
Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning

 , ,
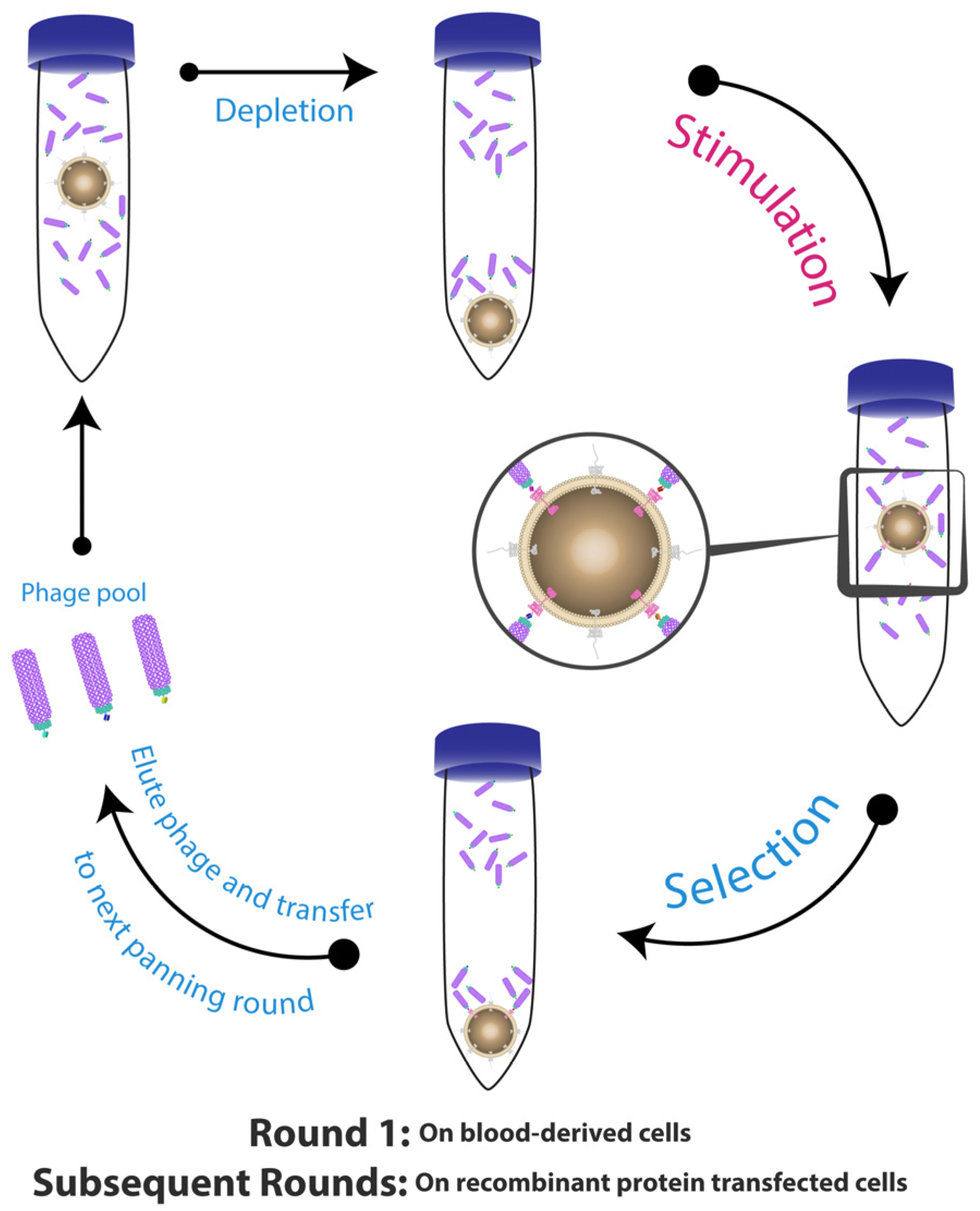
, , 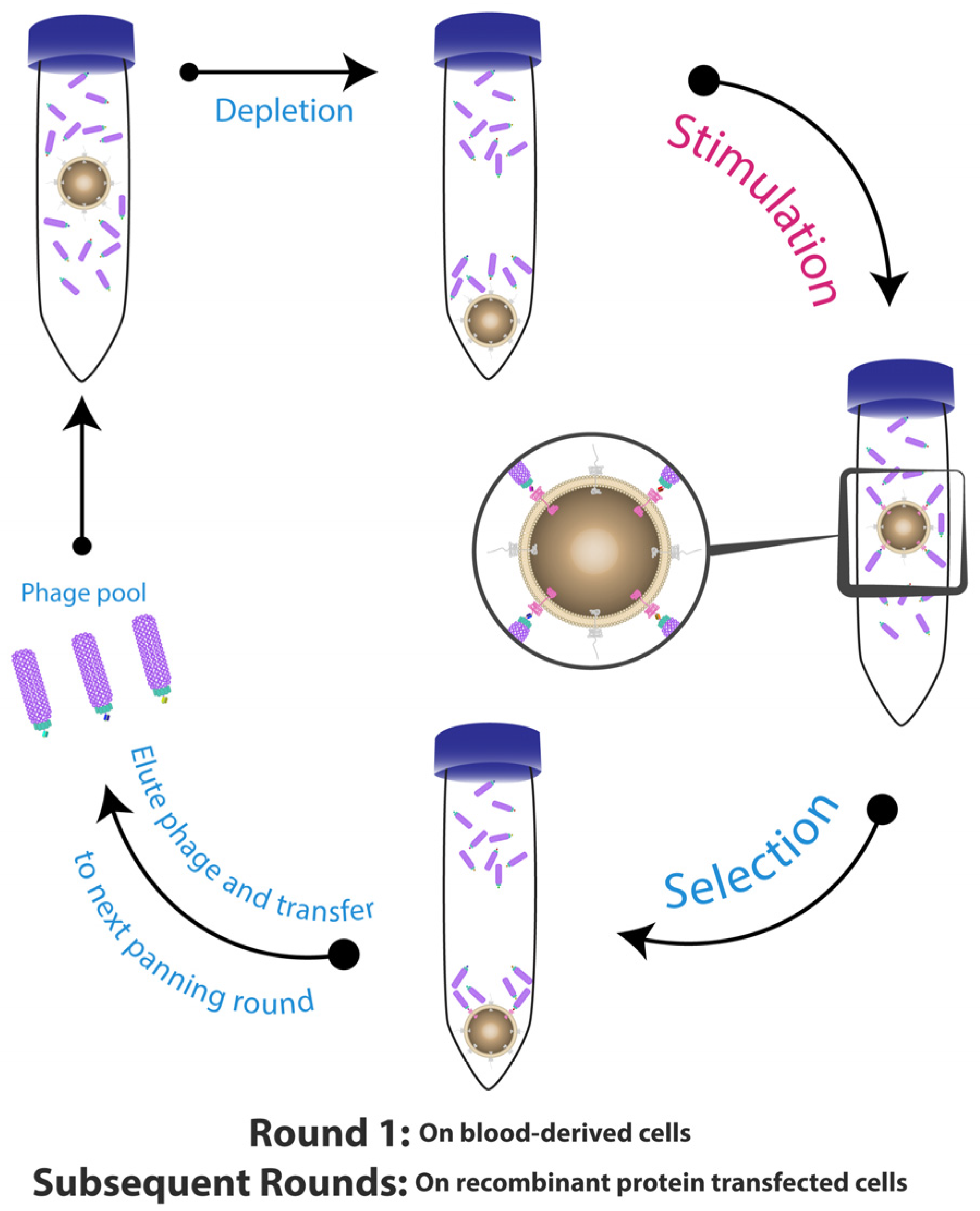{kind=link}
{kind=link}
{kind=link}
{kind=link}
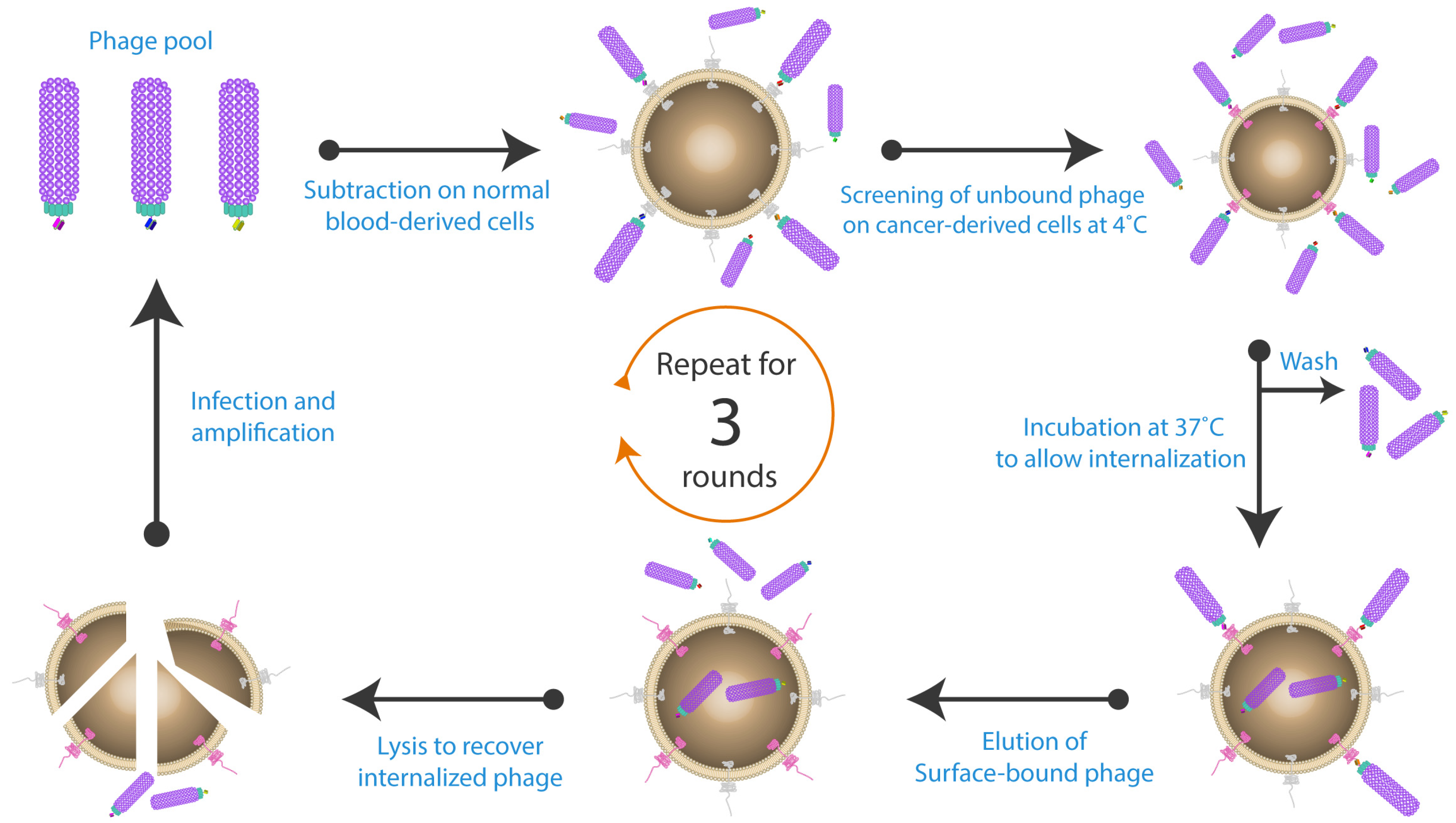{kind=link}
Abstract
:1. Monoclonal Antibodies
1.1. Generation of Monoclonal Antibodies Using Hybridoma Technology
1.2. Generation of Monoclonal Antibodies Using Phage Display Technology
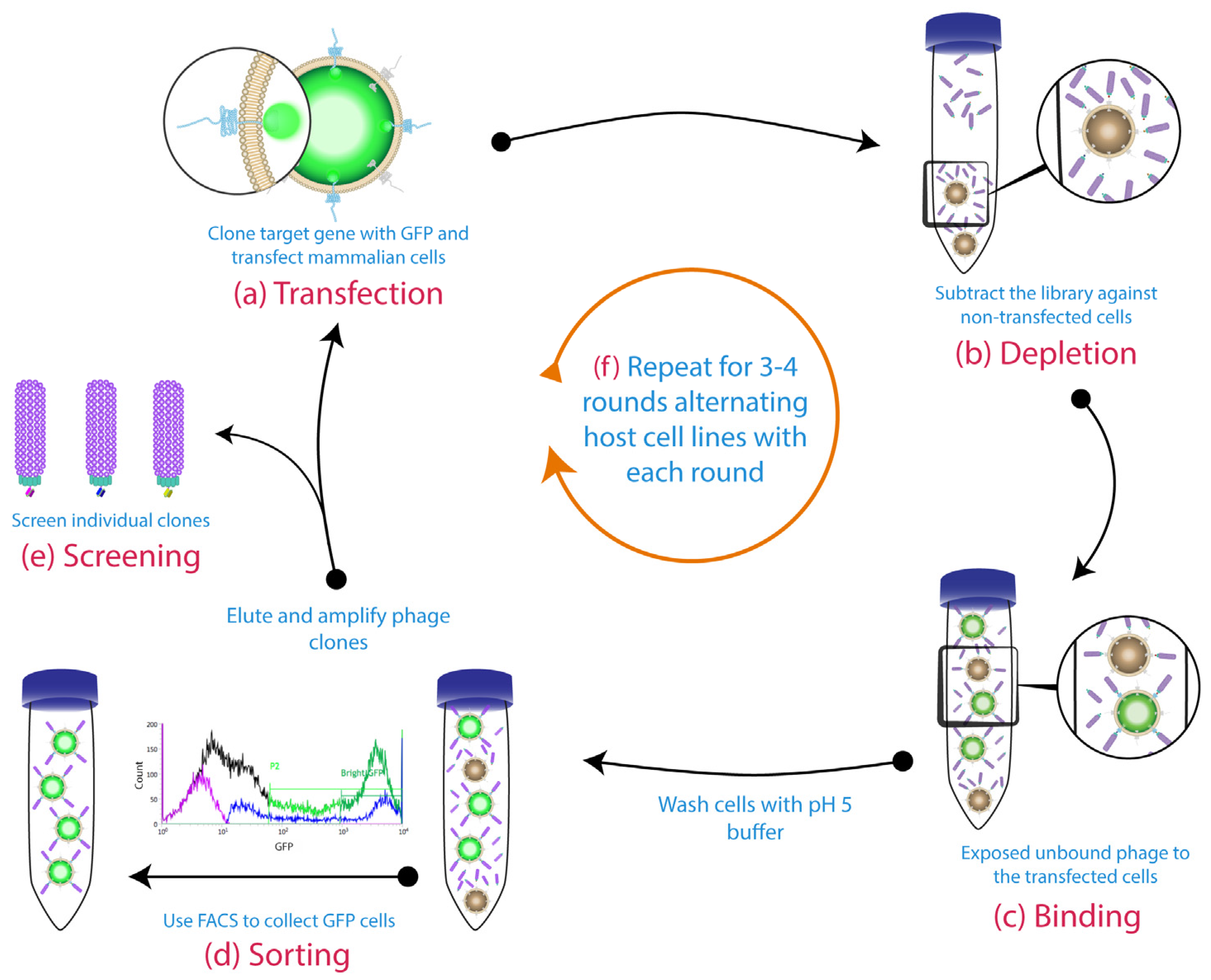
2. Whole Cell-Based Phage Display: A Monoclonal Antibody Discovery Platform for Membrane Proteins
3. Optimisation of Strategies for Phage Display with Cell-Based Panning
3.1. Optimising Cell Surface Antigen Presentation
3.2. Reducing Non-Specific Binding Events
3.3. Optimising Incubation Time and Temperature
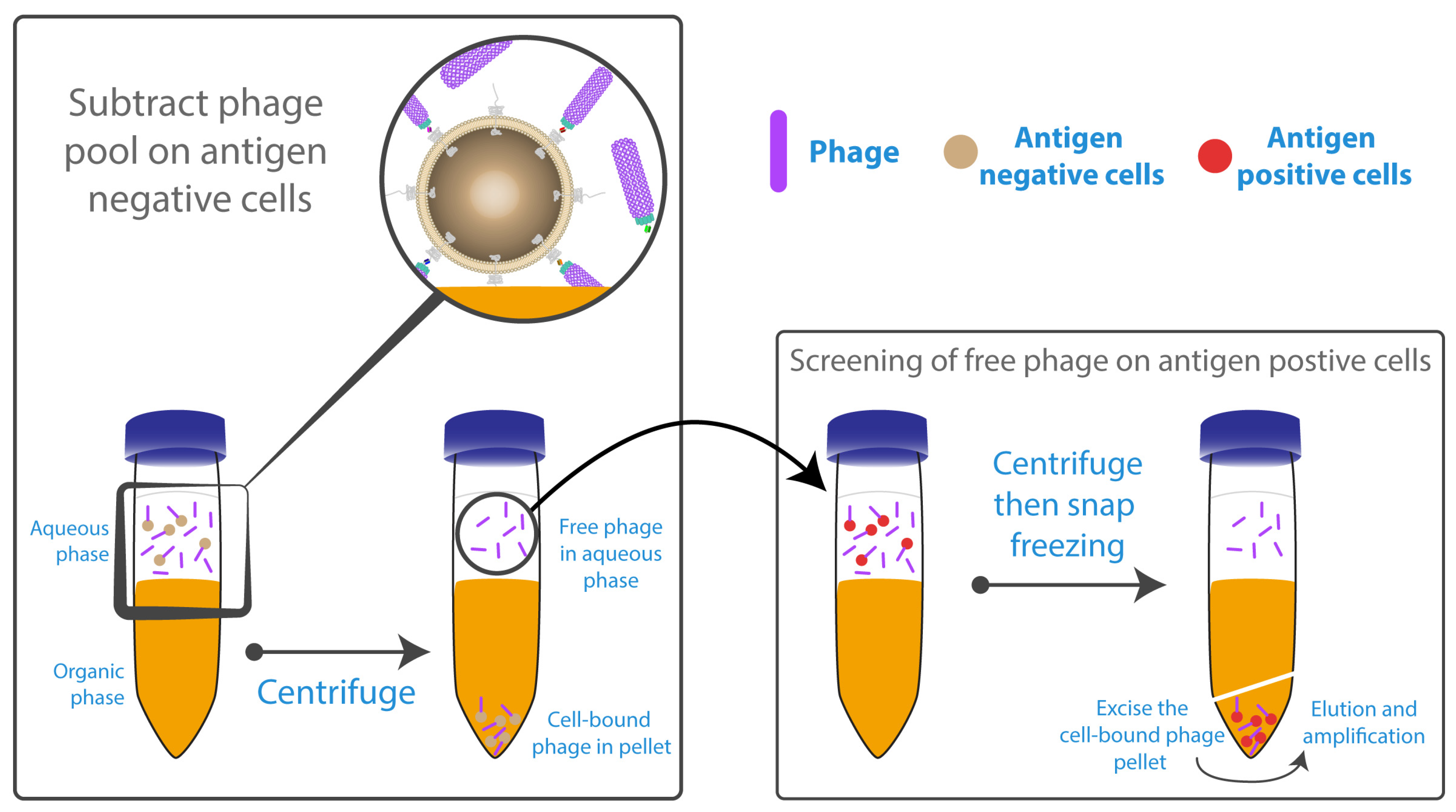
3.4. Improving Phage Recovery
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bradbury, A.; Pluckthun, A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Kong, X.; Hu, H.; Chen, J.; Yang, F.; Liang, H.; Chen, X.; Hu, Y. Research and development of therapeutic mAbs: An analysis based on pipeline projects. Hum. Vaccin. Immunother. 2015, 11, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Satoh, M. The current status and prospects of antibody engineering for therapeutic use: Focus on glycoengineering technology. J. Pharm. Sci. 2015, 104, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Valge-Archer, V.E. Development trends for monoclonal antibody cancer therapeutics. Nat. Rev. Drug Discov. 2007, 6, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Shankar, V.; Vafai, A. Therapeutic applications of monoclonal antibodies. Am. J. Med. Sci. 2002, 324, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Oi, V.T. Genetically engineered antibody molecules. Adv. Immunol. 1989, 44, 65–92. [Google Scholar] [PubMed]
- Studnicka, G.M.; Soares, S.; Better, M.; Williams, R.E.; Nadell, R.; Horwitz, A.H. Human-engineered monoclonal antibodies retain full specific binding activity by preserving non-CDR complementarity-modulating residues. Protein Eng. 1994, 7, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Lonberg, N. Human antibodies from transgenic animals. Nat. Biotechnol. 2005, 23, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Milstein, C. Man-made antibodies. Nature 1991, 349, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Tikunova, N.V.; Morozova, V.V. Phage display on the base of filamentous bacteriophages: Application for recombinant antibodies selection. Acta Nat. 2009, 1, 20–28. [Google Scholar]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Thie, H.; Meyer, T.; Schirrmann, T.; Hust, M.; Dubel, S. Phage display derived therapeutic antibodies. Curr. Pharm. Biotechnol. 2008, 9, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Barbas, C.F., 3rd; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of combinatorial antibody libraries on phage surfaces: The gene III site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Celer, V. The production and application of single-chain antibody fragments. Folia Microbiol. 2003, 48, 687–698. [Google Scholar] [CrossRef]
