2.2. Syntheses of Sugar-Based Azacrown Ethers
2.2.1. Methyl-4,6-O-benzylidene-β-d-galactopyranoside-2,3-ditosylate (4)
Methyl-4,6-O-benzylidene-β-d-galactopyranoside (3) (44.07 g, 156 mmol) was dissolved in 115 mL of dry pyridine. To this solution, tosyl chloride (89.30 g, 468 mmol) dissolved in 180 mL of dry CHCl3 -—was added dropwise, at such a rate that the internal temperature did not exceed 40 °C. The reaction was then stirred at 40 °C for 30 h, at which point it was complete according to thin-layer chromatography (TLC) (Dichloromethane (DCM)-MeOH 100:2). A total of 50 mL of ice-cold water was added to the mixture and after a short burst of stirring, the mixture was left to stand for an hour. Then, it was extracted with 650 mL of chloroform–diethyl ether 1:1 in multiple portions. The organic phase was then washed with 1 M HCl (4 × 150 mL), once with 150 mL of 10% sodium carbonate solution and once with water. The organic phase was dried with sodium sulfate, filtered, and concentrated in vacuum. The crude product—a yellowish foam—was dissolved in toluene and concentrated two times to remove traces of pyridine. During the process, compound 4 crystallized from the solution as a white solid, which was filtered.
Yield: 85% (78.7 g), white solid. Mp.: 178–180 °C
1H NMR (500 MHz, CDCl3) δ 7.78–7.77 (d, J = 6.3 Hz, 2H, ArH), 7.77–7.76 (d, J = 6.3 Hz, 2H, ArH), 7.44–7.38 (m, 2H, ArH), 7.38–7.32 (m, 3H, ArH), 7.31–7.22 (m, 4H, ArH), 5.33 (s, 1H, ArCH), 4.85 (dd, J = 9.9, 7.7 Hz, 1H, H-2), 4.64 (dd, J = 9.9, 3.7 Hz, 1H, H-3), 4.43 (d, J = 3.6 Hz, 1H, H-4), 4.29–4.20 (m, 2H, H-6a, H-1), 3.99 (dd, J = 12.5, 1.8 Hz, 1H, H-6b), 3.42 (s, 1H, H-5), 3.18 (s, 3H, OCH3), 2.41 (s, 3H, ArCH3), 2.39 (s, 3H, ArCH3).
13C NMR (126 MHz, CDCl3) δ 145.11 (ArCS), 144.40 (ArCS), 137.04 (ArCCH), 134.51 (ArC), 133.29 (ArC), 129.73 (ArC), 129.33 (ArC), 129.06 (ArC), 128.25 (ArC), 128.11 (ArC), 128.10 (ArC), 126.26 (ArC), 101.39 (ArCH), 100.97 (C-1OMe), 77.08 (CH), 76.22 (CH), 74.18 (CH), 68.52 (C-6), 65.92 (CH), 56.88 (OCH3), 21.70 (ArCCH3), 21.64 (ArCCH3).
2.2.2. Methyl-2,3-anhydro-4,6-O-benzylidene-β-d-talopyranoside (5)
Methyl-4,6-O-benzylidene-β-d-galactopyranoside-2,3-ditosylate (4) (73.70 g, 125 mmol) was dissolved in 1170 mL of dry 1,4-dioxane. Meanwhile, 220 mL of 3 M NaOMe solution in MeOH was freshly prepared in a separate round-bottom flask. When all the starting material dissolved in the dioxane, the NaOMe solution was added at once with vigorous stirring and the mixture was allowed to react for 4 h. During this time a yellowish-white precipitate was formed. After 4 hours, the reaction was stopped by pouring it into ice-cold water while stirring. The precipitate was filtered and kept, and the aqueous phase was extracted with chloroform (3 × 100 mL). The organic phase was then dried with sodium sulfate, filtered, and concentrated. The two crude fractions were combined and purified by recrystallization from MeOH. A multitude of crystallizations was necessary to purify all the crude product, since the solubility in MeOH, even at the boiling point, was quite low; around 200 mL of boiling MeOH was necessary to dissolve 1 g of crude anhydro sugar 5. Thus, the MeOH used was recovered and reused after each crystallization.
