Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine




Abstract
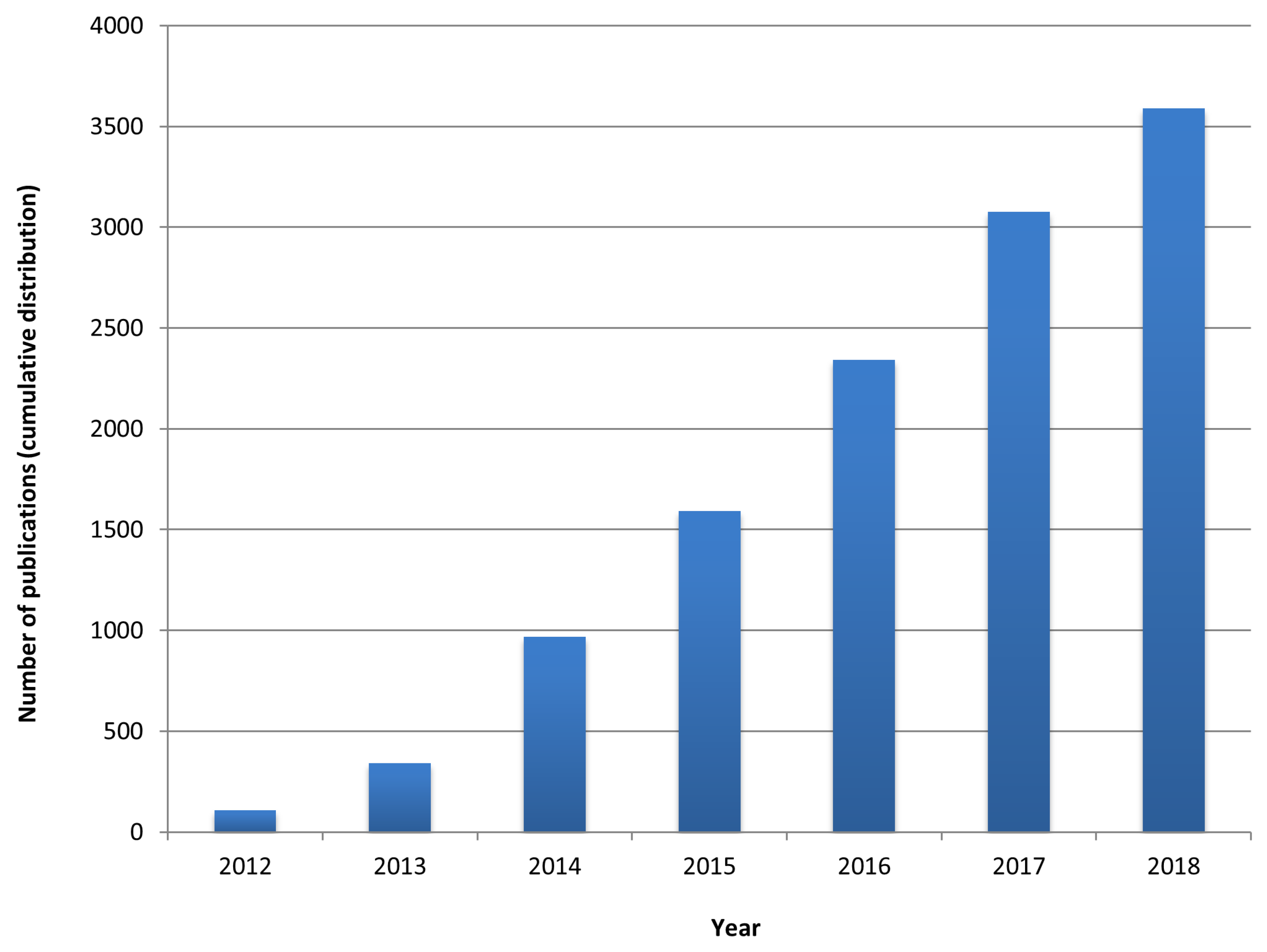
:1. Network Medicine: An Emergent Paradigm in Medicine
2. DIseAse MOdule Detection (DIAMOnD)
- Interactome reconstruction merges the most up-to-date information on protein–protein interactions, co-complex memberships, regulatory interactions and metabolic network maps in the tissue and cell line of interest.
- Disease gene (seed) identification collects the known disease-associated genes obtained from linkage analysis, genome-wide association studies or other sources, which serve as the seed of the disease module.
- In disease module identification, the seed genes are placed on the interactome, with the aim of identifying a subnetwork that contains most of the disease-associated components, exploiting both the functional and topological modularity of the network.
