Pathogen Species Identification from Metagenomes in Ancient Remains: The Challenge of Identifying Human Pathogenic Species of Trypanosomatidae via Bioinformatic Tools


Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Methods
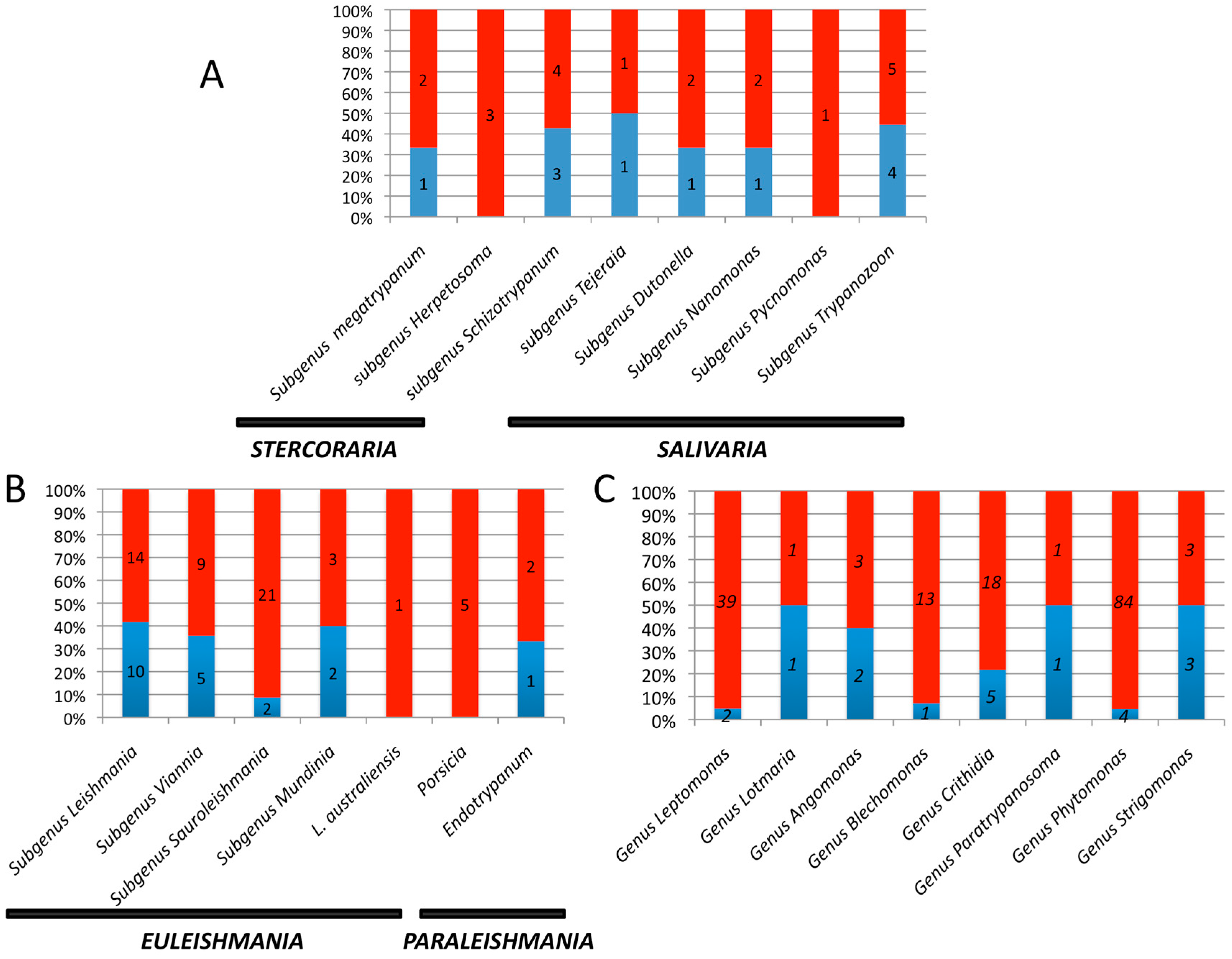
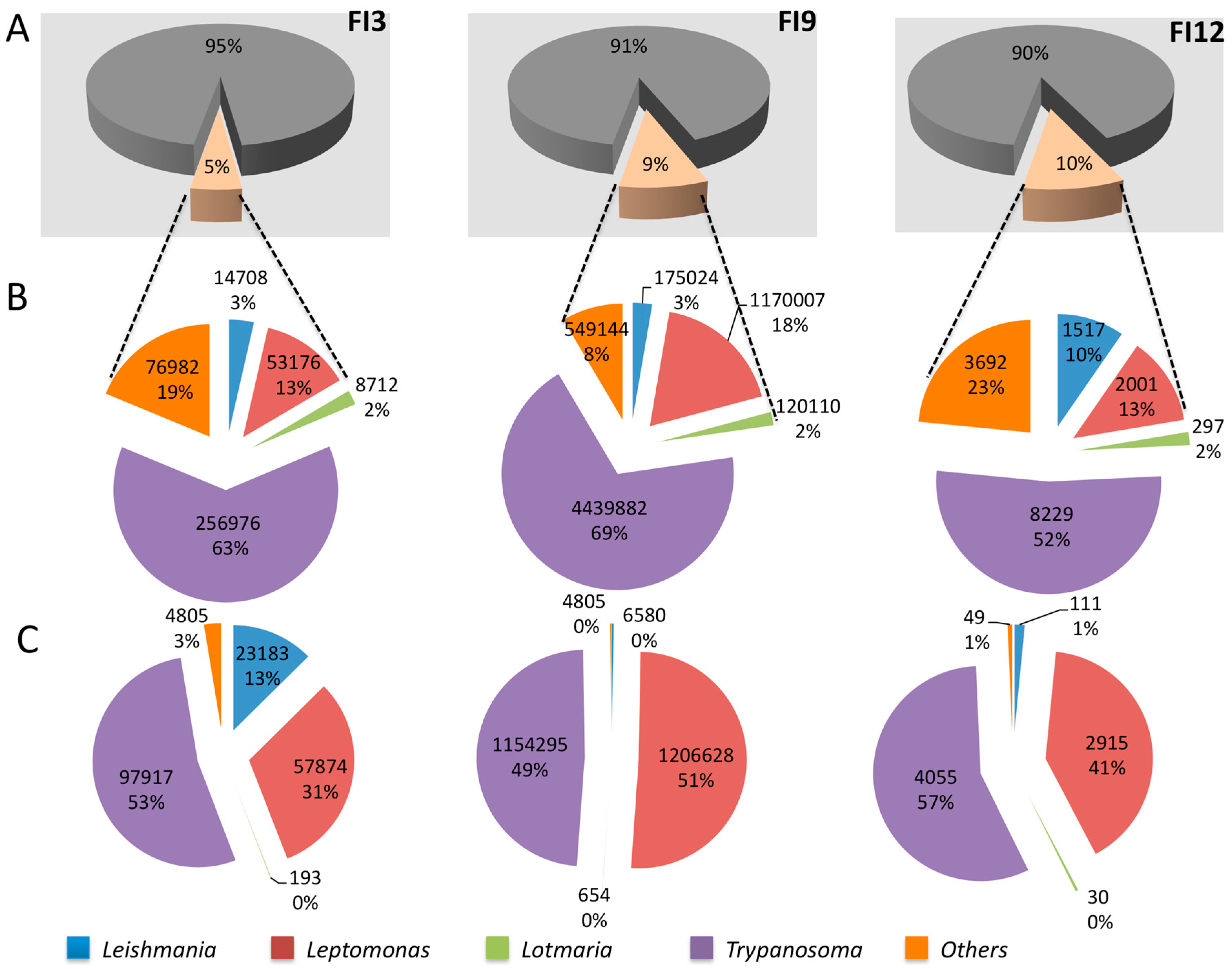
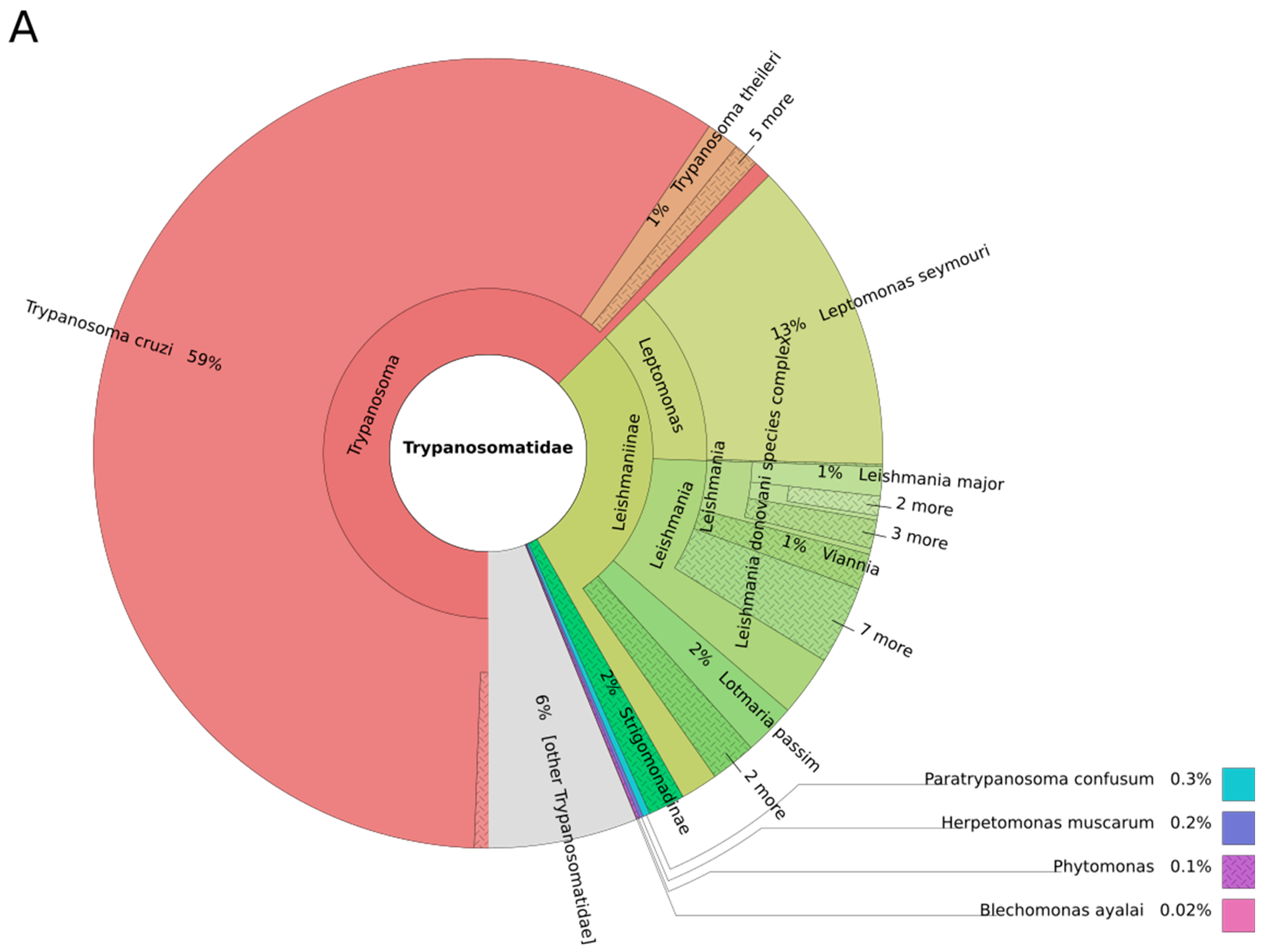
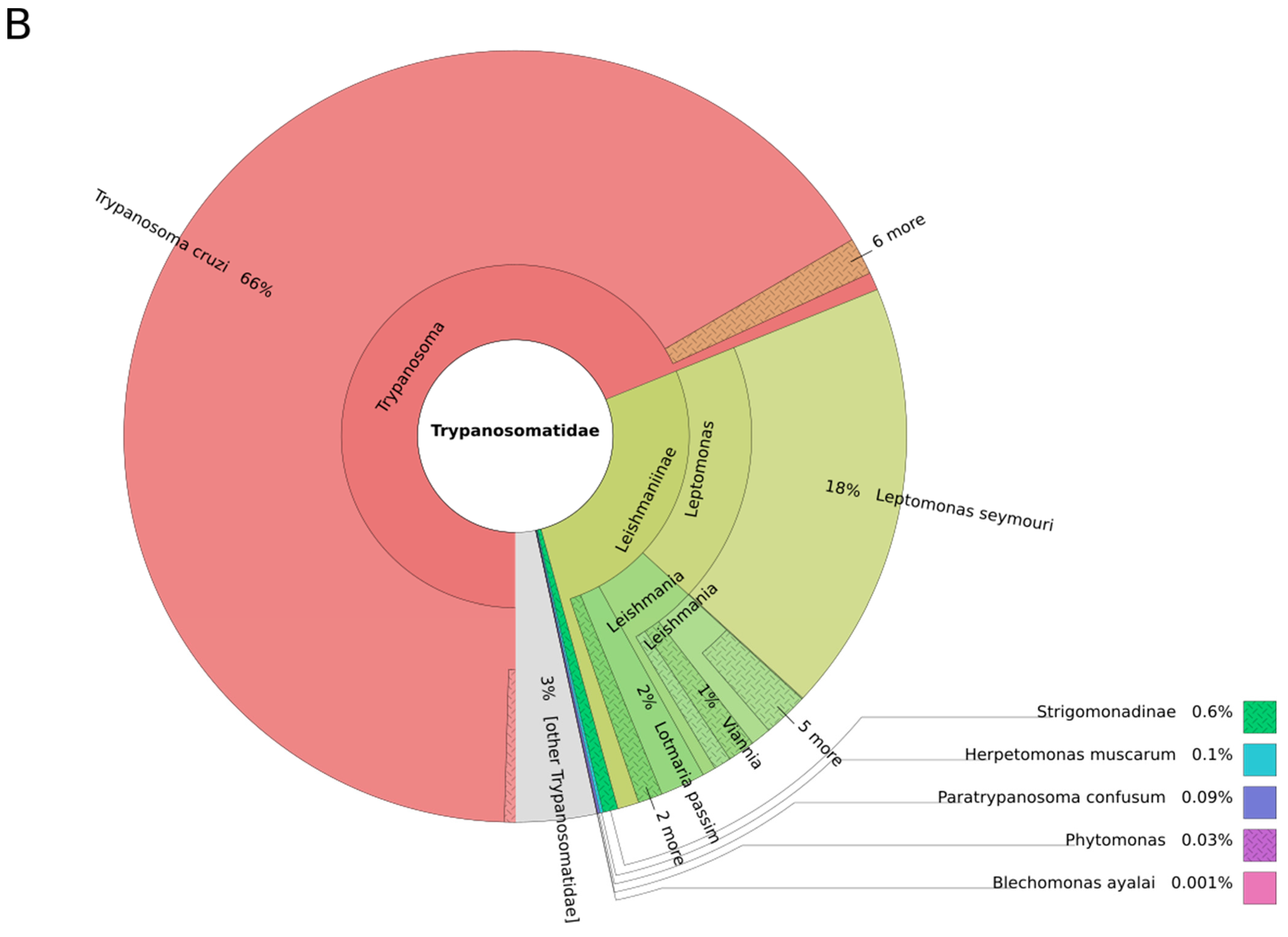
3. Results and Discussion
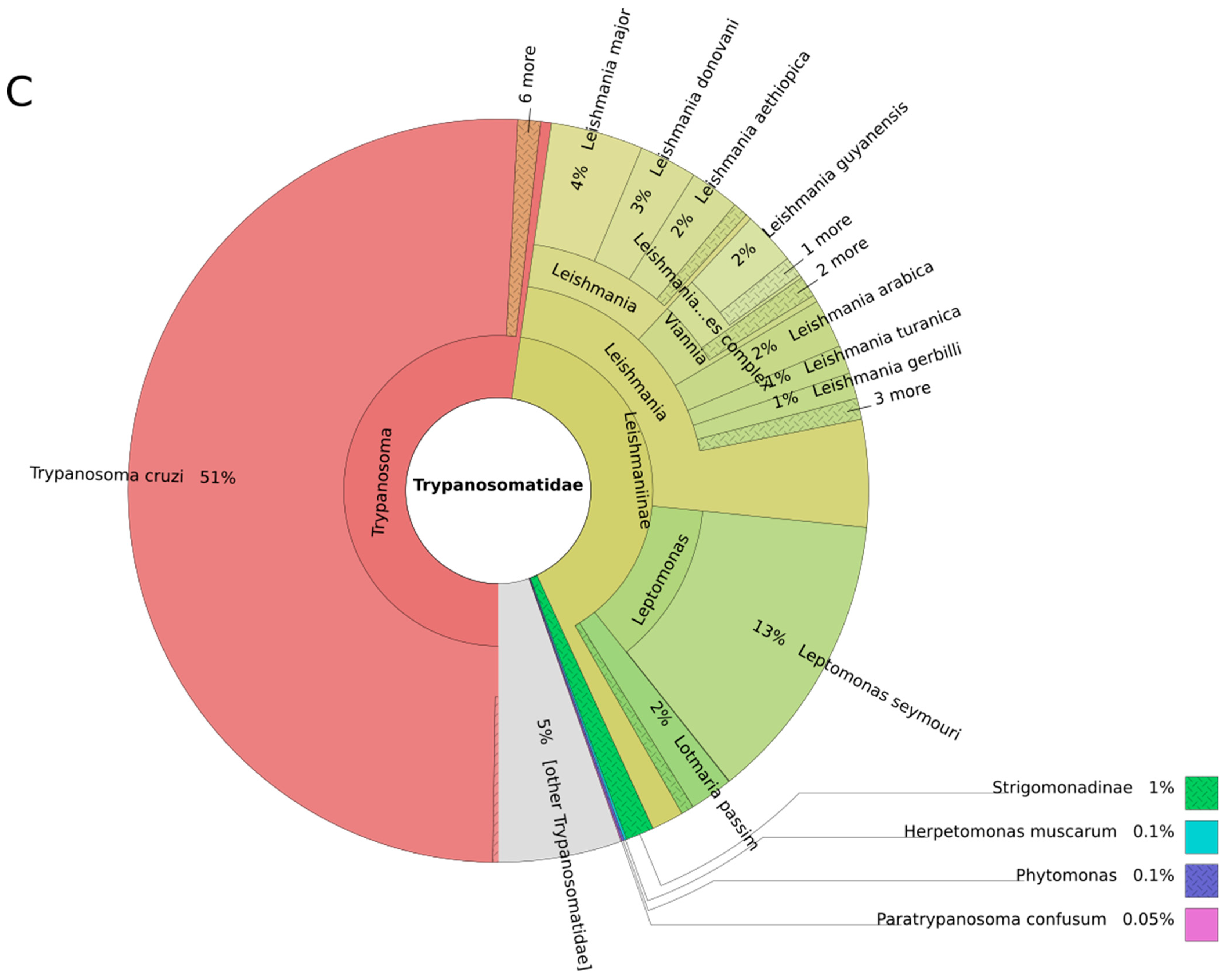
3.1. Trypanosomes and Chagas Disease
