Regulation of Germinal Center Reactions by B and T Cells

{kind=link}
{kind=link}
Abstract
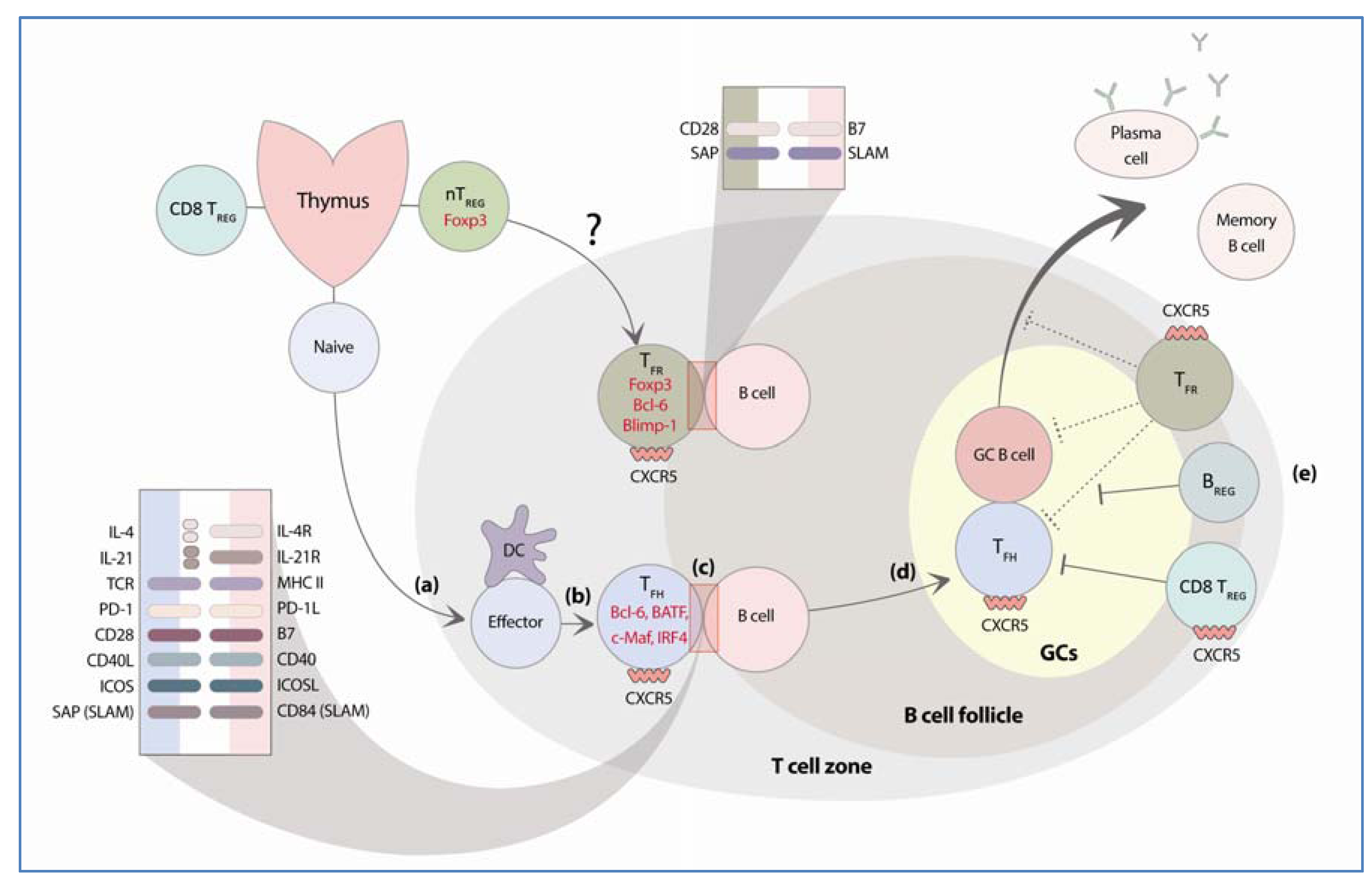
:1. Introduction
2. Follicular Helper T Cells
2.1. Transcription Factors and Molecules Required for the Differentiation of TFH Cells

2.1.1. Bcl-6
2.1.2. c-Maf
2.1.3. BATF
2.1.4. STAT3
2.1.5. IRF4
2.1.6. microRNAs
2.1.7. Cytokines
2.2. Molecular Requirements for TFH Cell Function
2.2.1. CXCR5
2.2.2. ICOS
2.2.3. CD40L–CD40
2.2.4. SAP:SLAM Family Receptor
2.2.5. IL-21
2.2.6. IL-4
2.3. TFH Cells and Autoimmunity
2.3.1. TFH Cells in Animal Models of Antibody-Mediated Autoimmunity
2.3.2. TFH Cells in Human Autoimmune Diseases
3. Regulatory Cells that Control GC B Cell Responses
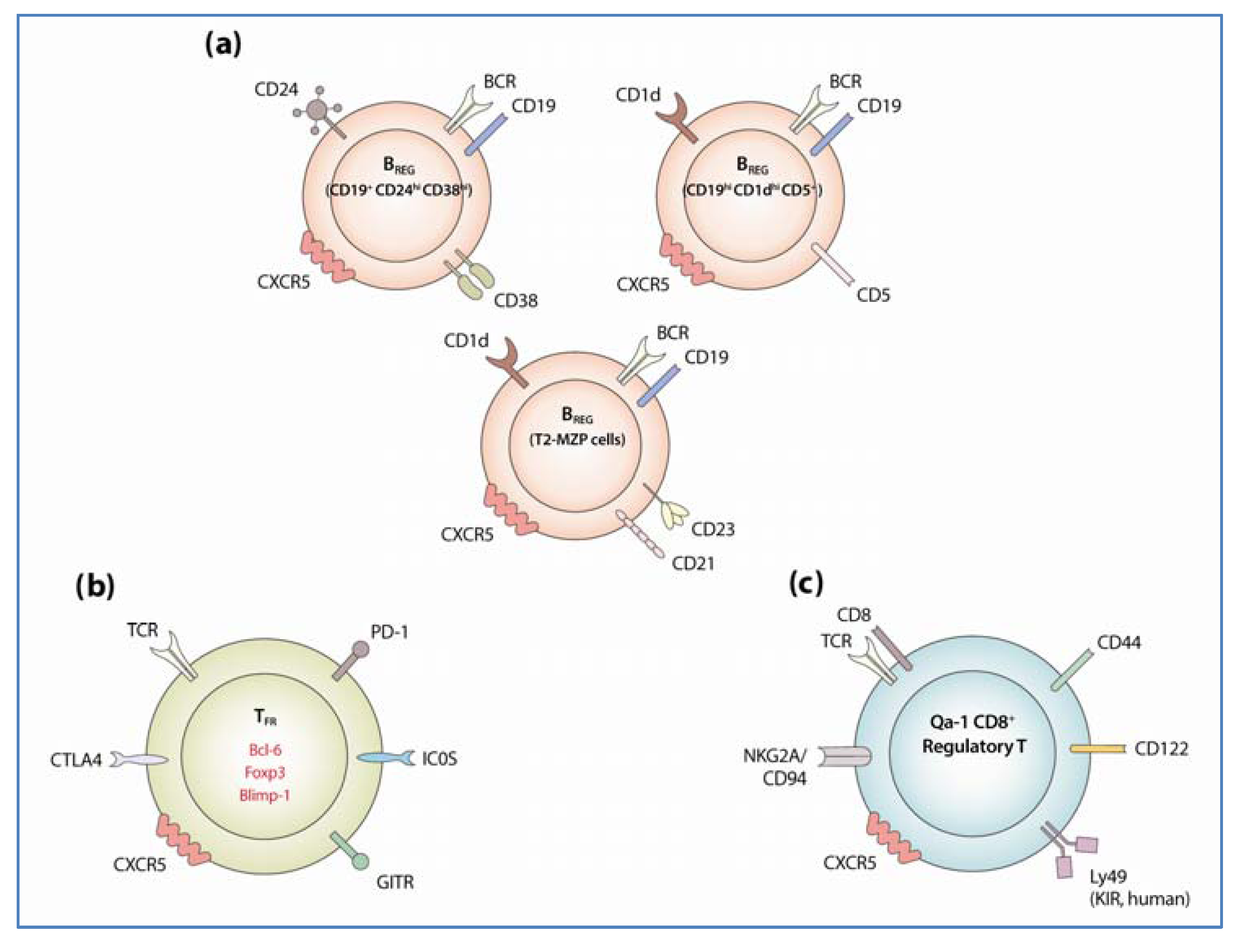
3.1. Regulatory B Cells
3.1.1. Identification and Development of Regulatory B Cells

3.1.2. Regulatory Mechanism of BREG Cells
3.1.3. BREG Cells in Animal Models of Autoimmune Diseases
3.2. Follicular Regulatory T Cells
3.2.1. Identification and Development of TFR
3.2.2. Mechanism of TFR Suppressive Activity
3.3. Qa-1 Restricted CD8+ T Cells
3.3.1. Identification of Qa-1 Restricted CD8+ TREG Cells
3.3.2. Mechanism of Qa-1 Restricted CD8+ TREG-Mediated Suppression on GC Reactions
4. Translational Potentials
4.1. Vaccine Design
4.2. Autoimmune Diseases
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Zhang, X.; Ing, S.; Fraser, A.; Chen, M.; Khan, O.; Zakem, J.; Davis, W.; Quinet, R. Follicular helper T cells: New insights into mechanisms of autoimmune diseases. Ochsner J. 2013, 13, 131–139. [Google Scholar]
- Fujio, K.; Okamura, T.; Sumitomo, S.; Yamamoto, K. Regulatory cell subsets in the control of autoantibody production related to systemic autoimmunity. Ann. Rheum Dis. 2013, 72, ii85–ii89. [Google Scholar] [CrossRef]
- Miller, J.F.; De Burgh, P.M.; Grant, G.A. Thymus and the production of antibody-plaque-forming cells. Nature 1965, 208, 1332–1334. [Google Scholar] [CrossRef]
- Breitfeld, D. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef]
- Schaerli, P. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 2000, 192, 1553–1562. [Google Scholar] [CrossRef]
- Kim, C.H. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center [mdash] localized subset of CXCR5+ T cells. J. Exp. Med. 2001, 193, 1373–1381. [Google Scholar] [CrossRef]
- Ansel, K.M.; McHeyzer-Williams, L.J.; Ngo, V.N.; McHeyzer-Williams, M.G.; Cyster, J.G. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J. Exp. Med. 1999, 190, 1123–1134. [Google Scholar] [CrossRef]
- King, I.L.; Mohrs, M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J. Exp. Med. 2009, 206, 1001–1007. [Google Scholar] [CrossRef]
- Yusuf, I. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J. Immunol. 2010, 185, 190–202. [Google Scholar] [CrossRef]
- Reinhardt, R.L.; Liang, H.E.; Locksley, R.M. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 2009, 10, 385–393. [Google Scholar] [CrossRef]
- Nurieva, R.I. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef]
- Johnston, R.J. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef]
- Nurieva, R.I. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef]
- Yu, D. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 2009, 31, 457–468. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; Yaari, G.; Patel, J.R.; Johnson, K.L.; Gonzalez, D.G.; Kleinstein, S.H.; Haberman, A.M. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 2011, 34, 947–960. [Google Scholar] [CrossRef]
- Leon, B.; Ballesteros-Tato, A.; Browning, J.L.; Dunn, R.; Randall, T.D.; Lund, F.E. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat. Immunol. 2012, 13, 681–690. [Google Scholar]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef]
- Shulman, Z.; Gitlin, A.D.; Targ, S.; Jankovic, M.; Pasqual, G.; Nussenzweig, M.C.; Victora, G.D. T follicular helper cell dynamics in germinal centers. Science 2013, 341, 673–677. [Google Scholar] [CrossRef]
- Nutt, S.L.; Tarlinton, D.M. Germinal center B and follicular helper T cells: Siblings, cousins or just good friends? Nat. Immunol. 2011, 12, 472–477. [Google Scholar] [CrossRef]
- Ma, C.S.; Deenick, E.K.; Batten, M.; Tangye, S.G. The origins, function, and regulation of T follicular helper cells. J. Exp. Med. 2012, 209, 1241–1253. [Google Scholar] [CrossRef]
- Chtanova, T. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 2004, 173, 68–78. [Google Scholar]
- Vinuesa, C.G. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458. [Google Scholar] [CrossRef]
- Rasheed, A.U.; Rahn, H.P.; Sallusto, F.; Lipp, M.; Muller, G. Follicular B helper T cell activity is confined to CXCR5hiICOShi CD4 T cells and is independent of CD57 expression. Eur. J. Immunol. 2006, 36, 1892–1903. [Google Scholar] [CrossRef]
- Haynes, N.M. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J. Immunol. 2007, 179, 5099–5108. [Google Scholar]
- Deenick, E.K. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity 2010, 33, 241–253. [Google Scholar] [CrossRef]
- Kroenke, M.A. Bcl6 and maf cooperate to instruct human follicular helper CD4 T cell differentiation. J. Immunol. 2012, 188, 3734–3744. [Google Scholar]