- Pathway identification can be used in instances in which the number of components contained in the ascertained disease module is so large that it cannot serve as a tractable starting point for further experimental work.
- Disease modules are tested for their functional and dynamic homogeneity.
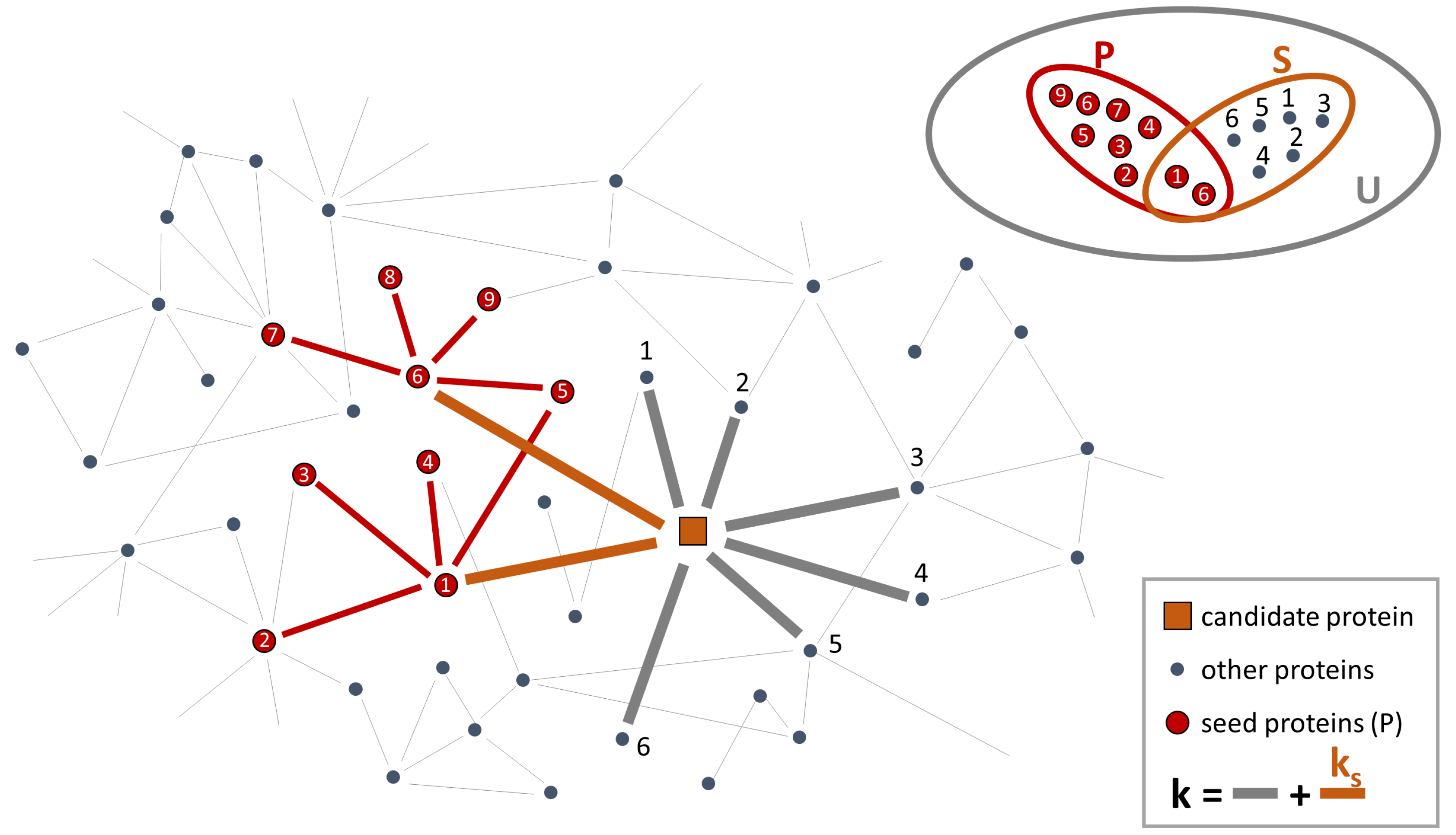
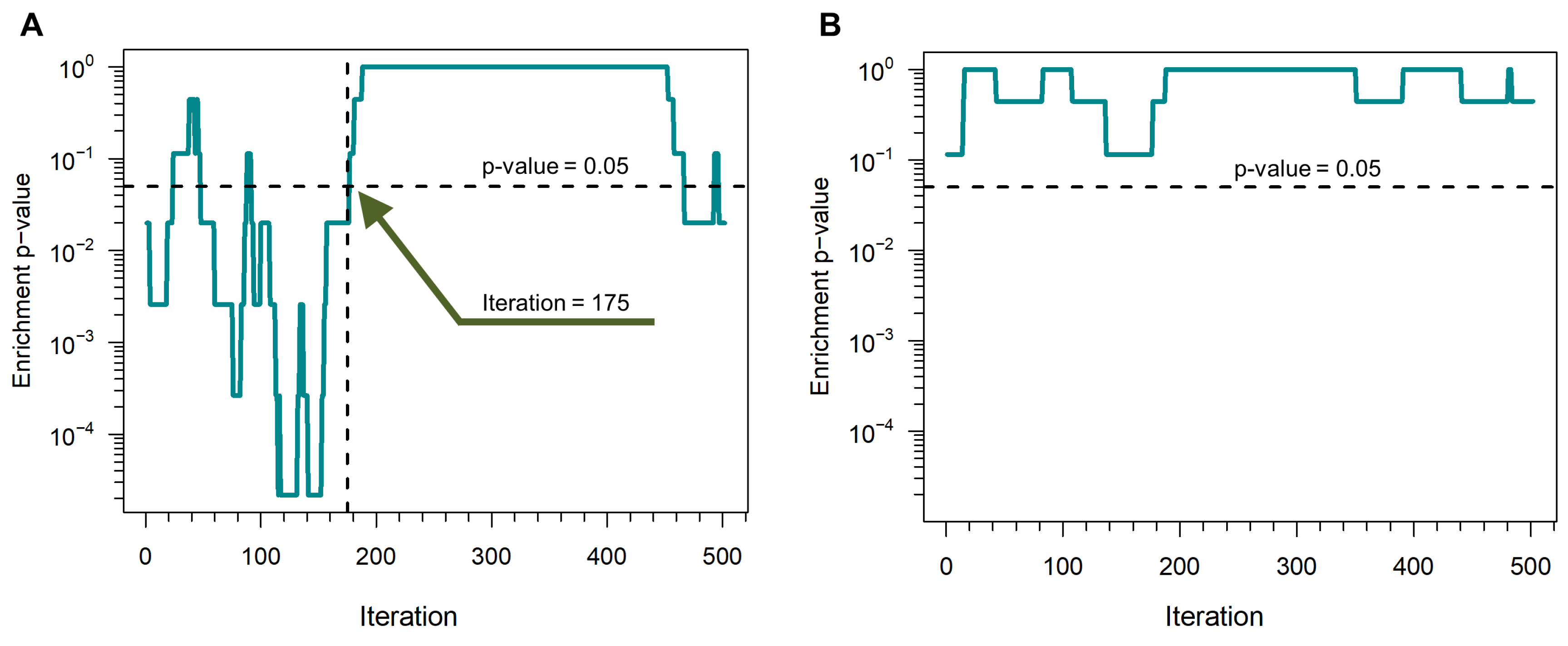
- For all proteins with at least one connection to any of the seed proteins, it calculates the “connectivity significance”. Specifically, DIAMOnD uses the hypergeometric distribution to calculate the statistical significance of having drawn seed proteins (out of k total draws) from a population of N proteins including seed proteins. The hypergeometric distribution is:and then, the “connectivity