Can the MerPAS Passive Air Sampler Discriminate Landscape, Seasonal, and Elevation Effects on Atmospheric Mercury? A Feasibility Study in Mississippi, USA

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Hg Passive Air Sampler
2.3. Deployment of PAS
2.4. Determination of Hg on the Sorbent and in the Soil and Sediment using a Direct Mercury Analyzer
2.5. Calculation of Atmospheric Hg Concentration
2.6. Statistical Analysis
3. Results and Discussion
3.1. Summary Statistics and Overall Trends
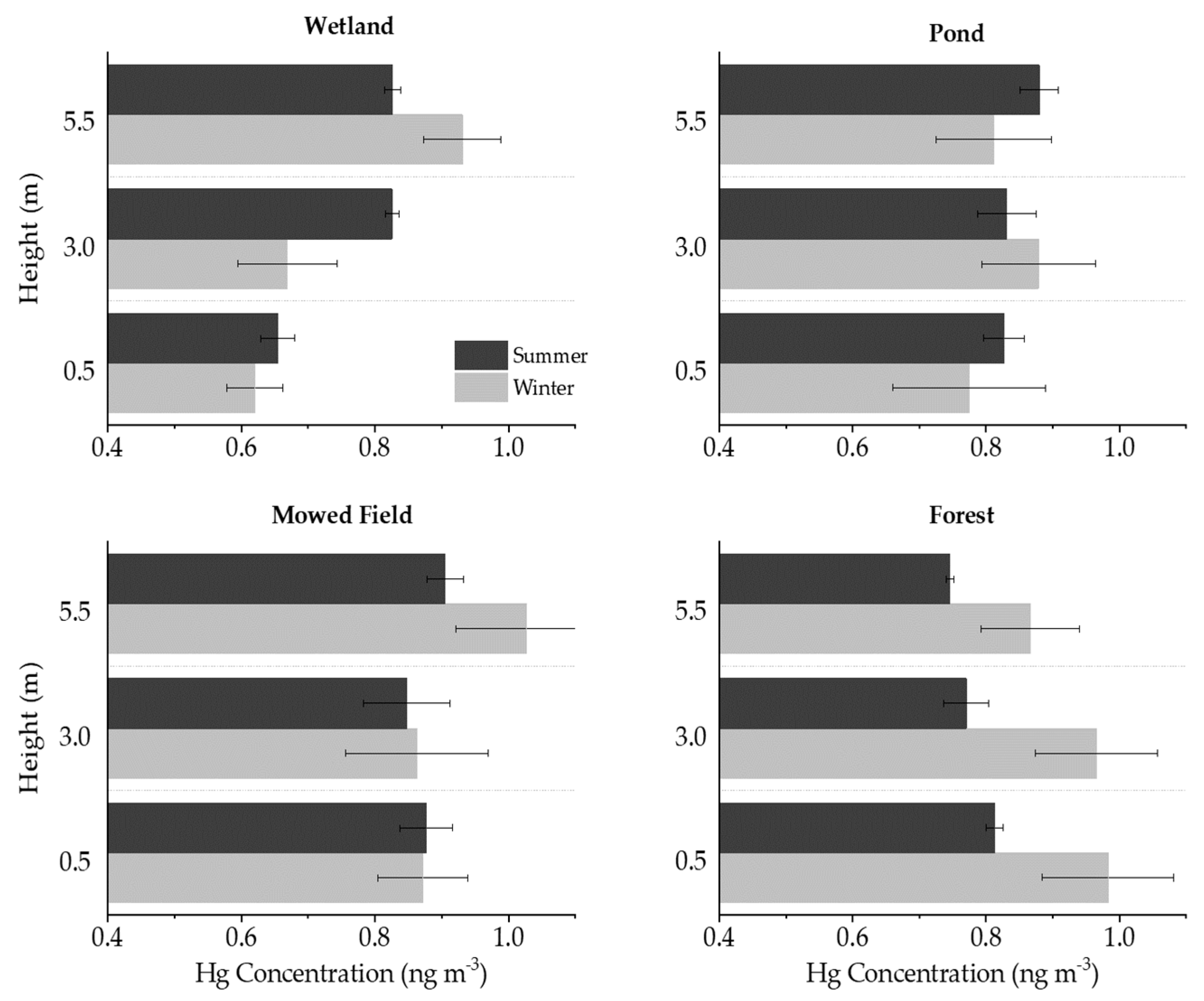
3.2. Wetland
3.3. Pond
3.4. Mowed Field
3.5. Forest
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Winter | Summer |
|---|---|---|
| Mean Temperature (°C) | 11.9 | 25.7 |
| Mean Wind Speed (m/s) | 0.16 | 0.08 |
| Accumulated Precipitation (mm) | 1.1 | 6.9 |
| Landscape | Hg in Soil or Sediment (ng g−1, dry weight, n = 6) | Height (m) | Summer | Winter | ||
|---|---|---|---|---|---|---|
| Mean (n = 4) | Standard Error | Mean (n = 3) | Standard Error | |||
| Wetland | 5.5 | 0.83 | 0.01 | 0.93 | 0.06 | |
| 45.5 ± 1.9 | 3.0 | 0.83 | 0.01 | 0.67 | 0.07 | |
| 0.5 | 0.65 | 0.03 | 0.62 | 0.04 | ||
| Pond | 5.5 | 0.88 (n = 10) | 0.03 | 0.81 | 0.09 | |
| Not Measured | 3.0 | 0.83 (n = 6) | 0.04 | 0.88 | 0.09 | |
| 0.5 | 0.83 (n = 8) | 0.03 | 0.77 | 0.11 | ||
| Mowed Field | 5.5 | 0.91 | 0.03 | 1.03 | 0.10 | |
| 21.2 ± 0.6 | 3.0 | 0.85 (n = 7) | 0.06 | 0.86 | 0.10 | |
| 0.5 | 0.88 (n = 6) | 0.04 | 0.87 | 0.07 | ||
| Forest | 5.5 | 0.75 | 0.01 | 0.87 | 0.07 | |
| 74.8 ± 2.8 | 3.0 | 0.77 | 0.03 | 0.97 | 0.09 | |
| 0.5 | 0.81 | 0.01 | 0.98 | 0.10 | ||
References
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Valente, R.J.; Shea, C.; Humes, K.L.; Tanner, R.L. Atmospheric mercury in the Great Smoky Mountains compared to regional and global levels. Atmos. Environ. 2007, 41, 1861–1873. [Google Scholar] [CrossRef]
- Skov, H.; Christensen, J.H.; Goodsite, M.E.; Heidam, N.Z.; Jensen, B.; Wåhlin, P.; Geernaert, G. Fate of Elemental Mercury in the Arctic during Atmospheric Mercury Depletion Episodes and the Load of Atmospheric Mercury to the Arctic. Environ. Sci. Technol. 2004, 38, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Gustin, M.; Jaffe, D. Reducing the Uncertainty in Measurement and Understanding of Mercury in the Atmosphere. Environ. Sci. Technol. 2010, 44, 2222–2227. [Google Scholar] [CrossRef] [PubMed]
- Keeler, G.; Glinsorn, G.; Pirrone, N. Particulate mercury in the atmosphere: Its significance, transport, transformation and sources. Water Air Soil Pollut. 1995, 80, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, C.T.; Mason, R.P.; Chan, H.M.; Jacob, D.J.; Pirrone, N. Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ. Sci. Technol. 2013, 47, 4967–4983. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Pehkonen, S.O. The chemistry of atmospheric mercury: A review. Atmos. Environ. 1999, 33, 2067–2079. [Google Scholar] [CrossRef]