- Cattoretti, G.; Chang, C.C.; Cechova, K.; Zhang, J.; Ye, B.H.; Falini, B.; Louie, D.C.; Offit, K.; Chaganti, R.S.; Dalla-Favera, R. BCL-6 protein is expressed in germinal-center B cells. Blood 1995, 86, 45–53. [Google Scholar]
- Kusam, S.; Toney, L.M.; Sato, H.; Dent, A.L. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J. Immunol. 2003, 170, 2435–2441. [Google Scholar]
- Ma, C.S. Early commitment of naive human CD4+ T cells to the T follicular helper (TFH) cell lineage is induced by IL-12. Immunol. Cell Biol. 2009, 87, 590–600. [Google Scholar] [CrossRef]
- Choi, Y.S.; Kageyama, R.; Eto, D.; Escobar, T.C.; Johnston, R.J.; Monticelli, L.; Lao, C.; Crotty, S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011, 34, 932–946. [Google Scholar] [CrossRef]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef]
- Ho, I.C.; Hodge, M.R.; Rooney, J.W.; Glimcher, L.H. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 1996, 85, 973–983. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Suto, A.; Kashiwakuma, D.; Kanari, H.; Kagami, S.; Ikeda, K.; Hirose, K.; Watanabe, N.; Grusby, M.J.; Iwamoto, I.; et al. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-beta inhibits c-Maf-induced IL-21 production in CD4+ T cells. J. Leukoc. Biol. 2010, 87, 703–712. [Google Scholar] [CrossRef]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef]
- Pot, C.; Jin, H.; Awasthi, A.; Liu, S.M.; Lai, C.Y.; Madan, R.; Sharpe, A.H.; Karp, C.L.; Miaw, S.C.; Ho, I.C.; et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009, 183, 797–801. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Schraml, B.U.; Hildner, K.; Ise, W.; Lee, W.L.; Smith, W.A.; Solomon, B.; Sahota, G.; Sim, J.; Mukasa, R.; Cemerski, S.; et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 2009, 460, 405–409. [Google Scholar]
- Betz, B.C.; Jordan-Williams, K.L.; Wang, C.; Kang, S.G.; Liao, J.; Logan, M.R.; Kim, C.H.; Taparowsky, E.J. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J. Exp. Med. 2010, 207, 933–942. [Google Scholar] [CrossRef]
- Ise, W.; Kohyama, M.; Schraml, B.U.; Zhang, T.; Schwer, B.; Basu, U.; Alt, F.W.; Tang, J.; Oltz, E.M.; Murphy, T.L.; et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011, 12, 536–543. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: Basic biology and implications for cancer and autoimmunity. Annu. Rev. Immunol. 2008, 26, 57–79. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.P.; Wang, G.; Qi, C.F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar]
- Kwon, H.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, H.P.; Oh, J.; Tunyaplin, C.; Carotta, S.; Donovan, C.E.; Goldman, M.L.; Tailor, P.; et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 2009, 31, 941–952. [Google Scholar] [CrossRef]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef]
- Eddahri, F.; Denanglaire, S.; Bureau, F.; Spolski, R.; Leonard, W.J.; Leo, O.; Andris, F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood 2009, 113, 2426–2433. [Google Scholar] [CrossRef]
- Eto, D. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One 2011, 6, e17739. [Google Scholar] [CrossRef]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef]
- Schmitt, N.; Morita, R.; Bourdery, L.; Bentebibel, S.E.; Zurawski, S.M.; Banchereau, J.; Ueno, H. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009, 31, 158–169. [Google Scholar] [CrossRef]
- Brustle, A.; Heink, S.; Huber, M.; Rosenplanter, C.; Stadelmann, C.; Yu, P.; Arpaia, E.; Mak, T.W.; Kamradt, T.; Lohoff, M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007, 8, 958–966. [Google Scholar] [CrossRef]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef]
- Bollig, N.; Brustle, A.; Kellner, K.; Ackermann, W.; Abass, E.; Raifer, H.; Camara, B.; Brendel, C.; Giel, G.; Bothur, E.; et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 8664–8669. [Google Scholar] [CrossRef]
- Glasmacher, E.; Agrawal, S.; Chang, A.B.; Murphy, T.L.; Zeng, W.; Vander Lugt, B.; Khan, A.A.; Ciofani, M.; Spooner, C.J.; Rutz, S.; et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 2012, 338, 975–980. [Google Scholar] [CrossRef]
- Li, P.; Spolski, R.; Liao, W.; Wang, L.; Murphy, T.L.; Murphy, K.M.; Leonard, W.J. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 2012, 490, 543–546. [Google Scholar] [CrossRef]
- Fanzo, J.C.; Yang, W.; Jang, S.Y.; Gupta, S.; Chen, Q.; Siddiq, A.; Greenberg, S.; Pernis, A.B. Loss of IRF-4-binding protein leads to the spontaneous development of systemic autoimmunity. J. Clin. Invest. 2006, 116, 703–714. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, W.; Gupta, S.; Biswas, P.; Smith, P.; Bhagat, G.; Pernis, A.B. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 2008, 29, 899–911. [Google Scholar] [CrossRef]
- Biswas, P.S.; Gupta, S.; Stirzaker, R.A.; Kumar, V.; Jessberger, R.; Lu, T.T.; Bhagat, G.; Pernis, A.B. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J. Exp. Med. 2012, 209, 581–596. [Google Scholar] [CrossRef]
- Kang, S.G.; Liu, W.H.; Lu, P.; Jin, H.Y.; Lim, H.W.; Shepherd, J.; Fremgen, D.; Verdin, E.; Oldstone, M.B.; Qi, H.; et al. MicroRNAs of the miR-17 approximately 92 family are critical regulators of T differentiation. Nat. Immunol. 2013, 14, 849–857. [Google Scholar] [CrossRef]