Yield: 62% (20.28 g), white fluffy solid. Mp.: 241–243 °C
1H NMR (500 MHz, DMSO-d6) δ 7.43–7.31 (m, 5H, ArH), 5.60 (s, 1H, ArCH), 4.76 (s, 1H, H-1), 4.31 (dd, J = 5.3, 2.6 Hz, 1H, H-4), 4.11 (dd, J = 12.9, 2.8 Hz, 1H, H-6a), 4.03 (d, J = 12.8 Hz, 1H, H-6b), 3.48 (t, J = 4.6 Hz, 1H, H-5), 3.43 (s, 3H, OCH3), 3.34–3.31 (m, 1H, H-3), 3.16 (d, J = 4.0 Hz, 1H, H-2).
13C NMR (126 MHz, DMSO-d6) δ 138.83 (ArCCH), 129.14 (ArC), 128.43 (ArC), 126.60 (ArC), 99.78 (ArCH), 99.46 (C-1OMe), 69.07 (C-6), 68.17 (CH), 67.74 (CH), 56.42 (OCH3), 51.20 (CH), 49.90 (CH).
2.2.3. Methyl-4,6-O-benzylidene-β-d-idopyranoside (6)
Anhydro sugar 5 (21.01 g, 80 mmol) was suspended in 630 mL of 2 M KOH solution, and this mixture was refluxed for 22 h. As the reaction proceeded, the mixture became more and more homogeneous. After completion of the reaction, the reaction mixture was carefully neutralized with 3 M sulfuric acid maintaining the internal temperature between 0–10 °C. The neutral aqueous phase was then extracted seven times with 80 mL of diethyl ether. The combined organic phase was dried using sodium sulfate, filtered, and concentrated. The crude product was purified by recrystallization from EtOH. Since the yield was low, to the ethanolic mother liquor diisopropyl ether was added, and the mixture was concentrated. During this step, crystals started to form in the flask: these were filtered, their mother liquor was concentrated, and the residue was recrystallized from EtOH. The crystalline fractions obtained this way were of sufficient purity according to TLC (DCM-MeOH 10:1).
Yield: 58% (13.06 g), white, crystalline solid. Mp.: 156–160 °C
1H NMR (500 MHz, CD3OD) δ 7.51–7.43 (m, 2H, ArH), 7.39–7.30 (m, 3H, ArH), 5.57 (s, 1H, ArCH), 4.72 (d, J = 1.1 Hz, 1H, H-1), 4.28 (dd, J = 12.6, 1.6 Hz, 1H, H-6a), 4.15 (dd, J = 12.6, 1.9 Hz, 1H, H-6b), 3.98 (d, J = 2.3 Hz, 2H, H-3, H-4), 3.80 (s, 1H, H-2), 3.55 (s, 3H, OCH3), 3.53 (br s, 1H, H-5).