- Blazek, D.; Celer, V.; Navratilova, I.; Skladal, P. Generation and characterization of single-chain antibody fragments specific against transmembrane envelope glycoprotein gp46 of maedi-visna virus. J. Virol. Methods 2004, 115, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Hawkins, N.J.; Papaioannou, A.; Fiddes, R.J.; Ward, R.L. Isolation of human anti-c-erbB-2 Fabs from a lymph node-derived phage display library. Clin. Exp. Immunol. 1997, 109, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Klimka, A.; Tur, M.K.; Pfitzner, T.; Huhn, M.; Sasse, S.; Mallmann, P.; Engert, A.; Barth, S. Construction of phage display libraries from reactive lymph nodes of breast carcinoma patients and selection for specifically binding human single chain Fv on cell lines. Int. J. Mol. Med. 2004, 14, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Imai, S.; Mukai, Y.; Nakagawa, S.; Abe, Y.; Kamada, H.; Tsunoda, S.; Tsutsumi, Y. Rapid isolation of intrabody candidates by using an optimized non-immune phage antibody library. Die Pharm. 2009, 64, 238–241. [Google Scholar]
- Lowman, H.B.; Bass, S.H.; Simpson, N.; Wells, J.A. Selecting high-affinity binding proteins by monovalent phage display. Biochemistry 1991, 30, 10832–10838. [Google Scholar] [CrossRef] [PubMed]
- Bass, S.; Greene, R.; Wells, J.A. Hormone phage: An enrichment method for variant proteins with altered binding properties. Proteins 1990, 8, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.S. Engineering M13 for phage display. Biomol. Eng. 2001, 18, 57–63. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Gera, N.; Hussain, M.; Rao, B.M. Protein selection using yeast surface display. Methods 2013, 60, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Raeeszadeh-Sarmazdeh, M.; Price, J.V. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 2012, 526, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Chao, G.; Lau, W.L.; Hackel, B.J.; Sazinsky, S.L.; Lippow, S.M.; Wittrup, K.D. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006, 1, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Kieke, M.C.; Cho, B.K.; Boder, E.T.; Kranz, D.M.; Wittrup, K.D. Isolation of anti-T cell receptor scFv mutants by yeast surface display. Protein Eng. 1997, 10, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Feldhaus, M.J.; Siegel, R.W.; Opresko, L.K.; Coleman, J.R.; Feldhaus, J.M.; Yeung, Y.A.; Cochran, J.R.; Heinzelman, P.; Colby, D.; Swers, J.; et al. Flow-cytometric isolation of human antibodies from a nonimmune saccharomyces cerevisiae surface display library. Nat. Biotechnol. 2003, 21, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Garg, P.; Holden, T.; Chao, G.; Webster, J.M.; Messer, A.; Ingram, V.M.; Wittrup, K.D. Development of a human light chain variable domain (VL) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J. Mol. Biol. 2004, 342, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Pastan, I. Display and selection of scFv antibodies on HEK-293T cells. Methods Mol. Biol. 2009, 562, 99–113. [Google Scholar] [PubMed]
- Ho, M.; Pastan, I. Mammalian cell display for antibody engineering. Methods Mol. Biol. 2009, 525, 337–352. [Google Scholar] [PubMed]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, Y.; Pakabunto, K.; Xu, Z.; Zhang, Y.; Tsurushita, N. Whole IgG surface display on mammalian cells: Application to isolation of neutralizing chicken monoclonal anti-IL-12 antibodies. J. Immunol. Methods 2007, 327, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Jacobsen, F.W.; Cai, L.; Chen, Q.; Shen, W.D. Development of a novel mammalian cell surface antibody display platform. mAbs 2010, 2, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar] [CrossRef] [PubMed]