- Baumjohann, D.; Kageyama, R.; Clingan, J.M.; Morar, M.M.; Patel, S.; de Kouchkovsky, D.; Bannard, O.; Bluestone, J.A.; Matloubian, M.; Ansel, K.M.; et al. The microRNA cluster miR-17 approximately 92 promotes T cell differentiation and represses subset-inappropriate gene expression. Nat. Immunol. 2013, 14, 840–848. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17. Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Suto, A.; Kashiwakuma, D.; Kagami, S.; Hirose, K.; Watanabe, N.; Yokote, K.; Saito, Y.; Nakayama, T.; Grusby, M.J.; Iwamoto, I.; et al. Development and characterization of IL-21-producing CD4+ T cells. J. Exp. Med. 2008, 205, 1369–1379. [Google Scholar] [CrossRef]
- Poholek, A.C.; Hansen, K.; Hernandez, S.G.; Eto, D.; Chandele, A.; Weinstein, J.S.; Dong, X.; Odegard, J.M.; Kaech, S.M.; Dent, A.L.; et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010, 185, 313–326. [Google Scholar] [CrossRef]
- Harker, J.A.; Lewis, G.M.; Mack, L.; Zuniga, E.I. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011, 334, 825–829. [Google Scholar] [CrossRef]
- Johnston, R.J.; Choi, Y.S.; Diamond, J.A.; Yang, J.A.; Crotty, S. STAT5 is a potent negative regulator of TFH cell differentiation. J. Exp. Med. 2012, 209, 243–250. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Mohn, S.E.; Weinmann, A.S. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat. Immunol. 2012, 13, 405–411. [Google Scholar] [CrossRef]
- Ballesteros-Tato, A.; Leon, B.; Graf, B.A.; Moquin, A.; Adams, P.S.; Lund, F.E.; Randall, T.D. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 2012, 36, 847–856. [Google Scholar] [CrossRef]
- Marshall, H.D.; Chandele, A.; Jung, Y.W.; Meng, H.; Poholek, A.C.; Parish, I.A.; Rutishauser, R.; Cui, W.; Kleinstein, S.H.; Craft, J.; et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 2011, 35, 633–646. [Google Scholar] [CrossRef]
- Nakayamada, S.; Kanno, Y.; Takahashi, H.; Jankovic, D.; Lu, K.T.; Johnson, T.A.; Sun, H.W.; Vahedi, G.; Hakim, O.; Handon, R.; et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity 2011, 35, 919–931. [Google Scholar] [CrossRef]
- Ma, C.S.; Avery, D.T.; Chan, A.; Batten, M.; Bustamante, J.; Boisson-Dupuis, S.; Arkwright, P.D.; Kreins, A.Y.; Averbuch, D.; Engelhard, D.; et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 2012, 119, 3997–4008. [Google Scholar] [CrossRef]
- Diehl, S.A.; Schmidlin, H.; Nagasawa, M.; Blom, B.; Spits, H. IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol. Cell Biol. 2012, 90, 802–811. [Google Scholar] [CrossRef]
- Hardtke, S.; Ohl, L.; Forster, R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood 2005, 106, 1924–1931. [Google Scholar] [CrossRef]
- Fazilleau, N.; Mark, L.; McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Follicular helper T cells: lineage and location. Immunity 2009, 30, 324–335. [Google Scholar] [CrossRef]
- Crotty, S.; Johnston, R.J.; Schoenberger, S.P. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 2010, 11, 114–120. [Google Scholar] [CrossRef]
- Arnold, C.N.; Campbell, D.J.; Lipp, M.; Butcher, E.C. The germinal center response is impaired in the absence of T cell-expressed CXCR5. Eur. J. Immunol. 2007, 37, 100–109. [Google Scholar] [CrossRef]
- Kitano, M. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity 2011, 34, 961–972. [Google Scholar] [CrossRef]
- Liu, X.; Yan, X.; Zhong, B.; Nurieva, R.I.; Wang, A.; Wang, X.; Martin-Orozco, N.; Wang, Y.; Chang, S.H.; Esplugues, E.; et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J. Exp. Med. 2012, 209, 1841–1852. [Google Scholar] [CrossRef]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef]
- Coyle, A.J.; Lehar, S.; Lloyd, C.; Tian, J.; Delaney, T.; Manning, S.; Nguyen, T.; Burwell, T.; Schneider, H.; Gonzalo, J.A.; et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 2000, 13, 95–105. [Google Scholar] [CrossRef]
- Ling, V.; Wu, P.W.; Finnerty, H.F.; Bean, K.M.; Spaulding, V.; Fouser, L.A.; Leonard, J.P.; Hunter, S.E.; Zollner, R.; Thomas, J.L.; et al. Cutting edge: identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J. Immunol. 2000, 164, 1653–1657. [Google Scholar]
- Wang, S.; Zhu, G.; Chapoval, A.I.; Dong, H.; Tamada, K.; Ni, J.; Chen, L. Costimulation of T cells by B7-H2, a B7-like molecule that binds ICOS. Blood 2000, 96, 2808–2813. [Google Scholar]
- Gigoux, M.; Shang, J.; Pak, Y.; Xu, M.; Choe, J.; Mak, T.W.; Suh, W.K. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 20371–20376. [Google Scholar]
- Chevalier, N. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J. Immunol. 2011, 186, 5556–5568. [Google Scholar] [CrossRef]
- Bossaller, L. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J. Immunol. 2006, 177, 4927–4932. [Google Scholar]
- Akiba, H.; Takeda, K.; Kojima, Y.; Usui, Y.; Harada, N.; Yamazaki, T.; Ma, J.; Tezuka, K.; Yagita, H.; Okumura, K. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J. Immunol. 2005, 175, 2340–2348. [Google Scholar]
- Rolf, J.; Bell, S.E.; Kovesdi, D.; Janas, M.L.; Soond, D.R.; Webb, L.M.; Santinelli, S.; Saunders, T.; Hebeis, B.; Killeen, N.; et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J. Immunol. 2010, 185, 4042–4052. [Google Scholar] [CrossRef]