13C NMR (126 MHz, CD3OD) δ 141.97 (ArCCH), 132.66 (ArC), 131.78 (ArC), 129.79 (ArC), 105.11 (ArCH), 103.90 (C-1OMe), 79.41 (CH), 73.30 (C-6), 73.28 (CH), 73.05 (CH), 70.59 (CH), 59.66 (OCH3).
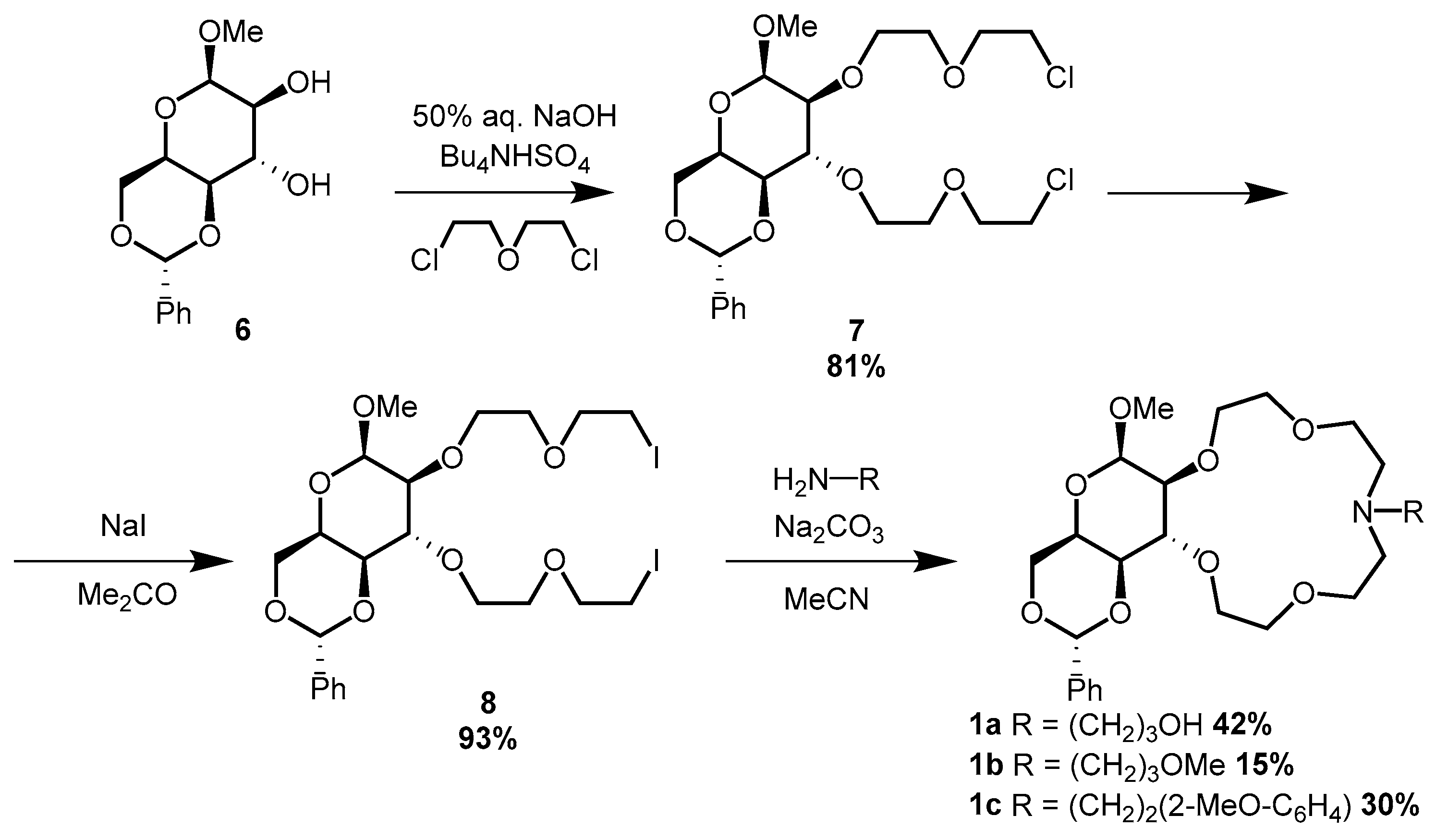
2.2.4. Methyl-4,6-O-benzylidene-2,3-bis-O-[(2-chloroethoxy) ethyl]-β-d-idopyranoside (7)
A two-necked round-bottom flask was fitted with a mechanical stirrer and was charged with methyl-4,6-O-benzylidene-β-d-idopyranoside (6) (13.06 g, 46 mmol) and bis(2-chloroethyl) ether (163 mL, 1390 mmol). To the resulting suspension, tetrabutylammonium hydrogensulfate (15.71 g, 46 mmol) and 50 m/m% NaOH solution (163 mL) were added. The resulting mixture was stirred vigorously for 12 h, after which it was diluted with water (250 mL) and dichloromethane (250 mL). The phases were separated, and the aqueous layer was extracted with dichloromethane (4 × 100 mL). Then the combined organic phase was washed with water (3 × 100 mL), dried over Na2SO4, and concentrated. The excess bis(2-chloroethyl) ether was removed by vacuum distillation and the crude product (23.03 g) was purified by column chromatography on silica gel (460 g) with gradient elution—DCM-MeOH 100:1 → 100:2.
Yield: 81% (18.64 g), colorless oil.
1H NMR (500 MHz, CDCl3) δ 7.58–7.53 (m, 2H, ArH), 7.38–7.33 (m, 3H, ArH), 5.51 (s, 1H, ArCH), 4.69 (d, J = 1.6 Hz, 1H, H-1), 4.38 (dd, J = 12.5, 1.5 Hz, 1H, H-6a), 4.10 (dd, J = 12.5, 2.1 Hz, 1H, H-6b), 4.01–3.92 (m, 2H, CH2Cl), 3.88 (dd, J = 3.9, 2.0 Hz, 1H, H-5), 3.86–3.73 (m, 5H, OCH2, H-2, CH2Cl), 3.75–3.64 (m, 9H, 4 × OCH2, H-3), 3.59 (dd, J = 3.9, 1.5 Hz, 1H, H-4), 3.57 (s, 3H, OCH3), 3.47 (t, J = 5.6 Hz, 2H, OCH2).