significance” is obtained as:In a network view, the population of N proteins corresponds to the nodes of the PPI-network and k are the nearest neighbors of a certain protein in the network. This set of nearest neighbors must include seed proteins. Thus, is the probability that a protein with a total of k links has exactly links to seed proteins and p-value is the probability that a protein with a total of k links has more connections to seed proteins than expected (Figure 2).
- It ranks the proteins according to their respective p-values. The protein with the highest rank (i.e., lowest p-value) is called “candidate protein”.
- It adds the candidate protein to the set of seed proteins, increasing their number from to .
- It iterates steps 1–3 with the expanded set of seed proteins, pulling one protein at a time into the growing disease module.
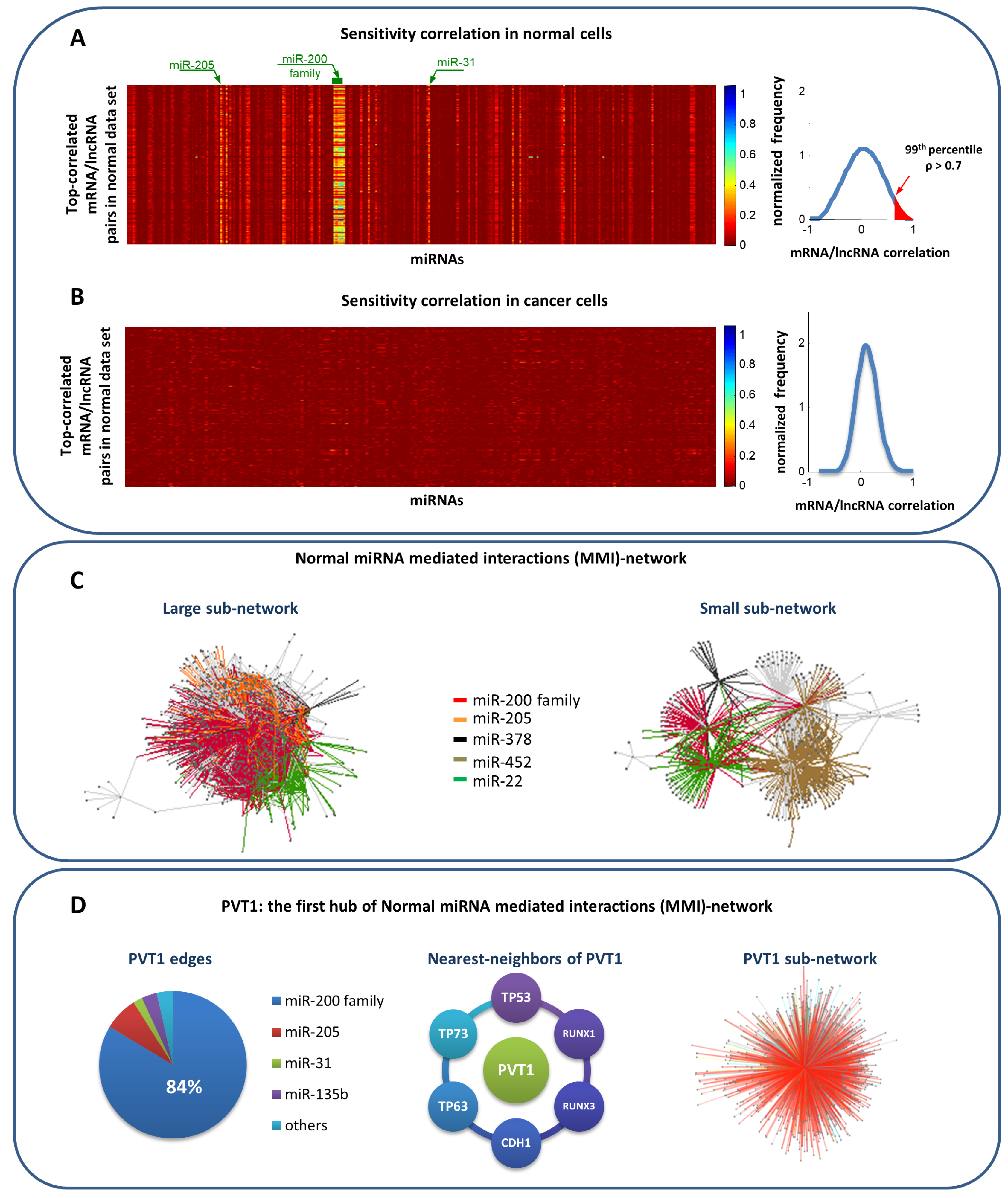
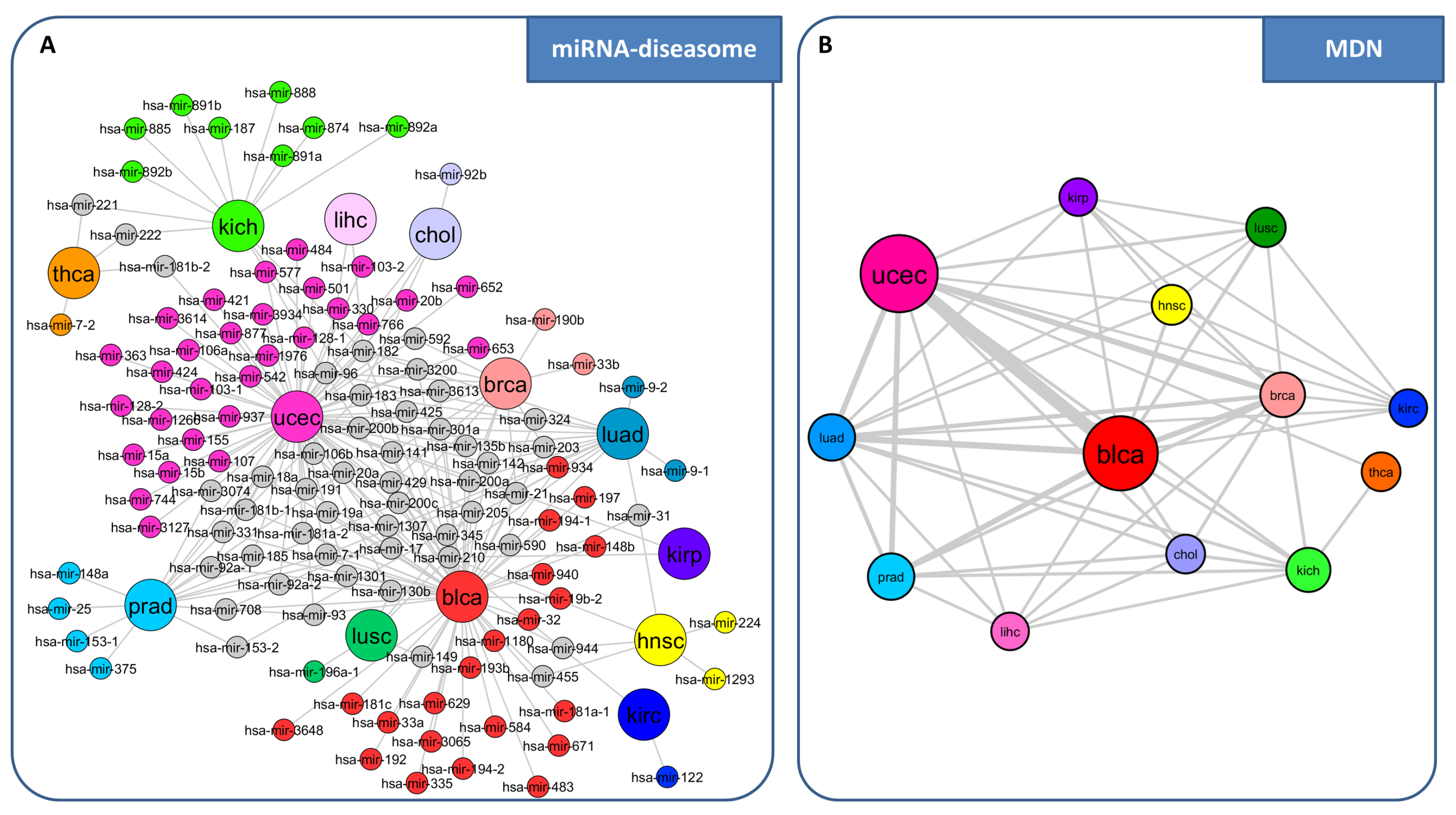
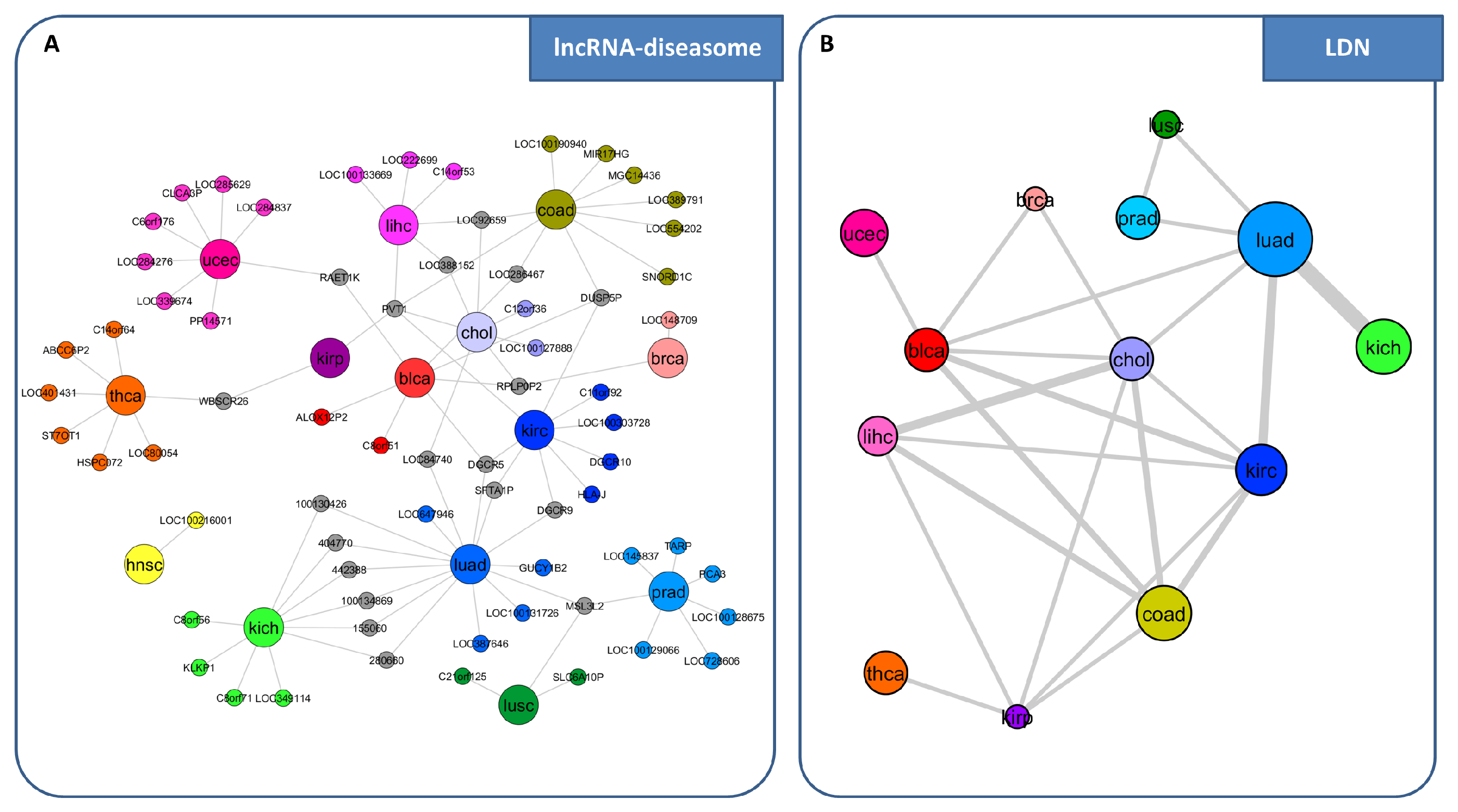
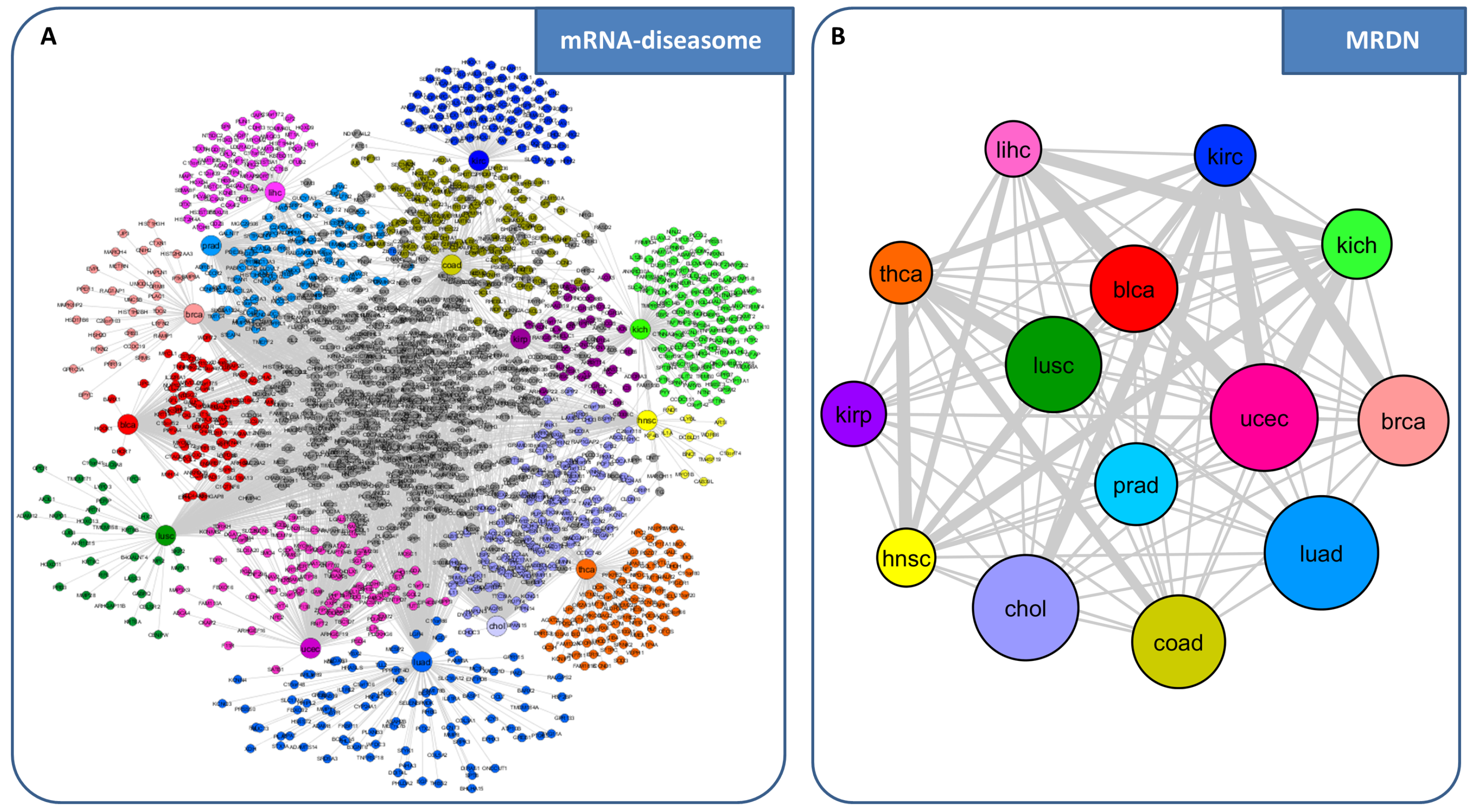
3. miRNA-Mediated Interactions Network: A Competing Endogenous RNA Model Exploiting the Topological Properties of Regulatory Networks
- RNAs competing for the same miRNA are marked by a highly positive correlation.
- Interaction between the RNAs competing for the same miRNA is indirect, i.e., mediated by miRNA.
- RNAs competing for the same miRNA harbor one or more MREs for the miRNA they sponge.
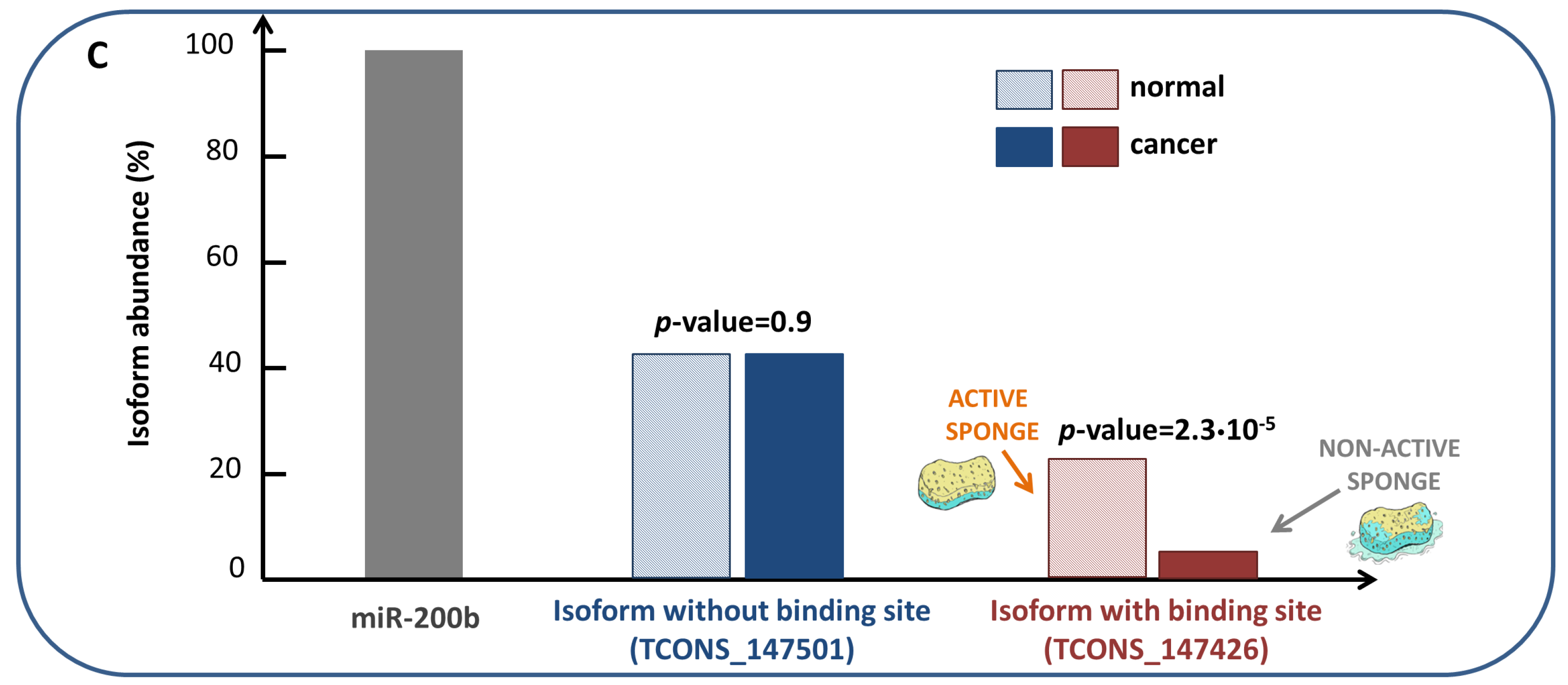
- Matching high values of the Pearson correlation between their expression profiles ( 0.7);
- Matching high values of the sensitivity correlation (S );
- Sharing binding sites for miRNAs (6-mer miRNA seed match).