- Boeke, J.D.; Model, P. A prokaryotic membrane anchor sequence: Carboxyl terminus of bacteriophage f1 gene III protein retains it in the membrane. Proc. Natl. Acad. Sci. USA 1982, 79, 5200–5204. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Feng, J.; Model, P. Filamentous phage are released from the bacterial membrane by a two-step mechanism involving a short C-terminal fragment of pIII. J. Mol. Biol. 1999, 289, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Loeb, T. Isolation of a bacteriophage specific for the F plus and Hfr mating types of escherichia coli K-12. Science 1960, 131, 932–933. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Bennett, N.J.; Spagnuolo, J.; Gagic, D.; Russel, M. Filamentous bacteriophage: Biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 2011, 13, 51–76. [Google Scholar] [PubMed]
- Specthrie, L.; Bullitt, E.; Horiuchi, K.; Model, P.; Russel, M.; Makowski, L. Construction of a microphage variant of filamentous bacteriophage. J. Mol. Biol. 1992, 228, 720–724. [Google Scholar] [CrossRef]
- Van Wezenbeek, P.M.; Hulsebos, T.J.; Schoenmakers, J.G. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: Comparison with phage fd. Gene 1980, 11, 129–148. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Weiss, G.A.; Wells, J.A. High copy display of large proteins on phage for functional selections. J. Mol. Biol. 2000, 296, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.; Marasco, W.A. Phage and yeast display. Microbiol. Spectr. 2015, 3, AID-0028-2014. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, J.W.; Kay, B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef]
- Vieira, J.; Messing, J. Production of single-stranded plasmid DNA. Methods Enzymol. 1987, 153, 3–11. [Google Scholar] [PubMed]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Fellouse, F.A.; Esaki, K.; Birtalan, S.; Raptis, D.; Cancasci, V.J.; Koide, A.; Jhurani, P.; Vasser, M.; Wiesmann, C.; Kossiakoff, A.A.; et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 2007, 373, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ura, M.; Hoey, R.J.; Kossiakoff, A.A. A new versatile immobilization tag based on the ultra high affinity and reversibility of the calmodulin-calmodulin binding peptide interaction. J. Mol. Biol. 2015, 427, 2707–2725. [Google Scholar] [CrossRef] [PubMed]
- Rader, C.; Barbas, C.F., 3rd. Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 1997, 8, 503–508. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Arce, J.; Sturgis, J.N.; Duneau, J.P. Dissecting membrane protein architecture: An annotation of structural complexity. Biopolymers 2009, 91, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Servais, E.L.; Colovos, C.; Rodriguez, L.; Bograd, A.J.; Nitadori, J.; Sima, C.; Rusch, V.W.; Sadelain, M.; Adusumilli, P.S. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2478–2489. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Feng, M.; Fisher, R.J.; Rader, C.; Pastan, I. A novel high-affinity human monoclonal antibody to mesothelin. Int. J. Cancer 2011, 128, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 2003, 278, 43496–43507. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Zhang, H.; Ludwig, D.; Persaud, A.; Jimenez, X.; Burtrum, D.; Balderes, P.; Liu, M.; Bohlen, P.; Witte, L.; et al. Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J. Biol. Chem. 2004, 279, 2856–2865. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Seldon, T.; Smede, M.; Linville, A.; Chin, D.Y.; Barnard, R.; Mahler, S.M.; Munster, D.; Hart, D.; Gray, P.P.; et al. A method for rapid, ligation-independent reformatting of recombinant monoclonal antibodies. J. Immunol. Methods 2010, 354, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.A.; Dodevski, I.; Kenig, M.; Dudli, S.; Mohr, A.; Hermans, E.; Pluckthun, A. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.E.; Navarro, P.; Sun, J. Adsorption-induced antigenic changes and their significance in ELISA and immunological disorders. Immunol. Investig. 1997, 26, 39–54. [Google Scholar] [CrossRef]