13C NMR (126 MHz, CDCl3) δ 138.23 (ArCCH), 128.94 (ArC), 128.02 (ArC), 126.61 (ArC), 101.26 (ArCH), 100.36 (C-1OMe), 77.70 (CH), 75.31 (CH), 73.46 (CH), 71.51 (OCH2), 71.48 (OCH2), 71.39 (OCH2), 70.82 (OCH2), 70.56 (OCH2), 69.91 (OCH2), 69.79 (C-6), 66.53 (CH), 56.60 (OCH3), 43.30 (CH2Cl), 42.89 (CH2Cl).
2.2.5. Methyl-4,6-O-benzylidene-2,3-bis-O-[(2-iodoethoxy) ethyl]-β-d-idopyranoside (8)
Bis-chloro compound 7 (18.64 g, 38 mmol) was dissolved in 180 mL of dry acetone along with 22.56 g (151 mmol) of dry sodium iodide and a small amount of dry sodium carbonate. This mixture was refluxed for 40 h while simultaneously, a white precipitate appeared. This was then filtered from the mixture and the acetone was distilled off. The crude product was dissolved in a mixture of 200 mL of dichloromethane and 150 mL of water, and the phases were separated. The aqueous phase was extracted with 50 mL of dichloromethane; then the combined organic phase was washed with water (3 × 50 mL), dried over sodium sulfate, filtered, and concentrated.
Yield: 93% (23.68 g), yellow oil.
1H NMR (500 MHz, CDCl3) δ 7.56–7.54 (m, 2H, ArH), 7.40–7.33 (m, 3H, ArH), 5.52 (s, 1H, ArCH), 4.70 (d, J = 1.6 Hz, 1H, H-1), 4.38 (dd, J = 12.5, 1.6 Hz, 1H, H-6a), 4.10 (dd, J = 12.5, 2.1 Hz, 1H, H-6b), 4.02–3.98 (m, 1H, H-2), 3.98–3.92 (m, 1H, H-3), 3.92–3.86 (m, 1H, H-5), 3.83–3.77 (m, 5H, 2 × OCH2, H-4), 3.74–3.65 (m, 8H, 4 × OCH2), 3.57 (s, 3H, OCH3), 3.29 (t, J = 6.6 Hz, 2H, CH2I), 3.12 (t, J = 6.6 Hz, 2H, CH2I).
13C NMR (126 MHz, CDCl3) δ 138.20 (ArCCH), 128.97 (ArC), 128.05 (ArC), 126.63 (ArC), 101.26 (ArCH), 100.32 (C-1OMe), 77.66 (CH), 75.28 (CH), 73.46 (CH), 71.96 (OCH2), 71.89 (OCH2), 71.31 (OCH2), 70.48 (OCH2), 70.13 (OCH2), 69.96 (OCH2), 69.79 (C-6), 66.52 (CH), 56.63 (OCH3), 4.12 (CH2I), 2.99 (CH2I).
2.2.6. Methyl-4,6-O-benzylidene-2,3-dideoxy-β-d-idopyranosido[2,3-h]-N-[3-hydroxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1a)
Bis-iodo compound 8 (3.00 g, 4 mmol) was dissolved in dry acetonitrile (50 mL) under Ar. To this solution, 2.80 g (26 mmol) of dry sodium carbonate was added. Next, 337 µL (4 mmol) of 3-aminopropanol was added, and the mixture was refluxed for 48 h. It was then filtered, and the solvent was distilled off. The residue was dissolved in dichloromethane (50 mL) and washed with water (3 × 30 mL). The combined aqueous phase was extracted once with dichloromethane, and then, the combined organic phase was dried over sodium sulfate, filtered, and concentrated. The crude product (2.7 g) was purified by chromatography (aluminium oxide, 82 g). Elution was isocratic with dichloromethane-MeOH 100:0.5.
Yield: 42% (0.92 g), yellow–orange viscous oil.