3.2. Leishmania and Leishmaniasis
3.3. Other Leishmaniinae (Crithidiatae)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Taxonomic and predicted metabolic profiles of the human gut microbiome in pre-Columbian mummies. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed]
- Sereno, D.; Akhoundi, M.; Dorkeld, F.; Oury, B.; Momen, H.; Perrin, P. What pre-Columbian mummies could teach us about South American leishmaniases? Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, E.; Allison, M.J.; Maguire, B. Paleopathology in South American mummies: A review and new findings. Pathobiology 2012, 79, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A.; Matheson, C.; Lachetta, L.; Llagostera, A.; Appenzeller, O. Ancient leishmaniasis in a highland desert of Northern Chile. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Herbig, A.; Mann, A.; Fellows Yates, J.A.; Weir, C.L.; Burbano, H.A.; Orlando, L.; Krause, J. A robust framework for microbial archaeology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 321–356. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Cooper, A.; Weyrich, L.S. Reply to Santiago-Rodriguez et al.: Proper authentication of ancient DNA is essential. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Weyrich, L.S. Proper authentication of ancient DNA is still essential. Genes 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Toranzos, G.A.; Santiago-Rodriguez, T.M.; Cano, R.J.; Fornaciari, G. Proper authentication of ancient DNA is essential, yes; but so are undogmatic approaches. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignment. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, F.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucl. Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Fact Sheet on Chagas Disease. Available online: http://www.who.int/chagas/resources/en/ (accessed on 15 April 2018).
- World Health Organization. Fact Sheet on Leishmaniasis. Available online: http://www.who.int/leishmaniasis/resources/en/ (accessed on 15 April 2018).