- Butler, J.E.; Ni, L.; Nessler, R.; Joshi, K.S.; Suter, M.; Rosenberg, B.; Chang, J.; Brown, W.R.; Cantarero, L.A. The physical and functional behavior of capture antibodies adsorbed on polystyrene. J. Immunol. Methods 1992, 150, 77–90. [Google Scholar] [CrossRef]
- Nagy, P.; Friedlander, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar] [PubMed]
- Lipes, B.D.; Chen, Y.H.; Ma, H.; Staats, H.F.; Kenan, D.J.; Gunn, M.D. An entirely cell-based system to generate single-chain antibodies against cell surface receptors. J. Mol. Biol. 2008, 379, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Kiss, M.; Batonick, M.; Weiner, M.; Kay, B. Generating recombinant antibodies to membrane proteins through phage display. Antibodies 2016, 5, 11. [Google Scholar] [CrossRef]
- Cattaneo, A. Tanezumab, a recombinant humanized mab against nerve growth factor for the treatment of acute and chronic pain. Curr. Opin. Mol. Ther. 2010, 12, 94–106. [Google Scholar] [PubMed]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Monne, M.; Chan, K.W.; Slotboom, D.J.; Kunji, E.R. Functional expression of eukaryotic membrane proteins in lactococcus lactis. Protein Sci. 2005, 14, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Siva, A.C.; Kirkland, R.E.; Lin, B.; Maruyama, T.; McWhirter, J.; Yantiri-Wernimont, F.; Bowdish, K.S.; Xin, H. Selection of anti-cancer antibodies from combinatorial libraries by whole-cell panning and stringent subtraction with human blood cells. J. Immunol. Methods 2008, 330, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Krag, D.N.; Shukla, G.S.; Shen, G.P.; Pero, S.; Ashikaga, T.; Fuller, S.; Weaver, D.L.; Burdette-Radoux, S.; Thomas, C. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006, 66, 7724–7733. [Google Scholar] [CrossRef] [PubMed]
- Dantas-Barbosa, C.; de Macedo Brigido, M.; Maranhao, A.Q. Antibody phage display libraries: Contributions to oncology. Int. J. Mol. Sci. 2012, 13, 5420–5440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qu, L.; Zhou, J.; Sun, Z.; Zou, H.; Chen, Y.Y.; Marks, J.D.; Zhou, Y. High throughput identification of monoclonal antibodies to membrane bound and secreted proteins using yeast and phage display. PLoS ONE 2014, 9, e111339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zou, H.; Zhang, S.; Marks, J.D. Internalizing cancer antibodies from phage libraries selected on tumor cells and yeast-displayed tumor antigens. J. Mol. Biol. 2010, 404, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Gorden, B.H.; Kim, J.H.; Sarver, A.L.; Frantz, A.M.; Breen, M.; Lindblad-Toh, K.; O’Brien, T.D.; Sharkey, L.C.; Modiano, J.F.; Dickerson, E.B. Identification of three molecular and functional subtypes in canine hemangiosarcoma through gene expression profiling and progenitor cell characterization. Am. J. Pathol. 2014, 184, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Song, J.M.; Ryu, C.J.; Kim, Y.G.; Lee, E.K.; Kang, S.; Kim, S.J. An efficient strategy for cell-based antibody library selection using an integrated vector system. BMC Biotechnol. 