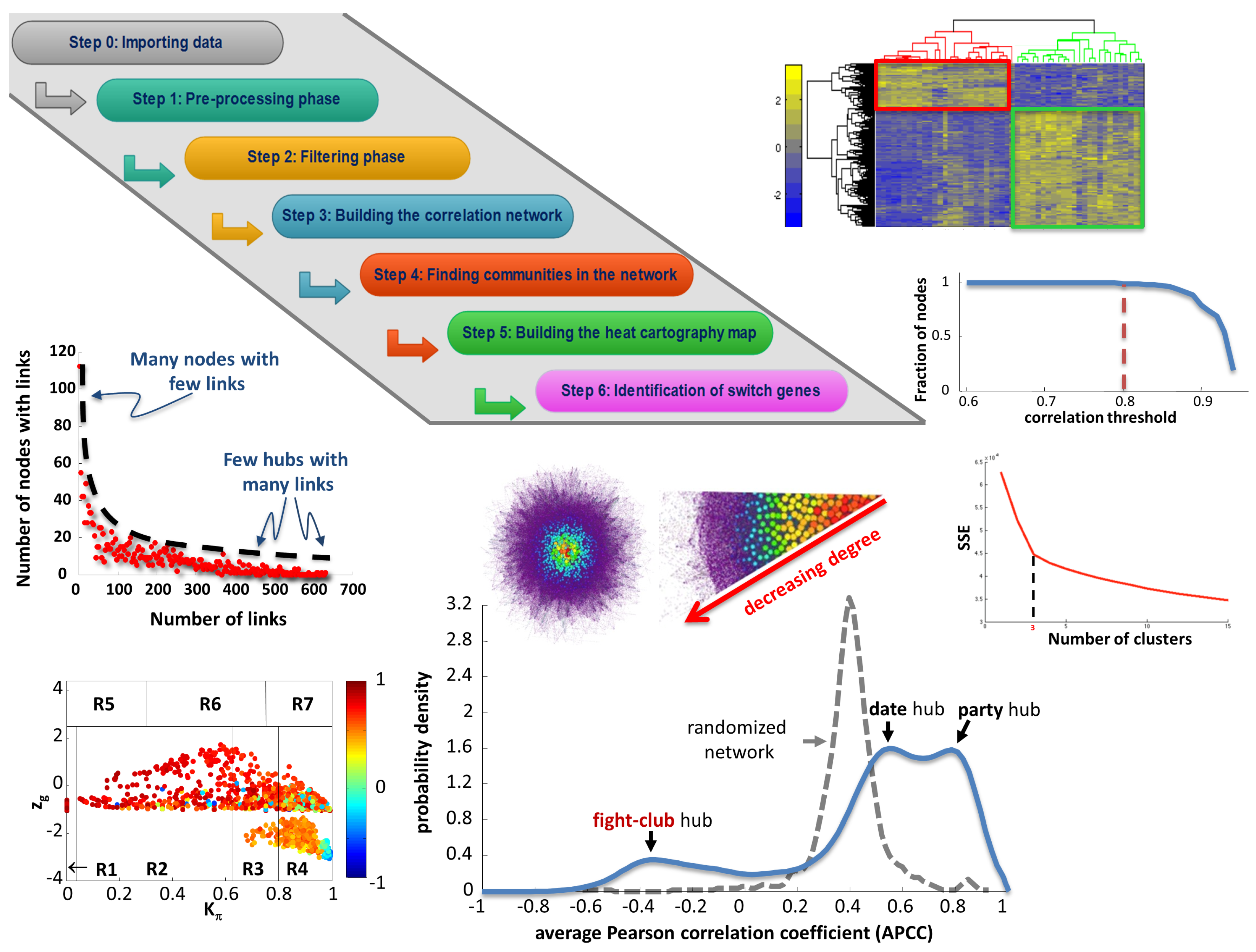
4. SWItchMiner (SWIM): A Tool Exploiting the Topological Properties of Gene Co-Expression Networks
4.1. SWIM Algorithm
4.1.1. Differential Gene Expression Analysis
4.1.2. Network Analysis
4.1.3. Role assignment to network nodes
- Being not a hub in their own cluster ();
- Having many links outside their own cluster ();
- Having a negative average weight of their incident links (APCC ).