- Pandey, S.K.; Kim, K.-H.; Brown, R.J. Measurement techniques for mercury species in ambient air. TrAC Trends Anal. Chem. 2011, 30, 899–917. [Google Scholar] [CrossRef]
- Osterwalder, S.; Eugster, W.; Feigenwinter, I.; Jiskra, M. First eddy covariance flux measurements of gaseous elemental mercury (Hg0) over a grassland. Atmos. Meas. Tech. Discuss. 2019. in review. [Google Scholar] [CrossRef]
- Gustin, M.; Lyman, S.; Kilner, P.; Prestbo, E. Development of a passive sampler for gaseous mercury. Atmos. Environ. 2011, 45, 5805–5812. [Google Scholar] [CrossRef]
- Landis, M.S.; Stevens, R.K.; Schaedlich, F.; Prestbo, E.M. Development and Characterization of an Annular Denuder Methodology for the Measurement of Divalent Inorganic Reactive Gaseous Mercury in Ambient Air. Environ. Sci. Technol. 2002, 36, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, W.; Petty, J.; May, T.; Huckins, J. A passive integrative sampler for mercury vapor in air and neutral mercury species in water. Chemosphere Glob. Chang. Sci. 2000, 2, 1–9. [Google Scholar] [CrossRef]
- Peterson, C.; Alishahi, M.; Gustin, M.S. Testing the use of passive sampling systems for understanding air mercury concentrations and dry deposition across Florida, USA. Sci. Total Environ. 2012, 424, 297–307. [Google Scholar] [CrossRef] [PubMed]
- McLagan, D.S.; Mitchell, C.P.J.; Huang, H.; Lei, Y.D.; Cole, A.S.; Steffen, A.; Hung, H.; Wania, F. A High-Precision Passive Air Sampler for Gaseous Mercury. Environ. Sci. Technol. Lett. 2016, 3, 24–29. [Google Scholar] [CrossRef]
- McLagan, D.S.; Mitchell, C.P.J.; Huang, H.; Hussain, B.A.; Lei, Y.D.; Wania, F. The effects of meteorological parameters and diffusive barrier reuse on the sampling rate of a passive air sampler for gaseous mercury. Atmos. Meas. Tech. 2017, 10, 3651–3660. [Google Scholar] [CrossRef] [Green Version]
- McLagan, D.S.; Mazur, M.E.E.; Mitchell, C.P.J.; Wania, F. Passive air sampling of gaseous elemental mercury: A critical review. Atmos. Chem. Phys. Discuss. 2016, 16, 3061–3076. [Google Scholar] [CrossRef]
- Sommar, J.; Zhu, W.; Lin, C.-J.; Feng, X. Field Approaches to Measure Hg Exchange Between Natural Surfaces and the Atmosphere—A Review. Crit. Rev. Environ. Sci. Technol. 2013, 43, 1657–1739. [Google Scholar] [CrossRef]
- Gustin, M.S.; Kolker, A.; Gårdfeldt, K. Transport and fate of mercury in the environment. Appl. Geochem. 2008, 23, 343–344. [Google Scholar] [CrossRef]
- Choi, H.-D.; Holsen, T.M. Gaseous mercury fluxes from the forest floor of the Adirondacks. Environ. Pollut. 2009, 157, 592–600. [Google Scholar] [CrossRef]
- Poissant, L.; Casimir, A. Water-air and soil-air exchange rate of total gaseous mercury measured at background sites. Atmos. Environ. 1998, 32, 883–893. [Google Scholar] [CrossRef]