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef]
- Mak, T.W.; Shahinian, A.; Yoshinaga, S.K.; Wakeham, A.; Boucher, L.M.; Pintilie, M.; Duncan, G.; Gajewska, B.U.; Gronski, M.; Eriksson, U.; et al. Costimulation through the inducible costimulator ligand is essential for both T helper and B cell functions in T cell-dependent B cell responses. Nat. Immunol. 2003, 4, 765–772. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; Peng, L.; Xu, C.; Liu, W.; et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013, 496, 523–527. [Google Scholar] [CrossRef]
- Foy, T.M.; Laman, J.D.; Ledbetter, J.A.; Aruffo, A.; Claassen, E.; Noelle, R.J. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J. Exp. Med. 1994, 180, 157–163. [Google Scholar] [CrossRef]
- Han, S.; Hathcock, K.; Zheng, B.; Kepler, T.B.; Hodes, R.; Kelsoe, G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7–2 in established germinal centers. J. Immunol. 1995, 155, 556–567. [Google Scholar]
- Takahashi, Y.; Dutta, P.R.; Cerasoli, D.M.; Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998, 187, 885–895. [Google Scholar] [CrossRef]
- Banchereau, J.; de Paoli, P.; Valle, A.; Garcia, E.; Rousset, F. Long-term human B cell lines dependent on interleukin-4 and antibody to CD40. Science 1991, 251, 70–72. [Google Scholar]
- Arpin, C.; Dechanet, J.; Van Kooten, C.; Merville, P.; Grouard, G.; Briere, F.; Banchereau, J.; Liu, Y.J. Generation of memory B cells and plasma cells in vitro. Science 1995, 268, 720–722. [Google Scholar] [CrossRef]
- Randall, T.D.; Heath, A.W.; Santos-Argumedo, L.; Howard, M.C.; Weissman, I.L.; Lund, F.E. Arrest of B lymphocyte terminal differentiation by CD40 signaling: Mechanism for lack of antibody-secreting cells in germinal centers. Immunity 1998, 8, 733–742. [Google Scholar]
- Kwakkenbos, M.J.; Diehl, S.A.; Yasuda, E.; Bakker, A.Q.; van Geelen, C.M.; Lukens, M.V.; van Bleek, G.M.; Widjojoatmodjo, M.N.; Bogers, W.M.; Mei, H.; et al. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat. Med. 2010, 16, 123–128. [Google Scholar] [CrossRef]
- Fillatreau, S.; Gray, D. T cell accumulation in B cell follicles is regulated by dendritic cells and is independent of B cell activation. J. Exp. Med. 2003, 197, 195–206. [Google Scholar] [CrossRef]
- van Essen, D.; Kikutani, H.; Gray, D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature 1995, 378, 620–623. [Google Scholar] [CrossRef]
- Grewal, I.S.; Xu, J.; Flavell, R.A. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature 1995, 378, 617–620. [Google Scholar] [CrossRef]
- Qi, H.; Cannons, J.L.; Klauschen, F.; Schwartzberg, P.L.; Germain, R.N. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 2008, 455, 764–769. [Google Scholar] [CrossRef]
- Cannons, J.L. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 2010, 32, 253–265. [Google Scholar] [CrossRef]
- Linterman, M.A. Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 2009, 206, 561–576. [Google Scholar] [CrossRef]
- Crotty, S.; Kersh, E.N.; Cannons, J.; Schwartzberg, P.L.; Ahmed, R. SAP is required for generating long-term humoral immunity. Nature 2003, 421, 282–287. [Google Scholar]
- Ma, C.S. Impaired humoral immunity in X-linked lymphoproliferative disease is associated with defective IL-10 production by CD4+ T cells. J. Clin. Invest. 2005, 115, 1049–1059. [Google Scholar]
- Ma, C.S.; Nichols, K.E.; Tangye, S.G. Regulation of cellular and humoral immune responses by the SLAM and SAP families of molecules. Annu Rev. Immunol 2007, 25, 337–379. [Google Scholar] [CrossRef]
- Cannons, J.L.; Wu, J.Z.; Gomez-Rodriguez, J.; Zhang, J.; Dong, B.; Liu, Y.; Shaw, S.; Siminovitch, K.A.; Schwartzberg, P.L. Biochemical and genetic evidence for a SAP-PKC-theta interaction contributing to IL-4 regulation. J. Immunol. 2010, 185, 2819–2827. [Google Scholar] [CrossRef]
- Tangye, S.G.; Ma, C.S.; Brink, R.; Deenick, E.K. The good, the bad and the ugly—TFH cells in human health and disease. Nat. Rev. Immunol. 2013, 13, 412–426. [Google Scholar] [CrossRef]
- Kumar, K.R.; Li, L.; Yan, M.; Bhaskarabhatla, M.; Mobley, A.B.; Nguyen, C.; Mooney, J.M.; Schatzle, J.D.; Wakeland, E.K.; Mohan, C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science 2006, 312, 1665–1669. [Google Scholar] [CrossRef]
- Kageyama, R. The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity 2012, 36, 986–1002. [Google Scholar] [CrossRef]
- Graham, D.B.; Bell, M.P.; McCausland, M.M.; Huntoon, C.J.; van Deursen, J.; Faubion, W.A.; Crotty, S.; McKean, D.J. Ly9 (CD229)-deficient mice exhibit T cell defects yet do not share several phenotypic characteristics associated with SLAM- and SAP-deficient mice. J. Immunol. 2006, 176, 291–300. [Google Scholar]
- McCausland, M.M.; Yusuf, I.; Tran, H.; Ono, N.; Yanagi, Y.; Crotty, S. SAP regulation of follicular helper CD4 T cell development and humoral immunity is independent of SLAM and Fyn kinase. J. Immunol. 2007, 178, 817–828. [Google Scholar]
- Linterman, M.A.; Beaton, L.; Yu, D.; Ramiscal, R.R.; Srivastava, M.; Hogan, J.J.; Verma, N.K.; Smyth, M.J.; Rigby, R.J.; Vinuesa, C.G. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010, 207, 353–363. [Google Scholar] [CrossRef]
