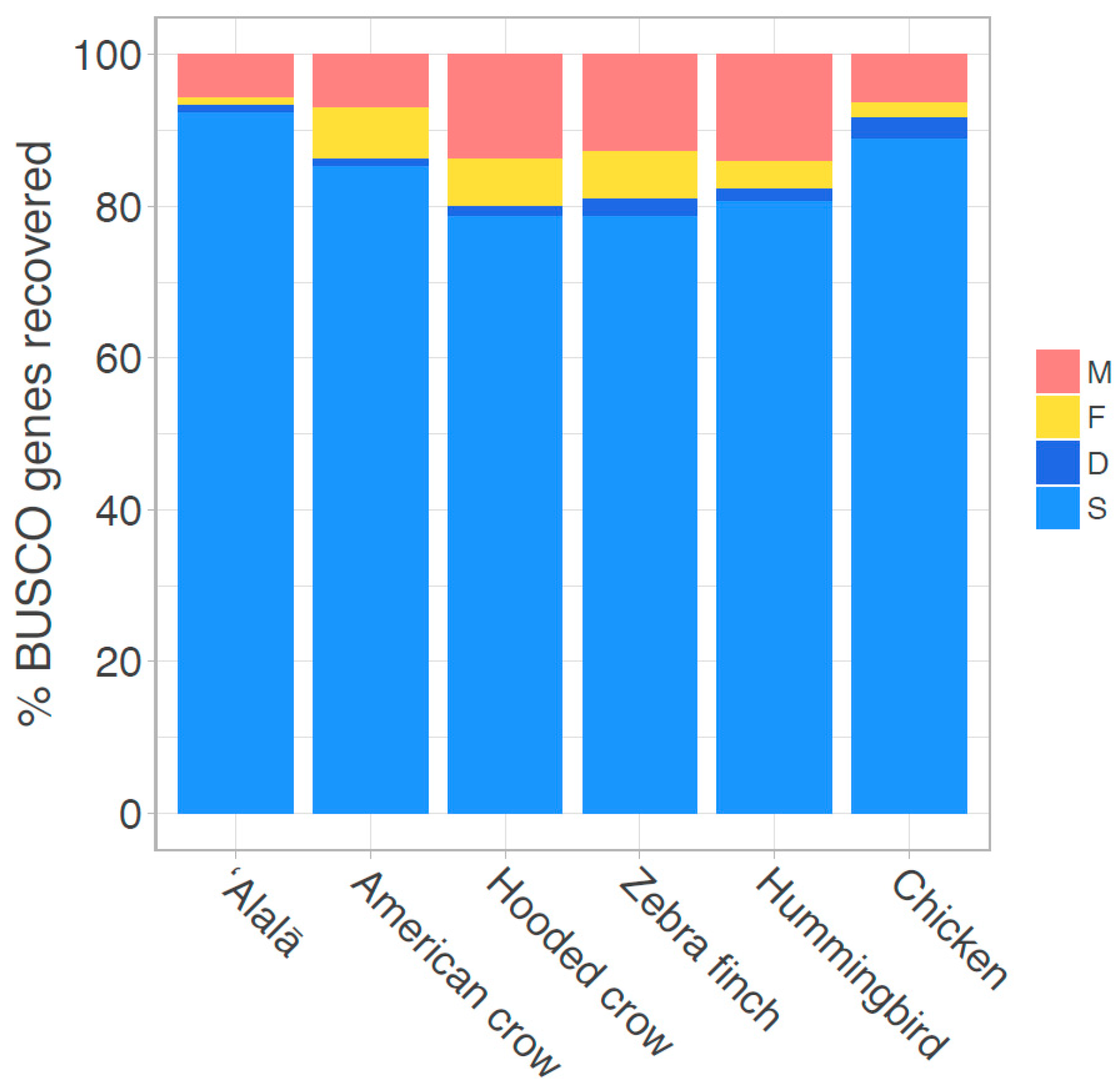
3.1. Genome Assembly and Quality
3.2. Mobile and Repetitive Elements
3.3. Candidate Gene Annotation and Analysis
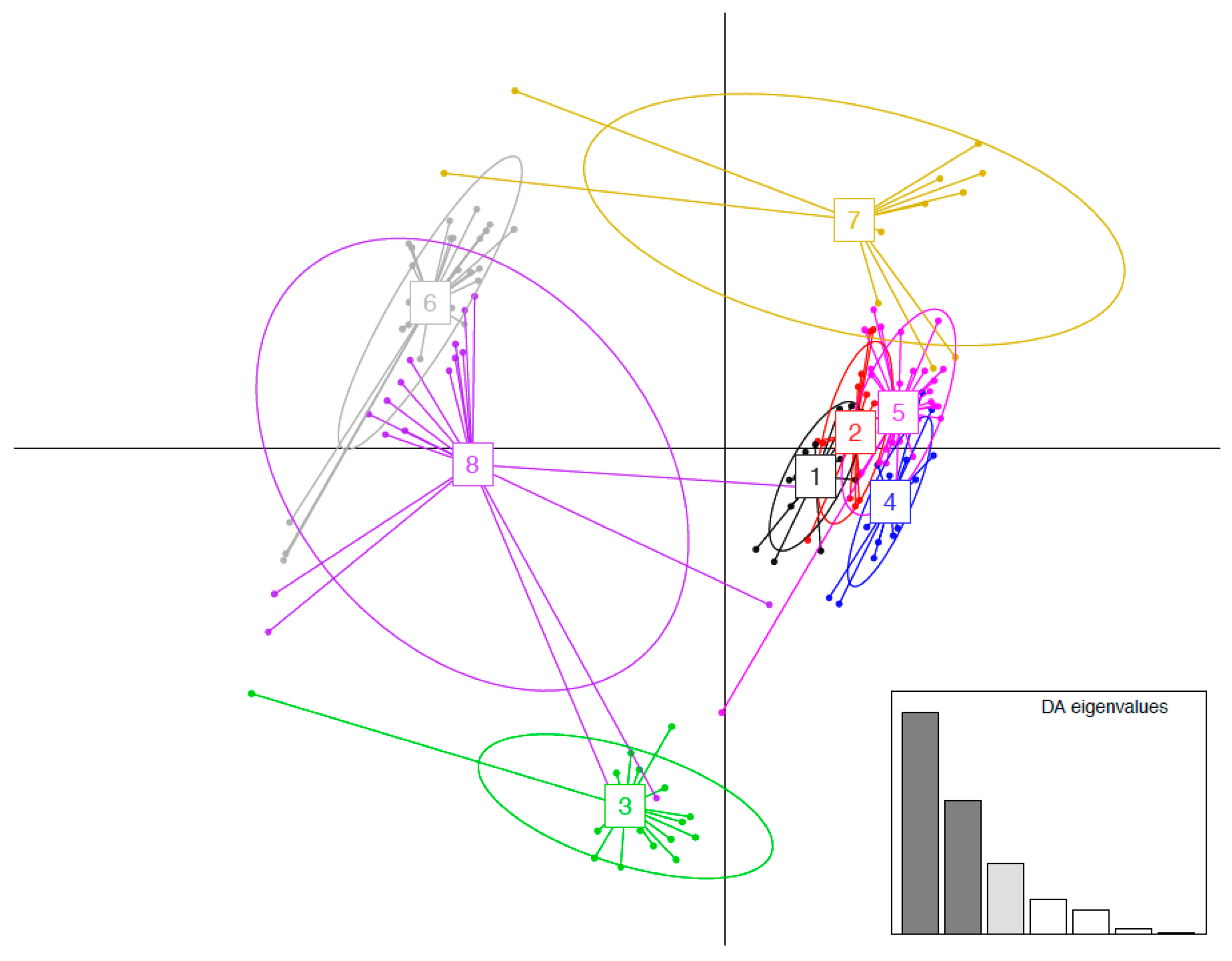
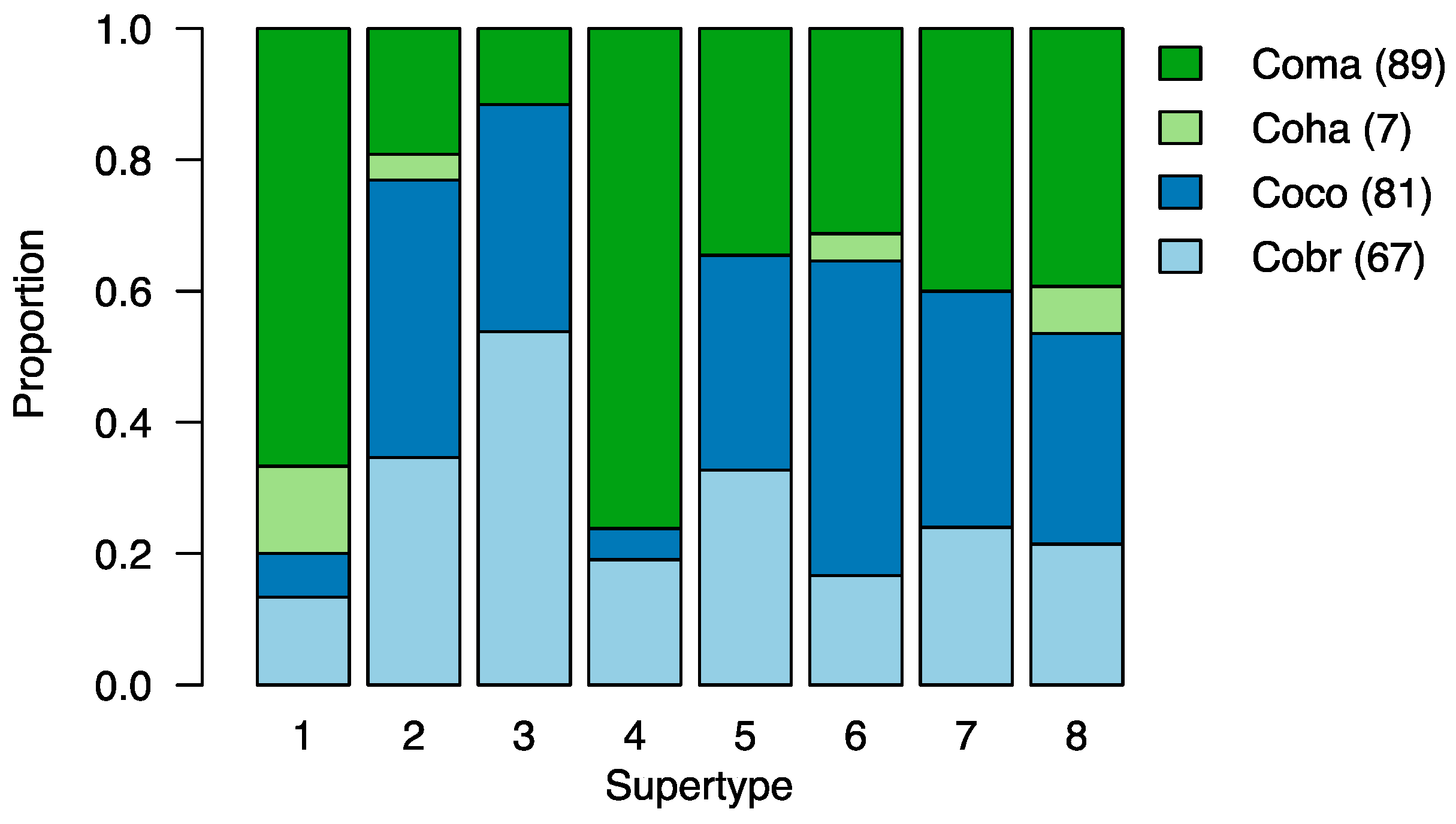
3.4. MHC Functional Supertypes
3.5. Runs of Homozygosity
4. Discussion
4.1. Candidate Immunity Genes
4.2. Runs of Homozygosity
4.3. Applications to ʻAlalā Conservation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ouborg, N.J.; Pertoldi, C.; Loeschcke, V.; Bijlsma, R.K.; Hedrick, P.W. Conservation genetics in transition to conservation genomics. Trends Genet. 2010, 26, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Grueber, C.E. Comparative genomics for biodiversity conservation. Comput. Struct. Biotechnol. J. 2015, 13, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.C.; Putnam, A.S.; Hoeck, P.E.; Ryder, O.A. Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 2013, 1, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.C. Technology: The $1,000 genome. Nature 2014, 507, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.W. Long-read sequence assembly of the gorilla genome. Science 2016, 352, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Shafer, A.B.; Wolf, J.B.