2012, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.A.; Robinson, M.K.; Simmons, H.H.; Russeva, M.; Adams, G.P. Isolation of anti-MISIIR scFv molecules from a phage display library by cell sorter biopanning. Cancer Immunol. Immunother. 2008, 57, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Xu, X.H.; Chen, G.Z.; Deng, X.L.; Li, J.; Yu, X.J.; Chen, M.Z. Production of a human single-chain variable fragment antibody against esophageal carcinoma. World J. Gastroenterol.: WJG 2004, 10, 2619–2623. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Garen, A. Anti-melanoma antibodies from melanoma patients immunized with genetically modified autologous tumor cells: Selection of specific antibodies from single-chain Fv fusion phage libraries. Proc. Natl. Acad. Sci. USA 1995, 92, 6537–6541. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, X.; Dong, N.; Xie, X.; Bai, X.; Shi, Y. Generation and selection of immunized Fab phage display library against human B cell lymphoma. Cell Res. 2007, 17, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Ng, E.; Kern, J.A.; Lee, J.; Brush, J.; Goddard, A.; Carter, P. Identification of a human anti-CD55 single-chain Fv by subtractive panning of a phage library using tumor and nontumor cell lines. Cancer Res. 1999, 59, 2718–2723. [Google Scholar] [PubMed]
- Keller, T.; Kalt, R.; Raab, I.; Schachner, H.; Mayrhofer, C.; Kerjaschki, D.; Hantusch, B. Selection of scFv antibody fragments binding to human blood versus lymphatic endothelial surface antigens by direct cell phage display. PLoS ONE 2015, 10, e0127169. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R.; Lutgerink, J.T.; Pelsers, M.M.; Rousch, M.J.; Coote, J.; Van Neer, N.; De Bruine, A.; Van Nieuwenhoven, F.A.; Glatz, J.F.; Arends, J.W. Selection-dominant and nonaccessible epitopes on cell-surface receptors revealed by cell-panning with a large phage antibody library. Eur. J. Biochem. 1999, 260, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, S.U.; Schwarz, M.; Bassler, N.; Peter, K. Subtractive single-chain antibody (scFv) phage-display: Tailoring phage-display for high specificity against function-specific conformations of cell membrane molecules. Nat. Protoc. 2007, 2, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Rottgen, P.; Takada, Y.; Le Gall, F.; Knackmuss, S.; Bassler, N.; Buttner, C.; Little, M.; Bode, C.; Peter, K. Single-chain antibodies for the conformation-specific blockade of activated platelet integrin αIIbβ3 designed by subtractive selection from naive human phage libraries. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1704–1706. [Google Scholar]
- Schwarz, M.; Meade, G.; Stoll, P.; Ylanne, J.; Bassler, N.; Chen, Y.C.; Hagemeyer, C.E.; Ahrens, I.; Moran, N.; Kenny, D.; et al. Conformation-specific blockade of the integrin GPIIb/IIIa: A novel antiplatelet strategy that selectively targets activated platelets. Circ. Res. 2006, 99, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, S.U.; Schwarz, M.; Schallner, N.; Soosairajah, J.; Bassler, N.; Huang, D.; Bode, C.; Peter, K. Generation of activation-specific human anti-alphambeta2 single-chain antibodies as potential diagnostic tools and therapeutic agents. Blood 2007, 109, 3521–3528. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Teesalu, T.; Sugahara, K.N.; Kotamrajua, V.R.; Adams, J.D.; Ferguson, B.S.; Gong, Q.; Oh, S.S.; Csordas, A.T.; et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. Proc. Natl. Acad. Sci. USA 2011, 108, 6909–6914. [Google Scholar] [CrossRef] [PubMed]