4.2. SWIM Applications
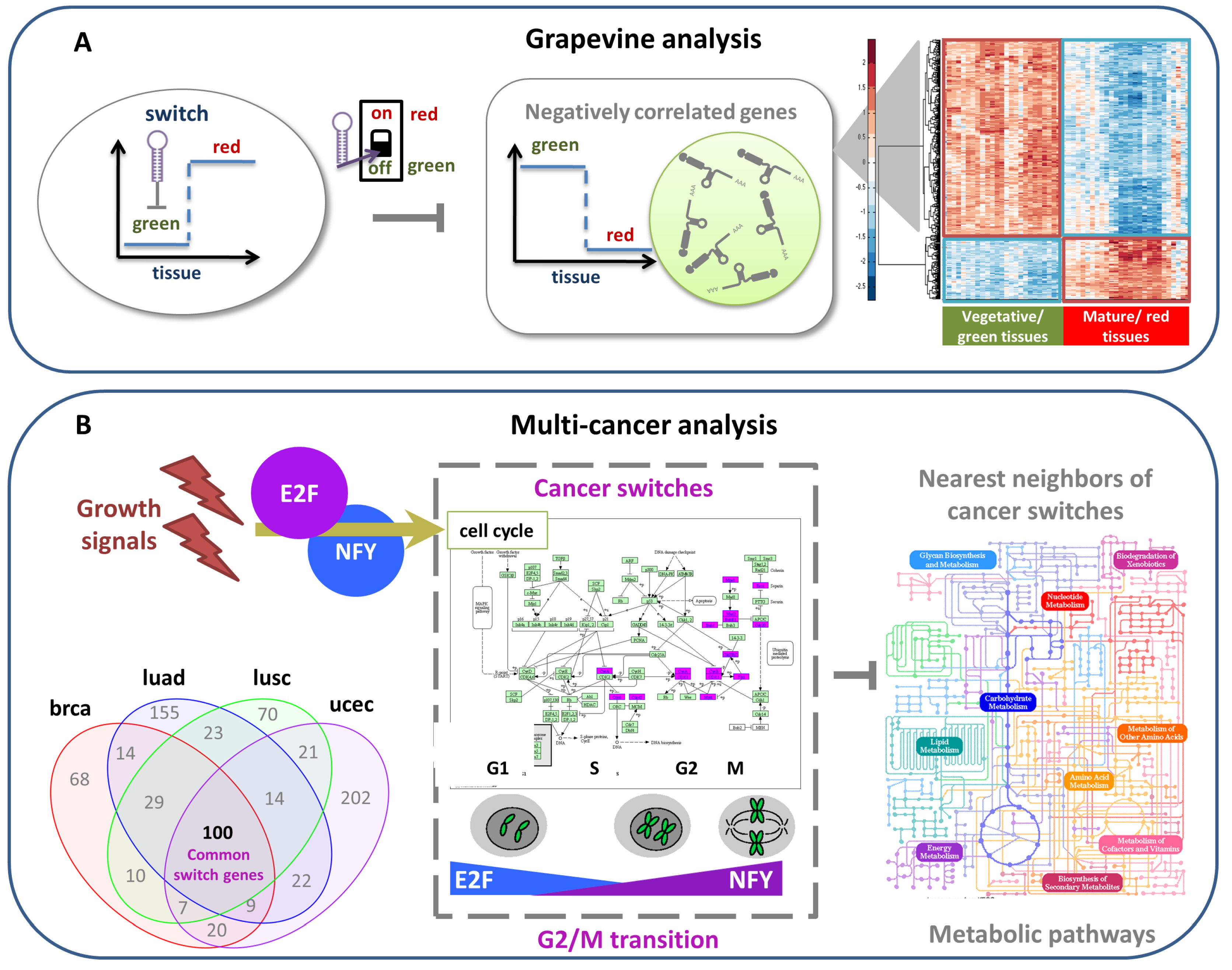
4.2.1. Grapevine Analysis
4.2.2. Multi-Cancer Analysis
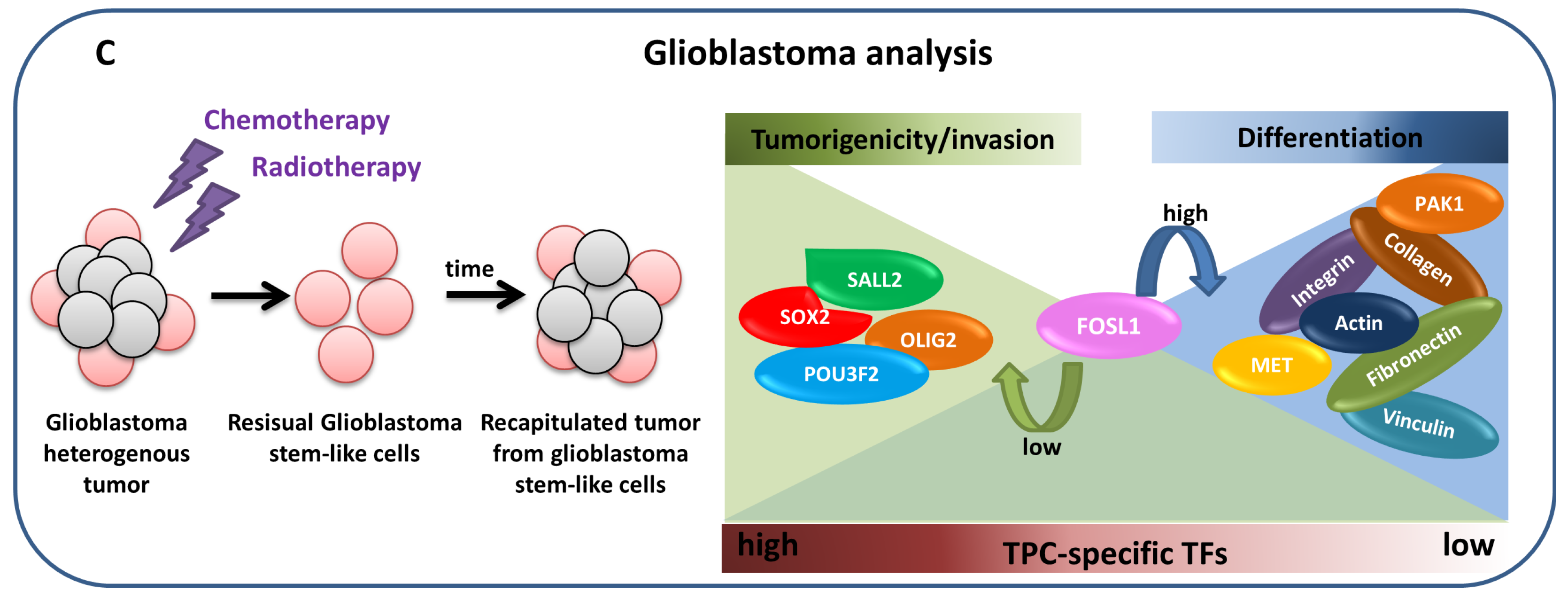
4.2.3. Glioblastoma analysis
4.3. SWIM Switch Genes towards DIAMOnD Disease Genes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A Network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein–protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Oti, M.; Snel, B.; Huynen, M.A.; Brunner, H.G. Predicting disease genes using protein–protein interactions. Am. J. Med. Genet. 2006, 43, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Chen, S.; Wu, X.; Tian, W. GenePANDA—A novel network-based gene prioritizing tool for complex diseases. Sci. Rep. 2017, 7, 43258. [Google Scholar] [CrossRef] [PubMed]
- Erten, S.; Bebek, G.; Ewing, R.M.; Koyutürk, M. DADA: Degree-aware algorithms for network-based disease gene prioritization. BioData Min. 2011, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Ghiassian, S.D.; Menche, J.; Barabási, A.L. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 2015, 11, e1004120. [Google Scholar] [CrossRef] [PubMed]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef] [PubMed]
- Mordelet, F.; Vert, J.P. ProDiGe: Prioritization Of Disease Genes with multitask machine learning from positive and unlabeled examples. BMC Bioinform. 2011, 12, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. The central role of RNA in human development and cognition. FEBS Lett. 2011, 585, 1600–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Stamatoyannopoulos, J.A.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowling, S.; Morris, K.V. Non-coding RNA and antisense RNA. Nature’s trash or treasure? Biochimie 2011, 93, 1922–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y. Genome Regulation by Long Non-Coding RNAs. Blood 2013, 122, SCI-29. [Google Scholar]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333. [Google Scholar] [CrossRef] [PubMed]
- Paci, P.; Colombo, T.; Farina, L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Le, T.D.; Zhang, J.; Liu, L.; Li, J. Computational methods for identifying miRNA sponge interactions. Brief. Bioinform. 2016, bbw042. [Google Scholar] [CrossRef] [PubMed]
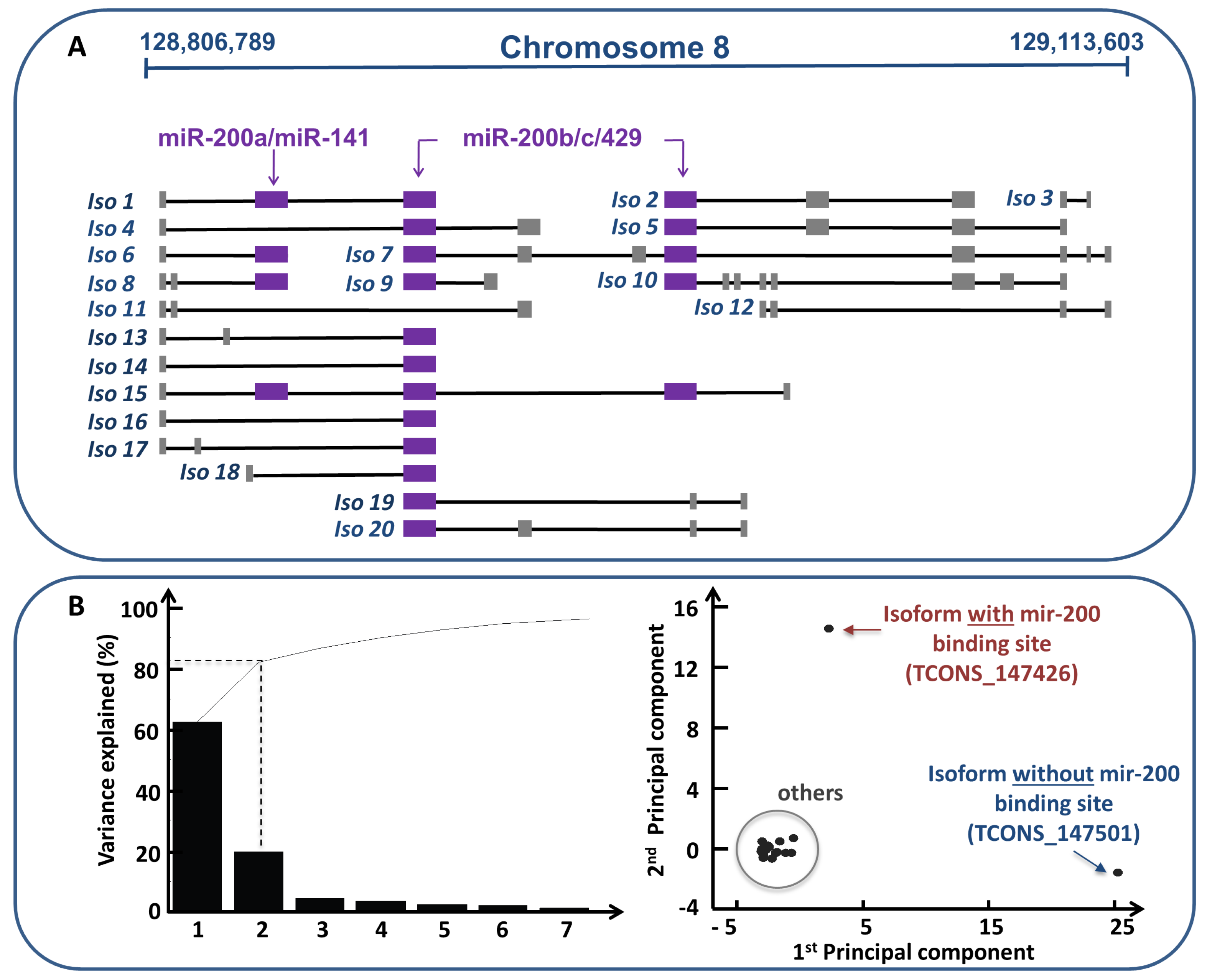
- Conte, F.; Fiscon, G.; Chiara, M.; Colombo, T.; Farina, L.; Paci, P. Role of the long non-coding RNA PVT1 in the dysregulation of the ceRNA-ceRNA network in human breast cancer. PLoS ONE 2017, 12, e0171661. [Google Scholar] [CrossRef] [PubMed]
- Paci, P.; Colombo, T.; Fiscon, G.; Gurtner, A.; Pavesi, G.; Farina, L. SWIM: A computational tool to unveiling crucial nodes in complex biological networks. Sci. Rep. 2017, 7, 44797. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, M.C.; Zenoni, S.; Fasoli, M.; Massonnet, M.; Farina, L.; Castiglione, F.; Pezzotti, M.; Paci, P. Integrated network analysis identifies fight-club nodes as a class of hubs encompassing key putative switch genes that induce major transcriptome reprogramming during grapevine development. Plant Cell 2014, 26, 4617–4635. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Conte, F.; Licursi, V.; Nasi, S.; Paci, P. Computational identification of specific genes for glioblastoma stem-like cells identity. Sci. Rep. 2018, 8, 7769. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Bauer, S.; Horn, D.; Robinson, P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008, 82, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Magger, O.; Berman, I.; Ruppin, E.; Sharan, R. PRINCIPLE: A tool for associating genes with diseases via network propagation. Bioinformatics 2011, 27, 3325–3326. [Google Scholar] [CrossRef] [PubMed]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.C. Passing messages between biological networks to refine predicted interactions. PLoS ONE 2013, 8, e64832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding tissue-specific gene regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Pandolfi, P. PTEN ceRNA networks in human cancer. Methods 2015, 77, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ergun, S.; Oztuzcu, S. Oncocers: ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways. Tumor Biol. 2015, 36, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, D.H.; Wu, N.; Xiao, J.H.; Wang, X.; Ma, W. ceRNA in cancer: Possible functions and clinical implications. Am. J. Med. Genet. 2015, 52, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, D.; Gao, L.; Liu, X.; Jin, Y.; Wang, D.; Wang, T.; Li, X. Competing endogenous RNA networks in human cancer: Hypothesis, validation, and perspectives. Oncotarget 2016, 7, 13479–13490. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82. [Google Scholar] [CrossRef] [PubMed]
- Iden, M.; Fye, S.; Li, K.; Chowdhury, T.; Ramchandran, R.; Rader, J. The lncRNA PVT1 contributes to the cervical cancer phenotype and associates with poor patient prognosis. PLoS ONE 2016, 11, e0156274. [Google Scholar] [CrossRef] [PubMed]
- Colombo, T.; Farina, L.; Macino, G.; Paci, P. PVT1: A rising star among oncogenic long noncoding RNAs. Biomed Res. Int. 2015, 2015, 304208. [Google Scholar] [CrossRef] [PubMed]