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, B.A.; Hand, B.K.; Amish, S.J.; Bernatchez, L.; Foster, J.T.; Miller, K.M.; Morin, P.A.; Narum, S.R.; O’Brien, S.J.; Roffler, G. Genomics in conservation: Case studies and bridging the gap between data and application. Trends Ecol. Evol. 2016, 31, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.R.; Dussex, N.; van Heezik, Y. Bridging the conservation genetics gap by identifying barriers to implementation for conservation practitioners. Glob. Ecol. Conserv. 2017, 10, 231–242. [Google Scholar] [CrossRef]
- Britt, M.; Haworth, S.E.; Johnson, J.B.; Martchenko, D.; Shafer, A.B. The importance of non-academic coauthors in bridging the conservation genetics gap. Biol. Conserv. 2018, 218, 118–123. [Google Scholar] [CrossRef]
- Huisman, J.; Kruuk, L.E.; Ellis, P.A.; Clutton-Brock, T.; Pemberton, J.M. Inbreeding depression across the lifespan in a wild mammal population. Proc. Natl. Acad. Sci. USA 2016, 113, 3585–3590. [Google Scholar] [CrossRef] [PubMed]
- Kardos, M.; Taylor, H.R.; Ellegren, H.; Luikart, G.; Allendorf, F.W. Genomics advances the study of inbreeding depression in the wild. Evol. Appl. 2016, 9, 1205–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerly, S.C.; Morrow, M.E.; Johnson, J.A. A comparison of pedigree-and DNA-based measures for identifying inbreeding depression in the critically endangered Attwater’s Prairie-chicken. Mol. Ecol. 2013, 22, 5313–5328. [Google Scholar] [CrossRef] [PubMed]