- Dorfmueller, S.; Tan, H.C.; Ngoh, Z.X.; Toh, K.Y.; Peh, G.; Ang, H.P.; Seah, X.Y.; Chin, A.; Choo, A.; Mehta, J.S.; et al. Isolation of a recombinant antibody specific for a surface marker of the corneal endothelium by phage display. Sci. Rep. 2016, 6, 21661. [Google Scholar] [CrossRef] [PubMed]
- Persson, J.; Augustsson, P.; Laurell, T.; Ohlin, M. Acoustic microfluidic chip technology to facilitate automation of phage display selection. FEBS J. 2008, 275, 5657–5666. [Google Scholar] [CrossRef] [PubMed]
- Levitan, B. Stochastic modeling and optimization of phage display. J. Mol. Biol. 1998, 277, 893–916. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.; Meyvantsson, I.; Ju, J.; Beebe, D.J. High-throughput microfluidics: Improved sample treatment and washing over standard wells. Lab Chip 2007, 7, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Chien, S.; Barakat, A.I.; Nerem, R.M. Endothelial cellular response to altered shear stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L529–L533. [Google Scholar] [PubMed]
- Nagel, T.; Resnick, N.; Atkinson, W.J.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. J. Clin. Investig. 1994, 94, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.D.; Agerholm, I.E.; Christensen, B.; Kolvraa, S.; Kristensen, P. Microselection--affinity selecting antibodies against a single rare cell in a heterogeneous population. J. Cell. Mol. Med. 2010, 14, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.D.; Kristensen, P. Selection of antibodies against a single rare cell present in a heterogeneous population using phage display. Nat. Protoc. 2011, 6, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.A.; Cooper, B.W.; Lazarus, H.M.; Mackay, W.; Moss, T.J.; Ciobanu, N.; Tallman, M.S.; Kennedy, M.J.; Davidson, N.E.; Sweet, D.; et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993, 82, 2605–2610. [Google Scholar] [PubMed]
- Sanchez-Martin, D.; Sorensen, M.D.; Lykkemark, S.; Sanz, L.; Kristensen, P.; Ruoslahti, E.; Alvarez-Vallina, L. Selection strategies for anticancer antibody discovery: Searching off the beaten path. Trends Biotechnol. 2015, 33, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C.; Srikant, C.B. Somatostatin receptors. Trends Endocrinol. Metab.: TEM 1997, 8, 398–405. [Google Scholar] [CrossRef]
- Daviet, L.; Buckland, R.; Puente Navazo, M.D.; McGregor, J.L. Identification of an immunodominant functional domain on human cd36 antigen using human-mouse chimaeric proteins and homologue-replacement mutagenesis. Biochem. J. 1995, 305 Pt 1, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, S.; Park, M.-K.; Zhang, Z.; Cheng, Z.; Petrenko, V.A.; Chin, B.A. Blocking agent optimization for nonspecific binding on phage based magnetoelastic biosensors. J. Electrochem. Soc. 2012, 159, B818–B823. [Google Scholar] [CrossRef]
- Menendez, A.; Scott, J.K. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal. Biochem. 2005, 336, 145–157. [Google Scholar] [CrossRef] [PubMed]
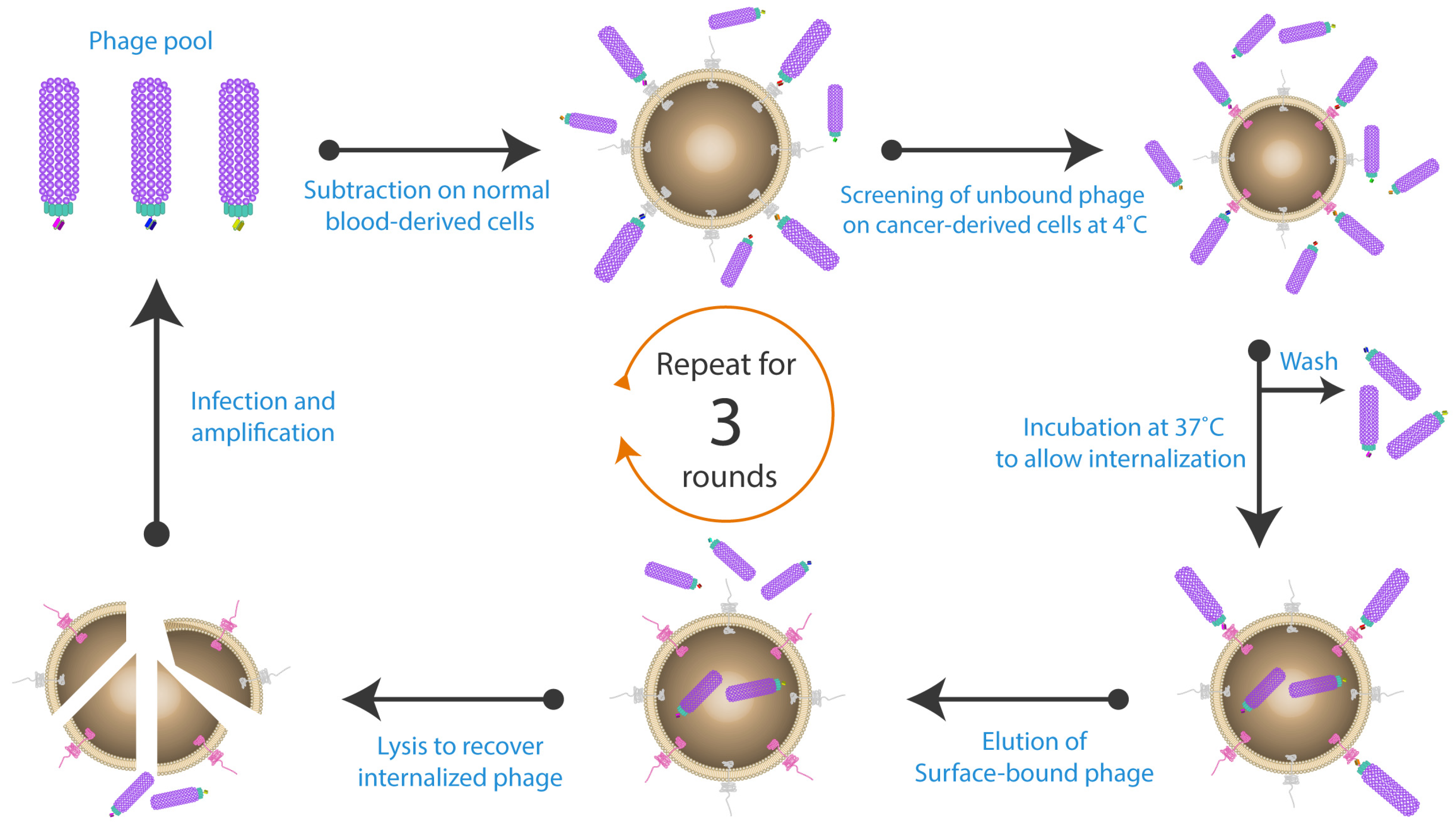
- Heitner, T.; Moor, A.; Garrison, J.L.; Marks, C.; Hasan, T.; Marks, J.D. Selection of cell binding and internalizing epidermal growth factor receptor antibodies from a phage display library. J. Immunol. Methods 2001, 248, 17–30. [Google Scholar] [CrossRef]
- Watters, J.M.; Telleman, P.; Junghans, R.P. An optimized method for cell-based phage display panning. Immunotechnol.: Int. J.Immunol. Eng. 1997, 3, 21–29. [Google Scholar] [CrossRef]