- Brenière, S.F.; Waleckx, E.; Barnabé, C. Over six thousand Trypanosoma cruzi strains classified into discrete typing units (DTUs): Attempt at an inventory. PLoS Negl. Trop. Dis. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Aufderheide, A.C.; Salo, W.; Madden, M.; Streitz, J.; Buikstra, J.; Guhl, F.; Arriaza, B.; Renier, C.; Wittmers, L.E., Jr.; Fornaciari, G.; et al. A 9000-year record of chagas’ disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, N.; Diosque, P. Evolution of Trypanosoma cruzi: Clarifying hybridisations, mitochondrial introgressions and phylogenetic relationships between major lineages. Mem. Inst. Oswaldo Cruz 2015, 110, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, M.; Downing, T.; Votypka, J.; Kuhls, K.; Lukes, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Badaro, R.; Barral-Netto, M., Jr.; Grimaldi, G.; Momen, H.; Carvalho, E.M. Isolation of Leishmania mexicana amazonensis from the bone marrow in a case of American visceral leishmaniasis. Am. J. Trop. Med. Hyg. 1986, 35, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Pedral-Sampaio, D., Jr.; Grimaldi, G.; Momen, H.; MacMahon-Pratt, D.; Ribeiro de Jesus, A.; Almeida, R.; Badaro, R.; Barral-Netto, M.; Carvalho, E.M.; et al. Leishmaniasis in Bahia, Brazil: Evidence that Leishmania amazonensis produces a wide spectrum of clinical diseases. Am. J. Trop. Med. Hyg. 1991, 44, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Kraeva, N.; Butenko, A.; Hlavacova, J.; Kostygov, A.; Myskova, J.; Grybchuk, D.; Lestinova, T.; Votypka, J.; Volf, P.; Opperdoes, F.; et al. Leptomonas seymouri: Adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with Leishmania donovani. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Banerjee, P.; Sarkar, A.; Datta, S.; Chatterjee, M. Coinfection of Leptomonas seymouri and Leishmania donovani in Indian leishmaniasis. J. Clin. Microbiol. 2012, 50, 2774–2778. [Google Scholar] [CrossRef] [PubMed]
- Tritschler, M.; Retschnig, G.; Yañez, O.; Williams, G.R.; Neumann, P. Host sharing by the honey bee parasites Lotmaria passim and Nosema ceranae. Ecol. Evol. 2017, 7, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.P.; Costa, F.B.; Soares, H.S.; Ramirez, D.G.; Mesquita, E.T.; Gennari, S.M.; Marcili, A. Trypanosoma cruzi and Leishmania infantum chagasi infection in wild mammals from Maranhão state, Brazil. Vector Borne Zoonotic Dis. 2015, 15, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Martínez, D.Y.; Verdonck, K.; Kaye, P.M.; Adaui, V.; Polman, K.; Llanos-Cuentas, A.; Dujardin, J.C.; Boelaert, M. Tegumentary leishmaniasis and coinfections other than HIV. PLoS Negl. Trop. Dis. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Dedet, J.P.; Pratlong, F. Leishmania, Trypanosoma and monoxenous Trypanosomatids as emerging opportunistic agents. J. Eukaryot. Microbiol. 2000, 47, 37–39. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leishmania | Viannia | Sauroleishmania | Mundinia | L. australiensis (L. macropodum) | Porcisia | Endotrypanum | |
|---|---|---|---|---|---|---|---|
| Number of species pathogenic for humans | 10 | 6 | 0 | 1 | 1 | 2 | 0 |
| Number of genomes available | 8 | 5 | 0 | 1 | 0 | 0 | 0 |
| Representativity in % | 80 | 83 | 0 | 100 | 0 | 0 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sereno, D.; Dorkeld, F.; Akhoundi, M.; Perrin, P. Pathogen Species Identification from Metagenomes in Ancient Remains: The Challenge of Identifying Human Pathogenic Species of Trypanosomatidae via Bioinformatic Tools. Genes 2018, 9, 418. https://doi.org/10.3390/genes9080418
Sereno D, Dorkeld F, Akhoundi M, Perrin P. Pathogen Species Identification from Metagenomes in Ancient Remains: The Challenge of Identifying Human Pathogenic Species of Trypanosomatidae via Bioinformatic Tools. Genes. 2018; 9(8):418. https://doi.org/10.3390/genes9080418
Chicago/Turabian StyleSereno, Denis, Franck Dorkeld, Mohammad Akhoundi, and Pascale Perrin. 2018. "Pathogen Species Identification from Metagenomes in Ancient Remains: The Challenge of Identifying Human Pathogenic Species of Trypanosomatidae via Bioinformatic Tools" Genes 9, no. 8: 418. https://doi.org/10.3390/genes9080418
APA StyleSereno, D., Dorkeld, F., Akhoundi, M., & Perrin, P. (2018). Pathogen Species Identification from Metagenomes in Ancient Remains: The Challenge of Identifying Human Pathogenic Species of Trypanosomatidae via Bioinformatic Tools. Genes, 9(8), 418. https://doi.org/10.3390/genes9080418