- Ivy, J.A.; Putnam, A.S.; Navarro, A.Y.; Gurr, J.; Ryder, O.A. Applying SNP-derived molecular coancestry estimates to captive breeding programs. J. Hered. 2016, 5, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hoeck, P.E.; Wolak, M.E.; Switzer, R.A.; Kuehler, C.M.; Lieberman, A.A. Effects of inbreeding and parental incubation on captive breeding success in Hawaiian crows. Biol. Conserv. 2015, 184, 357–364. [Google Scholar] [CrossRef]
- Rutz, C.; Klump, B.C.; Komarczyk, L.; Leighton, R.; Kramer, J.; Wischnewski, S.; Sugasawa, S.; Morrissey, M.B.; James, R.; St Clair, J.J.H.; Switzer, R.A.; Masuda, B.M. Discovery of species-wide tool use in the Hawaiian crow. Nature 2016, 537, 403–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culliney, S.; Pejchar, L.; Switzer, R.; Ruiz-Gutierrez, V. Seed dispersal by a captive corvid: The role of the ʻAlala (Corvus hawaiiensis) in shaping Hawaiʻi’s plant communities. Ecol. Appl. 2012, 22, 1718–1732. [Google Scholar] [CrossRef] [PubMed]
- U.S. Fish and Wildlife Service. Revised Recovery Plan for the ʻAlalā (Corvus hawaiiensis). 2009; pp. 1–104. Available online: https://www.fws.gov/pacific/ecoservices/documents/Alala_Revised_Recovery_Plan.pdf (accessed on 30 July 2018).
- Hedrick, P.W.; Hoeck, P.E.; Fleischer, R.C.; Farabaugh, S.; Masuda, B.M. The influence of captive breeding management on founder representation and inbreeding in the ʻAlalā, the Hawaiian crow. Conserv. Genet. 2016, 17, 369–378. [Google Scholar] [CrossRef]
- Fleischer, R. Genetic analysis of captive ʻAlalā (Corvus hawaiiensis). In Report to the U.S. Fish and Wildlife Service; Pacific Islands Fish and Wildlife Office: Honolulu, HI, USA, 2003; pp. 1–21. [Google Scholar]
- Jarvi, S.I.; Bianchi, K.R. Genetic Analyses of Captive ʻAlalā (Corvus hawaiiensis) Using AFLP Analyses; Open-File Report 2006-1349; US Geological Survey: Reston, VA, USA, 2006; pp. 1–40. [Google Scholar]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PacBio. Preparing > 30 kb SMRTbellTM Libraries Using the Megaruptor® Shearing and BluePippinTM Size-Selection System. Pacific Biosciences Unsupported Protocol. 2015. Available online: https://www.pacb.com/wp-content/uploads/Procedure-Checklist-Preparing-Greater-Than-30-kb-SMRTbell™-Libraries-Using-Megaruptor®-Shearing-and-BluePippin™-Size-Selection-on-Sequel®-and-RSII-Systems.pdf (accessed on 30 July 2018).