- Poul, M.A.; Becerril, B.; Nielsen, U.B.; Morisson, P.; Marks, J.D. Selection of tumor-specific internalizing human antibodies from phage libraries. J. Mol. Biol. 2000, 301, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.S.; Stryhn, A.; Hansen, B.E.; Fugger, L.; Engberg, J.; Buus, S. A recombinant antibody with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Proc. Natl. Acad. Sci. USA 1996, 93, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- De Kruif, J.; Terstappen, L.; Boel, E.; Logtenberg, T. Rapid selection of cell subpopulation-specific human monoclonal antibodies from a synthetic phage antibody library. Proc. Natl. Acad. Sci. USA 1995, 92, 3938–3942. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, E.; Vaccaro, P.; Anastasi, A.M.; Minenkova, O. Optimized selection of anti-tumor recombinant antibodies from phage libraries on intact cells. Mol. Immunol. 2014, 57, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Koopman, G.; Reutelingsperger, C.P.; Kuijten, G.A.; Keehnen, R.M.; Pals, S.T.; van Oers, M.H. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [PubMed]
- Neurauter, A.A.; Bonyhadi, M.; Lien, E.; Nokleby, L.; Ruud, E.; Camacho, S.; Aarvak, T. Cell isolation and expansion using dynabeads®. Adv. Biochem. Eng. Biot. 2007, 106, 41–73. [Google Scholar]
- Jain, J.; Veggiani, G.; Howarth, M. Cholesterol loading and ultrastable protein interactions determine the level of tumor marker required for optimal isolation of cancer cells. Cancer Res. 2013, 73, 2310–2321. [Google Scholar] [CrossRef] [PubMed]
- Berdichevsky, Y.; Ben-Zeev, E.; Lamed, R.; Benhar, I. Phage display of a cellulose binding domain from clostridium thermocellum and its application as a tool for antibody engineering. J. Immunol. Methods 1999, 228, 151–162. [Google Scholar] [CrossRef]
- Dziegiel, M.; Nielsen, L.K.; Andersen, P.S.; Blancher, A.; Dickmeiss, E.; Engberg, J. Phage display used for gene cloning of human recombinant antibody against the erythrocyte surface antigen, rhesus D. J. Immunol. Methods 1995, 182, 7–19. [Google Scholar] [CrossRef]
- Tur, M.K.; Huhn, M.; Sasse, S.; Engert, A.; Barth, S. Selection of scfv phages on intact cells under low pH conditions leads to a significant loss of insert-free phages. Biotechniques 2001, 30, 404–413. [Google Scholar] [PubMed]
- Mohan, K.; Weiss, G.A. Engineering chemically modified viruses for prostate cancer cell recognition. Mol. Biosyst. 2015, 11, 3264–3272. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.J.; Cardo-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.P.; Reis, C.F.; Morari, E.C.; Maia, Y.C.; Nascimento, R.; Bonatto, J.M.; de Souza, M.A.; Goulart, L.R.; Ward, L.S. A putative OTU domain-containing protein 1 deubiquitinating enzyme is differentially expressed in thyroid cancer and identifies less-aggressive tumours. Br. J. Cancer 2014, 111, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Gerstenbruch, S.; Brooks, C.L.; Kosma, P.; Brade, L.; Mackenzie, C.R.; Evans, S.V.; Brade, H.; Muller-Loennies, S. Analysis of cross-reactive and specific anti-carbohydrate antibodies against lipopolysaccharide from Chlamydophila psittaci. Glycobiology 2010, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, S.C.; Steinman, R.M.; Cohn, Z.A. Endocytosis. Annu. Rev. Biochem. 1977, 46, 669–722. [Google Scholar] [CrossRef] [PubMed]