- Zotos, D.; Coquet, J.M.; Zhang, Y.; Light, A.; D'Costa, K.; Kallies, A.; Corcoran, L.M.; Godfrey, D.I.; Toellner, K.M.; Smyth, M.J.; et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 2010, 207, 365–378. [Google Scholar] [CrossRef]
- Dienz, O.; Eaton, S.M.; Bond, J.P.; Neveu, W.; Moquin, D.; Noubade, R.; Briso, E.M.; Charland, C.; Leonard, W.J.; Ciliberto, G.; et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J. Exp. Med. 2009, 206, 69–78. [Google Scholar] [CrossRef]
- Batten, M.; Ramamoorthi, N.; Kljavin, N.M.; Ma, C.S.; Cox, J.H.; Dengler, H.S.; Danilenko, D.M.; Caplazi, P.; Wong, M.; Fulcher, D.A.; et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 2010, 207, 2895–2906. [Google Scholar] [CrossRef]
- Ettinger, R. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J. Immunol. 2005, 175, 7867–7879. [Google Scholar]
- Good, K.L.; Bryant, V.L.; Tangye, S.G. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J. Immunol. 2006, 177, 5236–5247. [Google Scholar]
- Bryant, V.L. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: Predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 2007, 179, 8180–8190. [Google Scholar]
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J. Immunol. 2007, 179, 5886–5896. [Google Scholar]
- Ozaki, K.; Spolski, R.; Feng, C.G.; Qi, C.F.; Cheng, J.; Sher, A.; Morse, H.C., 3rd; Liu, C.; Schwartzberg, P.L.; Leonard, W.J. A critical role for IL-21 in regulating immunoglobulin production. Science 2002, 298, 1630–1634. [Google Scholar] [CrossRef]
- Pene, J. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J. Immunol. 2004, 172, 5154–5157. [Google Scholar]
- Avery, D.T.; Bryant, V.L.; Ma, C.S.; de Waal Malefyt, R.; Tangye, S.G. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J. Immunol. 2008, 181, 1767–1779. [Google Scholar]
- Paul, W.E.; Ohara, J. B-cell stimulatory factor-1/interleukin 4. Annu. Rev. Immunol. 1987, 5, 429–459. [Google Scholar] [CrossRef]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. The IL-4 receptor: Signaling mechanisms and biologic functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef]
- Wurster, A.L.; Rodgers, V.L.; White, M.F.; Rothstein, T.L.; Grusby, M.J. Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J. Biol. Chem. 2002, 277, 27169–27175. [Google Scholar]
- Dufort, F.J.; Bleiman, B.F.; Gumina, M.R.; Blair, D.; Wagner, D.J.; Roberts, M.F.; Abu-Amer, Y.; Chiles, T.C. Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J. Immunol. 2007, 179, 4953–4957. [Google Scholar]
- Vinuesa, C.G.; Sanz, I.; Cook, M.C. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009, 9, 845–857. [Google Scholar] [CrossRef]
- Sweet, R.A.; Lee, S.K.; Vinuesa, C.G. Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr. Opin. Immunol. 2012, 24, 658–664. [Google Scholar]
- King, C.; Tangye, S.G.; Mackay, C.R. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008, 26, 741–766. [Google Scholar] [CrossRef]
- Hron, J.D.; Caplan, L.; Gerth, A.J.; Schwartzberg, P.L.; Peng, S.L. SH2D1A regulates T-dependent humoral autoimmunity. J. Exp. Med. 2004, 200, 261–266. [Google Scholar]
- Subramanian, S.; Tus, K.; Li, Q.Z.; Wang, A.; Tian, X.H.; Zhou, J.; Liang, C.; Bartov, G.; McDaniel, L.D.; Zhou, X.J.; et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA 2006, 103, 9970–9975. [Google Scholar] [CrossRef]
- Luzina, I.G.; Atamas, S.P.; Storrer, C.E.; daSilva, L.C.; Kelsoe, G.; Papadimitriou, J.C.; Handwerger, B.S. Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 2001, 70, 578–584. [Google Scholar]
- Hsu, H.C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef]
- Rankin, A.L.; Guay, H.; Herber, D.; Bertino, S.A.; Duzanski, T.A.; Carrier, Y.; Keegan, S.; Senices, M.; Stedman, N.; Ryan, M.; et al. IL-21 receptor is required for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. J. Immunol. 2012, 188, 1656–1667. [Google Scholar] [CrossRef]
- Pisitkun, P.; Deane, J.A.; Difilippantonio, M.J.; Tarasenko, T.; Satterthwaite, A.B.; Bolland, S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006, 312, 1669–1672. [Google Scholar] [CrossRef]
- Bubier, J.A.; Sproule, T.J.; Foreman, O.; Spolski, R.; Shaffer, D.J.; Morse, H.C., 3rd; Leonard, W.J.; Roopenian, D.C. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1518–1523. [Google Scholar] [CrossRef]
- Bertossi, A.; Aichinger, M.; Sansonetti, P.; Lech, M.; Neff, F.; Pal, M.; Wunderlich, F.T.; Anders, H.J.; Klein, L.; Schmidt-Supprian, M. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J. Exp. Med. 2011, 208, 1749–1756. [Google Scholar]
- Craft, J.E. Follicular helper T cells in immunity and systemic autoimmunity. Nat. Rev. Rheumatol. 2012, 8, 337–347. [Google Scholar] [CrossRef]
- William, J.; Euler, C.; Christensen, S.; Shlomchik, M.J. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 2002, 297, 2066–2070. [Google Scholar] [CrossRef]
- Odegard, J.M.; Marks, B.R.; DiPlacido, L.D.; Poholek, A.C.; Kono, D.H.; Dong, C.; Flavell, R.A.; Craft, J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 2008, 205, 2873–2886. [Google Scholar] [CrossRef]