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.W. The fragment assembly string graph. Bioinformatics 2005, 21, ii79–ii85. [Google Scholar] [CrossRef] [PubMed]
- Korlach, J.; Gedman, G.; Kingan, S.B.; Chin, C.-S.; Howard, J.T.; Audet, J.-N.; Cantin, L.; Jarvis, E.D. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience 2017, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S. The genome of a songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Poelstra, J.W.; Vijay, N.; Bossu, C.M.; Lantz, H.; Ryll, B.; Müller, I.; Baglione, V.; Unneberg, P.; Wikelski, M.; Grabherr, M.G. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 2014, 344, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F. University of Geneva. Personal communication, 2016. [Google Scholar]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013‒2015. Available online: www.repeatmasker.org (accessed on 2 May 2018).
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed]
- Genetic Information Research Institute (GIRI). Giri REPBASE. Available online: girinst.org (accessed on 2 May 2018).
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, C.N.; Ekblom, R.; Völker, M.; Westerdahl, H.; Godinez, R.; Kotkiewicz, H.; Burt, D.W.; Graves, T.; Griffin, D.K.; Warren, W.C. Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biol. 2010, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Eimes, J.A.; Townsend, A.K.; Jablonski, P.; Nishiumi, I.; Satta, Y. Early duplication of a single MHC IIB locus prior to the passerine radiations. PLoS ONE 2016, 11, e0163456. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, E.; Korber, B.; Sollars, C.; Kepler, T.B.; Hraber, P.T.; Hayes, E.; Funkhouser, R.; Fugate, M.; Theiler, J.; Hsu, Y.S.; et al. Advantage of rare HLA supertype in HIV disease progression. Nat. Med. 2003, 9, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Sepil, I.; Moghadam, H.K.; Huchard, E.; Sheldon, B.C. Characterization and 454 pyrosequencing of Major Histocompatibility Complex class I genes in the great tit reveal complexity in a passerine system. BMC Evol. Biol. 2012, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Schwensow, N.; Fietz, J.; Dausmann, K.H.; Sommer, S. Neutral versus adaptive genetic variation in parasite resistance: Importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity 2007, 99, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. In silico identification of supertypes for class II MHCs. J. Immunol. 2005, 174, 7085–7095. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Eimes, J.A.; Townsend, A.K.; Sepil, I.; Nishiumi, I.; Satta, Y. Patterns of evolution of MHC class II genes of crows (Corvus) suggest trans-species polymorphism. PeerJ 2015, 3, e853. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, M.H.; Pang, A.W.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Toll-Riera, M.; Castelo, R.; Bellora, N.; Alba, M.M. Evolution of primate orphan proteins. Biochem. Soc. Trans. 2009, 37, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Temperley, N.D.; Berlin, S.; Paton, I.R.; Griffin, D.K.; Burt, D.W. Evolution of the chicken Toll-like receptor gene family: A story of gene gain and gene loss. BMC Genom. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Edwards, S.V. Molecular evolution of the Toll-like receptor multigene family in birds. Mol. Biol. Evol. 2011, 28, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Grueber, C.E.; Knafler, G.J.; King, T.M.; Senior, A.M.; Grosser, S.; Robertson, B.; Weston, K.A.; Brekke, P.; Harris, C.L.; Jamieson, I.G. Toll-like receptor diversity in 10 threatened bird species: Relationship with microsatellite heterozygosity. Conserv. Genet. 2015, 16, 595–611. [Google Scholar] [CrossRef]