- McLagan, D.S.; Mitchell, C.P.J.; Steffen, A.; Hung, H.; Shin, C.; Stupple, G.W.; Olson, M.L.; Luke, W.T.; Kelley, P.; Howard, D.; et al. Global evaluation and calibration of a passive air sampler for gaseous mercury. Atmos. Chem. Phys. Discuss. 2018, 18, 5905–5919. [Google Scholar] [CrossRef] [Green Version]
- Feigis, M.; Mistry, S.; Snow, M.; Lei, Y.D.; Wania, F. Measuring Vertical Profiels of Gaseous Mercury Concentration using Passive Air Samplers. In Proceedings of the SETAC North America 39th Annual Meeting, Sacramento, CA, USA, 4–8 November 2018. [Google Scholar]
- Cizdziel, J.V.; Zhang, Y.; Nallamothu, D.; Brewer, J.S.; Gao, Z. Air/surface exchange of gaseous elemental mercury at different landscapes in Mississippi, USA. Atmosphere 2019, 10, 538. [Google Scholar] [CrossRef]
- Stupple, G.W.; McLagan, D.S.; Steffen, A. In situ reactive gaseous mercury uptake on Radiello diffusive barrier, cation exchange membrane and teflon filter membranes during atmospheric depletion events. In Proceedings of the International Conference on Mercury as a Global Pollutant, Krakow, Poland, 8–13 September 2019. [Google Scholar]
- Antón, M.A.L.; Yuan, Y.; Perry, R.; Maroto-Valer, M.M. Analysis of mercury species present during coal combustion by thermal desorption. Fuel 2010, 89, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Cizdziel, J.V.; Hinners, T.A.; Heithmar, E.M. Determination of Total Mercury in Fish Tissues using Combustion Atomic Absorption Spectrometry with Gold Amalgamation. Water Air Soil Pollut. 2002, 135, 355–370. [Google Scholar] [CrossRef]
- Chen, J.; Chakravarty, P.; Davidson, G.R.; Wren, D.G.; Locke, M.A.; Zhou, Y.; Brown, G.; Cizdziel, J.V. Simultaneous determination of mercury and organic carbon in sediment and soils using a direct mercury analyzer based on thermal decomposition–atomic absorption spectrophotometry. Anal. Chim. Acta 2015, 871, 9–17. [Google Scholar] [CrossRef] [PubMed]
- McLagan, D.S.; Huang, H.; Lei, Y.D.; Wania, F.; Mitchell, C.P. Application of sodium carbonate prevents sulphur poisoning of catalysts in automated total mercury analysis. Spectrochim. Acta Part B Spectrosc. 2017, 133, 60–62. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Dong, W.; Meyers, T. Transpiration of gaseous elemental mercury through vegetation in a subtropical wetland in Florida. Atmos. Environ. 2002, 36, 5207–5219. [Google Scholar] [CrossRef]
- Gustin, M.S.; Stamenković, J. Effect of Watering and Soil Moisture on Mercury Emissions from Soils. Biogeochemistry 2005, 76, 215–232. [Google Scholar] [CrossRef]
- Kuiken, T.; Gustin, M.; Zhang, H.; Lindberg, S.; Sedinger, B. Mercury emission from terrestrial background surfaces in the eastern USA. II: Air/surface exchange of mercury within forests from South Carolina to New England. Appl. Geochem. 2008, 23, 356–368. [Google Scholar] [CrossRef]