- Odegard, J.M.; DiPlacido, L.D.; Greenwald, L.; Kashgarian, M.; Kono, D.H.; Dong, C.; Flavell, R.A.; Craft, J. ICOS controls effector function but not trafficking receptor expression of kidney-infiltrating effector T cells in murine lupus. J. Immunol. 2009, 182, 4076–4084. [Google Scholar] [CrossRef]
- Tada, Y.; Koarada, S.; Tomiyoshi, Y.; Morito, F.; Mitamura, M.; Haruta, Y.; Ohta, A.; Nagasawa, K. Role of inducible costimulator in the development of lupus in MRL/lpr mice. Clin. Immunol. 2006, 120, 179–188. [Google Scholar] [CrossRef]
- Andrews, B.S.; Eisenberg, R.A.; Theofilopoulos, A.N.; Izui, S.; Wilson, C.B.; McConahey, P.J.; Murphy, E.D.; Roths, J.B.; Dixon, F.J. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978, 148, 1198–1215. [Google Scholar] [CrossRef]
- Hu, Y.L.; Metz, D.P.; Chung, J.; Siu, G.; Zhang, M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J. Immunol. 2009, 182, 1421–1428. [Google Scholar]
- Taylor, B.A.; Wnek, C.; Kotlus, B.S.; Roemer, N.; MacTaggart, T.; Phillips, S.J. Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mamm. Genome 1999, 10, 335–348. [Google Scholar] [CrossRef]
- Mountz, J.D.; Wang, J.H.; Xie, S.; Hsu, H.C. Cytokine regulation of B-cell migratory behavior favors formation of germinal centers in autoimmune disease. Discov. Med. 2011, 11, 76–85. [Google Scholar]
- Xie, S.; Li, J.; Wang, J.H.; Wu, Q.; Yang, P.; Hsu, H.C.; Smythies, L.E.; Mountz, J.D. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J. Immunol. 2010, 184, 2289–2296. [Google Scholar] [CrossRef]
- Ding, Y.; Li, J.; Wu, Q.; Yang, P.; Luo, B.; Xie, S.; Druey, K.M.; Zajac, A.J.; Hsu, H.C.; Mountz, J.D. IL-17RA Is Essential for Optimal Localization of Follicular Th Cells in the Germinal Center Light Zone To Promote Autoantibody-Producing B Cells. J. Immunol. 2013, 191, 1614–1624. [Google Scholar] [CrossRef]
- Cappione, A., 3rd; Anolik, J.H.; Pugh-Bernard, A.; Barnard, J.; Dutcher, P.; Silverman, G.; Sanz, I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J. Clin. Invest. 2005, 115, 3205–3216. [Google Scholar] [CrossRef]
- Simpson, N. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 234–244. [Google Scholar] [CrossRef]
- Morita, R. Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef]
- Li, X.Y. Role of the frequency of blood CD4+ CXCR5+ CCR6+ T cells in autoimmunity in patients with Sjogren's syndrome. Biochem. Biophys. Res. Commun. 2012, 422, 238–244. [Google Scholar] [CrossRef]
- Ma, J. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin. Dev. Immunol. 2012, 2012, 827–480. [Google Scholar]
- Zhang, Y.; Meyer-Hermann, M.; George, L.A.; Figge, M.T.; Khan, M.; Goodall, M.; Young, S.P.; Reynolds, A.; Falciani, F.; Waisman, A.; et al. Germinal center B cells govern their own fate via antibody feedback. J. Exp. Med. 2013, 210, 457–464. [Google Scholar] [CrossRef]
- Yang, M.; Rui, K.; Wang, S.; Lu, L. Regulatory B cells in autoimmune diseases. Cell. Mol. Immunol. 2013, 10, 122–132. [Google Scholar] [CrossRef]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A., Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Mauri, C.; Gray, D.; Mushtaq, N.; Londei, M. Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 2003, 197, 489–501. [Google Scholar] [CrossRef]
- Lenert, P.; Brummel, R.; Field, E.H.; Ashman, R.F. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J. Clin. Immunol. 2005, 25, 29–40. [Google Scholar] [CrossRef]
- Yanaba, K.; Bouaziz, J.D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef]
- Evans, J.G.; Chavez-Rueda, K.A.; Eddaoudi, A.; Meyer-Bahlburg, A.; Rawlings, D.J.; Ehrenstein, M.R.; Mauri, C. Novel suppressive function of transitional 2 B cells in experimental arthritis. J. Immunol. 2007, 178, 7868–7878. [Google Scholar]
- Duddy, M.E.; Alter, A.; Bar-Or, A. Distinct profiles of human B cell effector cytokines: A role in immune regulation? J. Immunol. 2004, 172, 3422–3427. [Google Scholar]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals butare functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef]
- Lundy, S.K. Killer B lymphocytes: the evidence and the potential. Inflamm. Res. 2009, 58, 345–357. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Hoehlig, K.; Roch, T.; Neves, P.; Calderon Gomez, E.; Sweenie, C.H.; Hao, Y.; Freitas, A.A.; Steinhoff, U.; Anderton, S.M.; et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J. Immunol. 2008, 180, 4763–4773. [Google Scholar]
- Bouaziz, J.D.; Calbo, S.; Maho-Vaillant, M.; Saussine, A.; Bagot, M.; Bensussan, A.; Musette, P. IL-10 produced by activated human B cells regulates CD4(+) T-cell activation in vitro. Eur. J. Immunol. 2010, 40, 2686–2691. [Google Scholar] [CrossRef]
- Tu, W.; Lau, Y.L.; Zheng, J.; Liu, Y.; Chan, P.L.; Mao, H.; Dionis, K.; Schneider, P.; Lewis, D.B. Efficient generation of human alloantigen-specific CD4+ regulatory T cells from naive precursors by CD40-activated B cells. Blood 2008, 112, 2554–2562. [Google Scholar] [CrossRef]
- Walters, S.; Webster, K.E.; Sutherland, A.; Gardam, S.; Groom, J.; Liuwantara, D.; Marino, E.; Thaxton, J.; Weinberg, A.; Mackay, F.; et al. Increased CD4+Foxp3+ T cells in BAFF-transgenic mice suppress T cell effector responses. J. Immunol. 2009, 182, 793–801. [Google Scholar]