- Huppi, K.; Siwarski, D.; Skurla, R.; Klinman, D.; Mushinski, J. Pvt-1 transcripts are found in normal tissues and are altered by reciprocal (6; 15) translocations in mouse plasmacytomas. Proc. Natl. Acad. Sci. USA 1990, 87, 6964–6968. [Google Scholar] [CrossRef] [PubMed]
- Huppi, K.; Siwarski, D. Chimeric transcripts with an open reading frame are generated as a result of translocation to the Pvt-1 region in mouse B-cell tumors. Int. J. Cancer 1994, 59, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Kuo, W.L.; Stilwell, J.L.; Takano, H.; Lapuk, A.V.; Fridlyand, J.; Mao, J.H.; Yu, M.; Miller, M.A.; Santos, J.L.; et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007, 13, 5745–5755. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.; Adams, J.M. Chromosome 8 breakpoint far 3’of the c-myc oncogene in a Burkitt’s lymphoma 2; 8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 1986, 5, 2845. [Google Scholar] [PubMed]
- Hodgson, G.; Hager, J.H.; Volik, S.; Hariono, S.; Wernick, M.; Moore, D.; Albertson, D.G.; Pinkel, D.; Collins, C.; Hanahan, D.; et al. Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas. Nat. Genet. 2001, 29, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.B.; Maia, A.T.; O’Reilly, M.; Ghoussaini, M.; Prathalingam, R.; Porter-Gill, P.; Ambs, S.; Prokunina-Olsson, L.; Carroll, J.; Ponder, B.A. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet. 2011, 7, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.H.; Tidswell, R.; Dooley, J.S.; Sandanayake, N.S.; Cerec, V.; Deheragoda, M.; Lee, A.J.; Swanton, C.; Andreola, F.; Pereira, S.P. Whole genome RNA expression profiling of endoscopic biliary brushings provides data suitable for biomarker discovery in cholangiocarcinoma. J. Hepatol. 2012, 56, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yuan, J.H.; Wang, S.B.; Yang, F.; Yuan, S.X.; Ye, C.; Yang, N.; Zhou, W.P.; Li, W.L.; Li, W.; et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology 2014, 60, 1278–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, C.; Li, J.; Liu, Y.; Chen, M.; Yuan, J.; Fu, X.; Zhan, Y.; Liu, L.; Lin, J.; Zhou, Q.; Xu, W.; Zhao, G.; Cai, Z.; Huang, W. Tetracycline-inducible shRNA targeting long non-coding RNA PVT1 inhibits cell growth and induces apoptosis in bladder cancer cells. Oncotarget 2015, 6, 41194–41203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chen, J.; Feng, J.; Wang, J. Long noncoding RNA PVT1 modulates thyroid cancer cell proliferation by recruiting EZH2 and regulating thyroid-stimulating hormone receptor (TSHR). Tumor Biol. 2016, 37, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Yu, C.H.; Liu, M.; Xia, Q.Q.; Zhang, Y.F.; Jiang, W.L. Long non-coding RNA PVT1 as a novel biomarker for diagnosis and prognosis of non-small cell lung cancer. Tumor Biol. 2016, 37, 4127–4134. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhou, H.; Liu, P.; Yan, L.; Yao, W.; Chen, K.; Zeng, J.; Li, H.; Hu, J.; Xu, H.; et al. lncRNA PVT1 and its splicing variant function as competing endogenous RNA to regulate clear cell renal cell carcinoma progression. Oncotarget 2017, 8, 85353. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhu, H.; Yin, L.; Wang, T.; Wu, J.; Xu, J.; Tao, H.; Liu, J.; He, X. lncRNA-PVT1 facilitates invasion through upregulation of MMP9 in nonsmall cell lung cancer cell. DNA Cell Biol. 2017, 36, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Hu, L.; Cheng, J.; Xu, J.; Zhong, Z.; Yang, Y.; Yuan, Z. lncRNA PVT1 promotes the angiogenesis of vascular endothelial cell by targeting miR-26b to activate CTGF/ANGPT2. Int. J. Mol. Med. 2018, 42, 489–496. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jing, Y.; Wei, F.; Tang, Y.; Yang, L.; Luo, J.; Yang, P.; Ni, Q.; Pang, J.; Liao, Q.; et al. Long non-coding RNA PVT1 predicts poor prognosis and induces radioresistance by regulating DNA repair and cell apoptosis in nasopharyngeal carcinoma. Cell Death Dis. 2018, 9, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houshmand, M.; Yazdi, N.; Kazemi, A.; Atashi, A.; Hamidieh, A.A.; Najemdini, A.A.; Pour, M.M.; Zarif, M.N. Long non-coding RNA PVT1 as a novel candidate for targeted therapy in hematologic malignancies. Int. J. Biochem. Cell Biol. 2018, 98, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, D.; Li, Y.; Li, X.; Zhao, L.; Zhang, J.; Song, Y. Effect of long non-coding RNA PVT1 on cell proliferation and migration in melanoma. Int. J. Mol. Med. 2018, 41, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 289–300. [Google Scholar]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A k-means clustering algorithm. J. R. Stat. Soc. Ser. B Stat. Methodol. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Meilă, M. The uniqueness of a good optimum for k-means. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 625–632. [Google Scholar]