- Stamenkovic, J.; Gustin, M.S. Nonstomatal versus Stomatal Uptake of Atmospheric Mercury. Environ. Sci. Technol. 2009, 43, 1367–1372. [Google Scholar] [CrossRef]
- Amyot, M.; McQueen, D.J.; Mierle, G.; Lean, D.R.S. Sunlight-Induced Formation of Dissolved Gaseous Mercury in Lake Waters. Environ. Sci. Technol. 1994, 28, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, D.; Ma, M. Mercury release flux and its influencing factors at the air-water interface in paddy field in Chongqing, China. Chin. Sci. Bull. 2013, 58, 266–274. [Google Scholar] [CrossRef]
- Nriagu, J.O. Mechanistic steps in the photoreduction of mercury in natural waters. Sci. Total. Environ. 1994, 154, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ericksen, J.; Gustin, M.; Xin, M.; Weisberg, P.; Fernandez, G. Air–soil exchange of mercury from background soils in the United States. Sci. Total. Environ. 2006, 366, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, J.; Gustin, M.; Schorran, D.; Johnson, D.; Lindberg, S.; Coleman, J. Accumulation of atmospheric mercury in forest foliage. Atmos. Environ. 2003, 37, 1613–1622. [Google Scholar] [CrossRef]
- Gustin, M.S.; Ericksen, J.A.; Schorran, D.E.; Johnson, D.W.; Lindberg, S.E.; Coleman, J.S. Application of Controlled Mesocosms for Understanding Mercury Air−Soil−Plant Exchange. Environ. Sci. Technol. 2004, 38, 6044–6050. [Google Scholar] [CrossRef]
- Feng, X.; Wang, S.; Qiu, G.; Hou, Y.; Tang, S. Total gaseous mercury emissions from soil in Guiyang, Guizhou, China: Mercury emission from soil in Guiyang. J. Geophys. Res. Atmos. 2005, 110. [Google Scholar] [CrossRef]
- Mazur, M.; Mitchell, C.; Eckley, C.; Eggert, S.; Kolka, R.; Sebestyen, S.; Swain, E.; Sebestyen, S. Gaseous mercury fluxes from forest soils in response to forest harvesting intensity: A field manipulation experiment. Sci. Total. Environ. 2014, 496, 678–687. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, B.; Cizdziel, J.V. Can the MerPAS Passive Air Sampler Discriminate Landscape, Seasonal, and Elevation Effects on Atmospheric Mercury? A Feasibility Study in Mississippi, USA. Atmosphere 2019, 10, 617. https://doi.org/10.3390/atmos10100617
Jeon B, Cizdziel JV. Can the MerPAS Passive Air Sampler Discriminate Landscape, Seasonal, and Elevation Effects on Atmospheric Mercury? A Feasibility Study in Mississippi, USA. Atmosphere. 2019; 10(10):617. https://doi.org/10.3390/atmos10100617
Chicago/Turabian StyleJeon, Byunggwon, and James V. Cizdziel. 2019. "Can the MerPAS Passive Air Sampler Discriminate Landscape, Seasonal, and Elevation Effects on Atmospheric Mercury? A Feasibility Study in Mississippi, USA" Atmosphere 10, no. 10: 617. https://doi.org/10.3390/atmos10100617