- Yang, M.; Sun, L.; Wang, S.; Ko, K.H.; Xu, H.; Zheng, B.J.; Cao, X.; Lu, L. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J. Immunol 2010, 184, 3321–3325. [Google Scholar] [CrossRef]
- Gray, M.; Miles, K.; Salter, D.; Gray, D.; Savill, J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14080–14085. [Google Scholar] [CrossRef]
- Blair, P.A.; Chavez-Rueda, K.A.; Evans, J.G.; Shlomchik, M.J.; Eddaoudi, A.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J. Immunol. 2009, 182, 3492–3502. [Google Scholar] [CrossRef]
- Tedder, T.F.; Matsushita, T. Regulatory B cells that produce IL-10: a breath of fresh air in allergic airway disease. J. Allergy Clin. Immunol. 2010, 125, 1125–1127. [Google Scholar] [CrossRef]
- Carter, N.A.; Vasconcellos, R.; Rosser, E.C.; Tulone, C.; Munoz-Suano, A.; Kamanaka, M.; Ehrenstein, M.R.; Flavell, R.A.; Mauri, C. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J. Immunol. 2011, 186, 5569–5579. [Google Scholar] [CrossRef]
- Amu, S.; Saunders, S.P.; Kronenberg, M.; Mangan, N.E.; Atzberger, A.; Fallon, P.G. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J. Allergy Clin. Immunol. 2010, 125, 1114–1124. [Google Scholar] [CrossRef]
- Carter, N.A.; Rosser, E.C.; Mauri, C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res. Ther. 2012, 14, R32. [Google Scholar] [CrossRef]
- Yang, M.; Deng, J.; Liu, Y.; Ko, K.H.; Wang, X.; Jiao, Z.; Wang, S.; Hua, Z.; Sun, L.; Srivastava, G.; et al. IL-10-producing regulatory B10 cells ameliorate collagen-induced arthritis via suppressing Th17 cell generation. Am. J. Pathol. 2012, 180, 2375–2385. [Google Scholar] [CrossRef]
- Lundy, S.K.; Boros, D.L. Fas ligand-expressing B-1a lymphocytes mediate CD4(+)-T-cell apoptosis during schistosomal infection: Induction by interleukin 4 (IL-4) and IL-10. Infect. Immun. 2002, 70, 812–819. [Google Scholar] [CrossRef]
- Klinker, M.W.; Reed, T.J.; Fox, D.A.; Lundy, S.K. Interleukin-5 Supports the Expansion of Fas Ligand-Expressing Killer B Cells that Induce Antigen-Specific Apoptosis of CD4(+) T Cells and Secrete Interleukin-10. PLoS One 2013, 8, e70131. [Google Scholar] [CrossRef]
- Lundy, S.K.; Fox, D.A. Reduced Fas ligand-expressing splenic CD5+ B lymphocytes in severe collagen-induced arthritis. Arthritis Res. Ther. 2009, 11, R128. [Google Scholar] [CrossRef]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Rheumatoid arthritis. Cell 1996, 85, 307–310. [Google Scholar] [CrossRef]
- Yanaba, K.; Bouaziz, J.D.; Matsushita, T.; Magro, C.M.; St Clair, E.W.; Tedder, T.F. B-lymphocyte contributions to human autoimmune disease. Immunol. Rev. 2008, 223, 284–299. [Google Scholar] [CrossRef]
- Haas, K.M.; Watanabe, R.; Matsushita, T.; Nakashima, H.; Ishiura, N.; Okochi, H.; Fujimoto, M.; Tedder, T.F. Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J. Immunol. 2010, 184, 4789–4800. [Google Scholar] [CrossRef]
- Matsushita, T.; Horikawa, M.; Iwata, Y.; Tedder, T.F. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J. Immunol. 2010, 185, 2240–2252. [Google Scholar] [CrossRef]
- Watanabe, R.; Ishiura, N.; Nakashima, H.; Kuwano, Y.; Okochi, H.; Tamaki, K.; Sato, S.; Tedder, T.F.; Fujimoto, M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J. Immunol. 2010, 184, 4801–4809. [Google Scholar] [CrossRef]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef]
- Lim, H.W.; Hillsamer, P.; Kim, C.H. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J. Clin. Invest. 2004, 114, 1640–1649. [Google Scholar]
- Lim, H.W.; Hillsamer, P.; Banham, A.H.; Kim, C.H. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J. Immunol. 2005, 175, 4180–4183. [Google Scholar]
- Gotot, J.; Gottschalk, C.; Leopold, S.; Knolle, P.A.; Yagita, H.; Kurts, C.; Ludwig-Portugall, I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 10468–10473. [Google Scholar]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Chung, Y. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Linterman, M.A. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef]
- Wollenberg, I. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef]
- Sage, P.T.; Francisco, L.M.; Carman, C.V.; Sharpe, A.H. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat. Immunol. 2012, 14, 152–161. [Google Scholar]
- Alexander, C.M.; Tygrett, L.T.; Boyden, A.W.; Wolniak, K.L.; Legge, K.L.; Waldschmidt, T.J. T regulatory cells participate in the control of germinal centre reactions. Immunology 2011, 133, 452–468. [Google Scholar]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Noble, A.; Zhao, Z.S.; Cantor, H. Suppression of immune responses by CD8 cells. II. Qa-1 on activated B cells stimulates CD8 cell suppression of T helper 2 responses. J. Immunol. 1998, 160, 566–571. [Google Scholar]
- Hu, D.; Ikizawa, K.; Lu, L.; Sanchirico, M.E.; Shinohara, M.L.; Cantor, H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol 2004, 5, 516–523. [Google Scholar]
- Lu, L.; Ikizawa, K.; Hu, D.; Werneck, M.B.; Wucherpfennig, K.W.; Cantor, H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity 2007, 26, 593–604. [Google Scholar] [CrossRef]