- Cormican, P.; Lloyd, A.T.; Downing, T.; Connell, S.J.; Bradley, D.; O’Farrelly, C. The avian Toll-Like receptor pathway—Subtle differences amidst general conformity. Dev. Comp. Immunol. 2009, 33, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Grueber, C.E.; Wallis, G.P.; King, T.M.; Jamieson, I.G. Variation at innate immunity Toll-like receptor genes in a bottlenecked population of a New Zealand robin. PLoS ONE 2012, 7, e45011. [Google Scholar] [CrossRef] [PubMed]
- Knafler, G.; Grueber, C.E.; Sutton, J.T.; Jamieson, I.G. Differential patterns of diversity at microsatellite, MHC, and TLR loci in South Island saddleback populations impacted by translocation and disease. N. Z. J. Ecol. 2017, 41, 98–106. [Google Scholar]
- Hartmann, S.A.; Schaefer, H.M.; Segelbacher, G. Genetic depletion at adaptive but not neutral loci in an endangered bird species. Mol. Ecol. 2014, 23, 5712–5725. [Google Scholar] [CrossRef] [PubMed]
- Bainová, H.; Králová, T.; Bryjová, A.; Albrecht, T.; Bryja, J.; Vinkler, M. First evidence of independent pseudogenization of Toll-like receptor 5 in passerine birds. Dev. Comp. Immunol. 2014, 45, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Burri, R.; Hirzel, H.N.; Salamin, N.; Roulin, A.; Fumagalli, L. Evolutionary patterns of MHC class II B in owls and their implications for the understanding of avian MHC evolution. Mol. Biol. Evol. 2008, 25, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.C.; Lambert, D.M. Gene duplication and gene conversion in class II MHC genes of New Zealand robins (Petroicidae). Immunogenetics 2004, 56, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Zagalska-Neubauer, M.; Babik, W.; Stuglik, M.; Gustafsson, L.; Cichoń, M.; Radwan, J. 454 sequencing reveals extreme complexity of the class II Major Histocompatibility Complex in the collared flycatcher. BMC Evol. Biol. 2010, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Ono, H.; Klein, D.; O’hUigin, C. The Accordion Model of Mhc Evolution. In Progress in Immunology Vol. VIII, Proceedings of the 8th International Congress of Immunology, Budapest, Hungary, 1992; Gergely, J., Benczúr, M., Erdei, A., Falus, A., Füst, G., Medgyesi, G., Petrányi, G., Rajnavölgyi, E., Eds.; Springer: Berlin\Heidelberg, Germany, 1993; pp. 137–143. [Google Scholar]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Künstner, A.; Mäkinen, H.; Nadachowska-Brzyska, K.; Qvarnström, A. The genomic landscape of species divergence in Ficedula flycatchers. Nature 2012, 491, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callicrate, T.E. Population Declines and Genetic Variation: Effects of Serial Bottlenecks; University of Maryland: College Park, MD, USA, 2015. [Google Scholar]
- Aslam, M.L.; Bastiaansen, J.W.; Elferink, M.G.; Megens, H.-J.; Crooijmans, R.P.; Blomberg, L.A.; Fleischer, R.C.; Van Tassell, C.P.; Sonstegard, T.S.; Schroeder, S.G. Whole genome SNP discovery and analysis of genetic diversity in Turkey (Meleagris gallopavo). BMC Genom. 2012, 13, 391. [Google Scholar] [CrossRef] [PubMed]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralls, K.; Ballou, J.D.; Rideout, B.A.; Frankham, R. Genetic management of chondrodystrophy in California condors. Anim. Conserv. Forum 2000, 3, 145–153. [Google Scholar] [CrossRef]
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Ralls, K.; Ballou, J.D. Genetic status and management of California condors. Condor 2004, 106, 215–228. [Google Scholar] [CrossRef]
- Romanov, M.N.; Koriabine, M.; Nefedov, M.; de Jong, P.J.; Ryder, O.A. Construction of a California condor BAC library and first-generation chicken–condor comparative physical map as an endangered species conservation genomics resource. Genomics 2006, 88, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Romanov, M.N.; Tuttle, E.M.; Houck, M.L.; Modi, W.S.; Chemnick, L.G.; Korody, M.L.; Mork, E.M.; Otten, C.A.; Renner, T.; Jones, K.C. The value of avian genomics to the conservation of wildlife. BMC Genom. 2009, 10, S10. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Garcia-Dorado, A. Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol. Evol. 2016, 31, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Oven, I.; Rus, K.R.; Dušanić, D.; Benčina, D.; Keeler, C.L.; Narat, M. Diacylated lipopeptide from Mycoplasma synoviae mediates TLR15 induced innate immune responses. Vet. Res. 2013, 44, 99. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chen, H.; Wei, P. Marek’s disease virus can infect chicken brain microglia and promote the transcription of toll-like receptor 15 and 1LB genes. Chin. J. Virol. 2011, 27, 18–25. [Google Scholar]