- Fitting, J.; Blume, T.; Ten Haaf, A.; Blau, W.; Gattenlohner, S.; Tur, M.K.; Barth, S. Phage display-based generation of novel internalizing antibody fragments for immunotoxin-based treatment of acute myeloid leukemia. mAbs 2015, 7, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Becerril, B.; Poul, M.A.; Marks, J.D. Toward selection of internalizing antibodies from phage libraries. Biochem. Biophys. Res. Commun. 1999, 255, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, S.A.; Kelly, K.A.; Niedre, M.; Weissleder, R. Near infrared fluorescence-based bacteriophage particles for ratiometric pH imaging. Bioconj. Chem. 2008, 19, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Raghunand, N.; Garcia-Martin, M.L.; Gatenby, R.A. pH imaging. A review of ph measurement methods and applications in cancers. IEEE Eng. Med. Biol. Mag.:Quart. Mag. Eng. Med. Biol. Soc. 2004, 23, 57–64. [Google Scholar] [CrossRef]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Michon, I.N.; Hauer, A.D.; von der Thusen, J.H.; Molenaar, T.J.; van Berkel, T.J.; Biessen, E.A.; Kuiper, J. Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo. Biochim. Biophys. Acta. 2002, 1591, 87–97. [Google Scholar] [CrossRef]
- Rangel, R.; Dobroff, A.S.; Guzman-Rojas, L.; Salmeron, C.C.; Gelovani, J.G.; Sidman, R.L.; Pasqualini, R.; Arap, W. Targeting mammalian organelles with internalizing phage (iPhage) libraries. Nat. Protoc. 2013, 8, 1916–1939. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, U.B.; Gunnarsson, L.; Geraudie, S.; Scheffler, U.; Griep, R.A.; Reiersen, H.; Duncan, A.R.; Kiprijanov, S.M. Fully human antagonistic antibodies against CCR4 potently inhibit cell signaling and chemotaxis. PLoS ONE 2014, 9, e103776. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, H.E.; Ramaekers, F.C.; van Muijen, G.N.; Ooms, E.C.; Ruiter, D.J. Distribution of cytokeratin polypeptides in epithelia of the adult human urinary tract. Histochemistry 1989, 91, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, E.V.; Slobbe, R.; Wasterval, P.; Ramaekers, F.C.; van Eys, G.J. Selection of phage-displayed antibodies specific for a cytoskeletal antigen by competitive elution with a monoclonal antibody. J. Mol. Biol. 1994, 244, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, E.V.; Nieland, L.J.; Debie, W.H.; Ramaekers, F.C.; van Eys, G.J. Phage displayed antibodies specific for a cytoskeletal antigen. Selection by competitive elution with a monoclonal antibody. Hum. Antib. Hybrid. 1995, 6, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Helwick, C. Will necitumumab be cost-effective? Am. Health Drug Benefits 2015, 8, 11. [Google Scholar] [PubMed]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. mAbs 2014, 6, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfaleh, M.A.; Jones, M.L.; Howard, C.B.; Mahler, S.M. Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning. Antibodies 2017, 6, 10. https://doi.org/10.3390/antib6030010
Alfaleh MA, Jones ML, Howard CB, Mahler SM. Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning. Antibodies. 2017; 6(3):10. https://doi.org/10.3390/antib6030010
Chicago/Turabian StyleAlfaleh, Mohamed A., Martina L. Jones, Christopher B. Howard, and Stephen M. Mahler. 2017. "Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning" Antibodies 6, no. 3: 10. https://doi.org/10.3390/antib6030010
APA StyleAlfaleh, M. A., Jones, M. L., Howard, C. B., & Mahler, S. M. (2017). Strategies for Selecting Membrane Protein-Specific Antibodies using Phage Display with Cell-Based Panning. Antibodies, 6(3), 10. https://doi.org/10.3390/antib6030010