- Kim, H.J.; Verbinnen, B.; Tang, X.; Lu, L.; Cantor, H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 2010, 467, 328–332. [Google Scholar]
- Cantor, H.; Boyse, E.A. Functional subclasses of T-lymphocytes bearing different Ly antigens. I. The generation of functionally distinct T-cell subclasses is a differentiative process independent of antigen. J. Exp. Med. 1975, 141, 1376–1389. [Google Scholar]
- Cantor, H.; Shen, F.W.; Boyse, E.A. Separation of helper T cells from suppressor T cells expressing different Ly components. II. Activation by antigen: after immunization, antigen-specific suppressor and helper activities are mediated by distinct T-cell subclasses. J. Exp. Med. 1976, 143, 1391–1340. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. Regulation of self-tolerance by Qa-1-restricted CD8(+) regulatory T cells. Semin. Immunol. 2011, 23, 446–452. [Google Scholar] [CrossRef]
- Jiang, H.; Canfield, S.M.; Gallagher, M.P.; Jiang, H.H.; Jiang, Y.; Zheng, Z.; Chess, L. HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J. Clin. Invest. 2010, 120, 3641–3650. [Google Scholar]
- Correale, J.; Villa, A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J. Neuroimmunol. 2008, 195, 121–134. [Google Scholar] [CrossRef]
- Lu, L.; Kim, H.J.; Werneck, M.B.; Cantor, H. Regulation of CD8+ regulatory T cells: Interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc. Natl. Acad. Sci. USA 2008, 105, 19420–19425. [Google Scholar]
- Kim, H.J.; Wang, X.; Radfar, S.; Sproule, T.J.; Roopenian, D.C.; Cantor, H. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2010–2015. [Google Scholar]
- Leavenworth, J.W.; Tang, X.; Kim, H.J.; Wang, X.; Cantor, H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J. Clin. Invest. 2013, 123, 1382–1389. [Google Scholar] [CrossRef]
- Mingari, M.C.; Ponte, M.; Cantoni, C.; Vitale, C.; Schiavetti, F.; Bertone, S.; Bellomo, R.; Cappai, A.T.; Biassoni, R. HLA-class I-specific inhibitory receptors in human cytolytic T lymphocytes: molecular characterization, distribution in lymphoid tissues and co-expression by individual T cells. Int. Immunol. 1997, 9, 485–491. [Google Scholar]
- Anfossi, N.; Pascal, V.; Vivier, E.; Ugolini, S. Biology of T memory type 1 cells. Immunol. Rev. 2001, 181, 269–278. [Google Scholar]
- Wilson-Welder, J.H.; Torres, M.P.; Kipper, M.J.; Mallapragada, S.K.; Wannemuehler, M.J.; Narasimhan, B. Vaccine adjuvants: Current challenges and future approaches. J. Pharm. Sci. 2009, 98, 1278–1316. [Google Scholar]
- Moon, J.J.; Suh, H.; Li, A.V.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA 2012, 109, 1080–1085. [Google Scholar]
- Su, C.; Duan, X.; Zheng, J.; Liang, L.; Wang, F.; Guo, L. IFN-alpha as an Adjuvant for Adenovirus-Vectored FMDV Subunit Vaccine through Improving the Generation of T Follicular Helper Cells. PLoS One 2013, 8, e66134. [Google Scholar]
- Zheng, D.; Sun, Q.; Su, Z.; Kong, F.; Shi, X.; Tong, J.; Shen, P.; Peng, T.; Wang, S.; Xu, H. Enhancing specific-antibody production to the ragB vaccine with GITRL that expand Tfh, IFN-gamma(+) T cells and attenuates Porphyromonas gingivalis infection in mice. PLoS One 2013, 8, e59604. [Google Scholar]
- Tone, M.; Tone, Y.; Adams, E.; Yates, S.F.; Frewin, M.R.; Cobbold, S.P.; Waldmann, H. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15059–15064. [Google Scholar]
- Hu, J.; Havenar-Daughton, C.; Crotty, S. Modulation of SAP dependent T:B cell interactions as a strategy to improve vaccination. Curr. Opin. Virol. 2013, 3, 363–370. [Google Scholar] [CrossRef]
- Aldhamen, Y.A.; Appledorn, D.M.; Seregin, S.S.; Liu, C.J.; Schuldt, N.J.; Godbehere, S.; Amalfitano, A. Expression of the SLAM family of receptors adapter EAT-2 as a novel strategy for enhancing beneficial immune responses to vaccine antigens. J. Immunol. 2011, 186, 722–732. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Ahmed, R. Manipulating the PD-1 pathway to improve immunity. Curr. Opin. Immunol. 2013, 25, 381–388. [Google Scholar] [CrossRef]
- Hams, E.; McCarron, M.J.; Amu, S.; Yagita, H.; Azuma, M.; Chen, L.; Fallon, P.G. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J. Immunol. 2011, 186, 5648–5655. [Google Scholar] [CrossRef]
- Butler, N.S.; Moebius, J.; Pewe, L.L.; Traore, B.; Doumbo, O.K.; Tygrett, L.T.; Waldschmidt, T.J.; Crompton, P.D.; Harty, J.T. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 2012, 13, 188–195. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, Y.U.; Liu, X.; Tanaka, S.; Tran, D.Q.; Chung, Y. Regulation of Germinal Center Reactions by B and T Cells. Antibodies 2013, 2, 554-586. https://doi.org/10.3390/antib2040554
Kim YU, Liu X, Tanaka S, Tran DQ, Chung Y. Regulation of Germinal Center Reactions by B and T Cells. Antibodies. 2013; 2(4):554-586. https://doi.org/10.3390/antib2040554
Chicago/Turabian StyleKim, Young Uk, Xindong Liu, Shinya Tanaka, Dat Quoc Tran, and Yeonseok Chung. 2013. "Regulation of Germinal Center Reactions by B and T Cells" Antibodies 2, no. 4: 554-586. https://doi.org/10.3390/antib2040554
APA StyleKim, Y. U., Liu, X., Tanaka, S., Tran, D. Q., & Chung, Y. (2013). Regulation of Germinal Center Reactions by B and T Cells. Antibodies, 2(4), 554-586. https://doi.org/10.3390/antib2040554