- Lisboa, P.J.; Etchells, T.A.; Jarman, I.H.; Chambers, S.J. Finding reproducible cluster partitions for the k-means algorithm. BMC Bioinform. 2013, 14, S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.D.J.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Guimera, R.; Amaral, L.A.N. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Niu, F.; Humburg, B.A.; Liao, K.; Bendi, S.; Callen, S.; Fox, H.S.; Buch, S. Molecular mechanisms of long noncoding RNAs and their role in disease pathogenesis. Oncotarget 2018, 9, 18648. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Targeted Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, G.; Weller, M. Glioblastoma stem cells. Cell Tissue Res. 2011, 343, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Günther, H.S.; Phillips, H.S.; Kemming, D.; Martens, T.; Kharbanda, S.; Soriano, R.H.; Modrusan, Z.; Zapf, S.; Westphal, M.; et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 2011, 59, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Galvagni, F.; Orlandini, M.; Oliviero, S. Role of the AP-1 transcription factor FOSL1 in endothelial cells adhesion and migration. Cell Adhes. Migr. 2013, 7, 408–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucl. Acids Res. 2010, 8, e178. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucl. Acids Res. 2011, 33, 514–517. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method/Tool | Brief Description | Availability | Reference |
|---|---|---|---|
| Protein–Protein Interaction Network | |||
| Oti et al. | It identifies new candidate disease genes by searching for disease proteins having interaction partners located within loci associated with the same disease | Prediction results available | [7] |
| GenePANDA (Gene Prioritizing Approach using Network Distance Analysis) | It identifies new candidate disease genes based on their relative distance to known disease genes in a functional association network | Prediction results available | [8] |
| DADA (Degree-Aware Disease Gene Prioritization) | It prioritizes candidate disease genes with respect to a disease of interest based on network proximity measure, calculated by using Random Walk with Restarts algorithm [32] with some statistical adjustment | MATLAB software package | [9] |
| DIAMOnD (DIseAse MOdule Detection) | It identifies full disease modules around a set of known disease proteins by performing a systematic analysis of the PPI-network that exploits the “connectivity significance” instead of local connection density | Python software package | [10] |
| PRINCE (PRIoritizatioN and Complex Elucidation) | It prioritizes genes related to a query disease based on their closeness, in the PPI-network, to genes causing phenotypically similar disorders to the query disease | Cytoscape Plug-in | [11,33] |
| ProDiGe (Prioritization Of Disease Genes) | It implements a novel machine learning strategy for gene prioritization based on learning from a set of positive examples (e.g., known disease genes) and unlabeled examples (e.g., candidate genes), allowing heterogeneous data integration | MATLAB software package | [12] |
| Regulatory Network | |||
| MMI-network (MiRNA-Mediated Interactions network) | ceRNA model based on partial association to investigate the role of lncRNAs as miRNA sponges in human breast cancer. It computes for each triplet (lncRNA, miRNA, messenger RNA (mRNA)) the difference between Pearson correlation of (lncRNA, mRNA) and partial correlation (lncRNA, mRNA|miRNA) to examine the contribution of the miRNA into the lncRNA/mRNA relationship | Prediction results available | [25,27] |
| PANDA (Passing Attributes between Networks for Data Assimilation) | It implements a message-passing model using multiple sources of information to predict regulatory relationships, and used it to integrate protein–protein interaction, gene expression, and sequence motif data to reconstruct genome-wide, condition-specific regulatory networks in yeast as a model | MATLAB/ R/Python software packages | [34] |
| Sonawane et al. | It uses PANDA to infer gene regulatory networks for 38 different tissues by integrating GTEx RNA-sequencing (RNA-seq) data with a canonical set of transcription factors to target gene edges and protein–protein interactions | Prediction results available | [35] |
| Co-Expression Network | |||
| SWIM (Switch Miner) | Wizard-like software that integrates gene expression data with network topological properties for identifying a small pool of genes (i.e., switch genes) critically associated with drastic changes in cell phenotype | MATLAB software package | [28,29,30] |
| WGCNA (Weighted Correlation Network Analysis) | Collection of R functions for performing weighted correlation network analysis of large data sets, including functions for network construction, module identification, topological properties calculation, data manipulation and visualization | R software package | [31] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiscon, G.; Conte, F.; Farina, L.; Paci, P. Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine. Genes 2018, 9, 437. https://doi.org/10.3390/genes9090437
Fiscon G, Conte F, Farina L, Paci P. Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine. Genes. 2018; 9(9):437. https://doi.org/10.3390/genes9090437
Chicago/Turabian StyleFiscon, Giulia, Federica Conte, Lorenzo Farina, and Paola Paci. 2018. "Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine" Genes 9, no. 9: 437. https://doi.org/10.3390/genes9090437
APA StyleFiscon, G., Conte, F., Farina, L., & Paci, P. (2018). Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine. Genes, 9(9), 437. https://doi.org/10.3390/genes9090437