- Ruan, W.; Wu, Y.; An, J.; Cui, D.; Li, H.; Zheng, S. Toll-like receptor 2 type 1 and type 2 polymorphisms in different chicken breeds. Poult. Sci. 2012, 91, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhao, C.; Hu, Q.; Sun, J.; Peng, X. Roles of Toll-like receptors 2 and 6 in the inflammatory response to Mycoplasma gallisepticum infection in DF-1 cells and in chicken embryos. Dev. Comp. Immunol. 2016, 59, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zou, H.; Qin, A.; Qian, K.; Shao, H.; Ye, J. Activation of Toll-like receptor 3 inhibits Marek’s disease virus infection in chicken embryo fibroblast cells. Arch. Virol. 2016, 161, 521–528. [Google Scholar]
- Karaffová, V.; Marcinková, E.; Bobíková, K.; Herich, R.; Revajová, V.; Stašová, D.; Kavuľová, A.; Levkutová, M.; Levkut, M.; Lauková, A. TLR4 and TLR21 expression, MIF, IFN-β, MD-2, CD14 activation, and sIgA production in chickens administered with EFAL41 strain challenged with Campylobacter jejuni. Folia Microbiol. 2017, 62, 89–97. [Google Scholar]
- St. Paul, M.; Paolucci, S.; Sharif, S. Treatment with ligands for toll-like receptors 2 and 5 induces a mixed T-helper 1-and 2-like response in chicken splenocytes. J. Interferon Cytokine Res. 2012, 32, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Liu, L.; Qi, Y.; Sun, Y.; Yang, N.; Xu, G.; Zhou, H.; Li, X. Splenic gene expression profiling in White Leghorn layer inoculated with the Salmonella enterica serovar Enteritidis. Anim. Genet. 2015, 46, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, Z.; Cao, L.; Hu, S.; Zhang, Z.; Qin, B.; Guo, Z.; Nie, K. Upregulation of chicken TLR4, TLR15 and MyD88 in heterophils and monocyte-derived macrophages stimulated with Eimeria tenella in vitro. Exp. Parasitol. 2013, 133, 427–433. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | PacBio-Based Primary Haplotype | PacBio-Based Secondary Haplotype |
|---|---|---|
| ʻAlalā (this study) | ||
| Number of contigs | 671 | 2082 |
| Contig N50 | 7,737,654 bp | 455,082 bp |
| Total size | 1,064,991,496 bp | 432,637,353 bp |
| Zebra finch [29] | ||
| Number of contigs | 1159 | 2188 |
| Contig N50 | 5,807,022 bp | 2,740,176 bp |
| Total size | 1,138,770,338 bp | 843,915,757 bp |
| Anna’s hummingbird [29] | ||
| Number of contigs | 1076 | 4895 |
| Contig N50 | 5,366,327 bp | 1,073,631 bp |
| Total size | 1,007,374,986 bp | 1,013,746,550 bp |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutton, J.T.; Helmkampf, M.; Steiner, C.C.; Bellinger, M.R.; Korlach, J.; Hall, R.; Baybayan, P.; Muehling, J.; Gu, J.; Kingan, S.; et al. A High-Quality, Long-Read De Novo Genome Assembly to Aid Conservation of Hawaiiʻs Last Remaining Crow Species. Genes 2018, 9, 393. https://doi.org/10.3390/genes9080393
Sutton JT, Helmkampf M, Steiner CC, Bellinger MR, Korlach J, Hall R, Baybayan P, Muehling J, Gu J, Kingan S, et al. A High-Quality, Long-Read De Novo Genome Assembly to Aid Conservation of Hawaiiʻs Last Remaining Crow Species. Genes. 2018; 9(8):393. https://doi.org/10.3390/genes9080393
Chicago/Turabian StyleSutton, Jolene T., Martin Helmkampf, Cynthia C. Steiner, M. Renee Bellinger, Jonas Korlach, Richard Hall, Primo Baybayan, Jill Muehling, Jenny Gu, Sarah Kingan, and et al. 2018. "A High-Quality, Long-Read De Novo Genome Assembly to Aid Conservation of Hawaiiʻs Last Remaining Crow Species" Genes 9, no. 8: 393. https://doi.org/10.3390/genes9080393