1H NMR (500 MHz, CDCl3) δ 7.59–7.53 (m, 2H ArH), 7.37–7.31 (m, 3H, ArH), 5.51 (s, 1H, ArCH), 4.74 (s, 1H, H-1), 4.33 (d, J = 12.5 Hz, 1H, H-6a), 4.07 (dd, J = 12.6, 4.6 Hz, 2H, H-6b, H-5), 4.02–3.97 (m, 1H, H-3), 3.95 (s, 1H, H-2), 3.89–3.73 (m, 6H, 3 × OCH2), 3.72–3.57 (m, 9H, 4 × OCH2, H-4), 3.55 (s, 3H, OCH3), 2.88–2.72 (m, 2H, NCH2), 2.72–2.60 (m, 4H, 2 × NCH2), 1.76–1.60 (m, 2H, CH2CH2CH2).
13C NMR (126 MHz, CDCl3) δ 138.33 (ArCCH), 128.88 (ArC), 128.03 (ArC), 126.70 (ArC), 101.24 (ArCH), 99.61 (C-1OMe), 78.11 (CH), 76.23 (CH), 74.27 (CH), 71.83 (OCH2), 70.98 (OCH2), 70.88 (OCH2), 69.93(OCH2), 69.43 (C-6), 69.28 (OCH2), 69.14 (OCH2), 66.02 (CH), 64.08 (CH2OH), 56.07 (OCH3), 55.84 (NCH2), 54.56 (NCH2), 54.18 (NCH2), 28.42 (CH2CH2CH2).
2.2.7. Methyl-4,6-O-benzylidene-2,3-dideoxy-β-d-idopyranosido[2,3-h]-N-[3-methoxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1b)
Synthesis was carried out according to the previous procedure (
Section 2.2.6) with the following quantities: 3.00 g (4 mmol) of bis-iodo compound
8 dissolved in 50 mL of dry acetonitrile, 2.81 g (26 mmol) of dry sodium carbonate, and 450 µL (4 mmol) of 3-methoxypropylamine.
After work-up, the crude product (2.7 g) was purified by column chromatography (85 g of silica gel) with gradient elution (dichloromethane-MeOH 100:4 → 100:4.5).
Yield: 15% (0.33 g), yellow-orange viscous oil.
1H NMR (500 MHz, CD3OD) δ 7.60–7.53 (m, 2H, ArH), 7.38–7.33 (m, 3H, ArH), 5.61 (s, 1H, ArCH), 4.76 (d, J = 1.8 Hz, 1H, H-1), 4.25 (d, J = 13.3 Hz, 1H, H-6a), 4.17 (dd, J = 12.6, 2.0 Hz, 1H, H-6b), 4.11 (d, J = 1.9 Hz, 1H, H-5), 4.07–4.02 (m, 1H, H-3), 3.99–3.88 (m, 2H, H-2, H-4), 3.85–3.61 (m, 14H, 7 × OCH2), 3.55 (s, 3H, OCH3), 3.53–3.43 (m, 2H, NCH2), 3.35 (s, 3H, OCH3), 3.30–2.94 (m, 4H, 2 × NCH2), 1.95–1.91 (m, 2H, CH2CH2CH2).
13C NMR (126 MHz, CD3OD) δ 138.58 (ArCCH), 128.56 (ArC), 127.65 (ArC), 126.38 (ArC), 101.13 (ArCH), 99.85 (C-1OMe), 78.04 (CH), 76.25 (CH), 73.63 (CH), 71.65 (OCH2), 70.63 (OCH2), 70.45 (OCH2), 69.38 (C-6), 68.63 (CH2OH), 66.30 (CH), 57.92 (OCH3), 55.13 (OCH3), 53.78 (NCH2), 27.57 (CH2CH2CH2).
2.2.8. Methyl-4,6-O-benzylidene-2,3-dideoxy-β-d-idopyranosido[2,3-h]-N-[2-(2-methoxyphenyl)ethyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (1c)
Synthesis was carried out according to the previous procedure (
Section 2.2.6) with the following quantities: 4.00 g (6 mmol) of bis-iodo compound
8 dissolved in 66 mL of dry acetonitrile, 3.75 g (35 mmol) of dry sodium carbonate, and 864 µL (6 mmol) of 2-(2-methoxyphenyl)ethylamine.
After work-up, the crude product (4.2 g) was purified by column chromatography (150 g of silica gel) with gradient elution (dichloromethane-MeOH 100:3 → 100:4)
Yield: 30% (1.02 g), yellow–orange viscous oil.
1H NMR (500 MHz, CDCl3) δ 7.61–7.51 (m, 2H, ArH), 7.36–7.30 (m, 3H, ArH), 7.27–7.17 (m, 2H, ArH), 6.91 (t, J = 7.4 Hz, 1H, ArH), 6.86 (d, J = 8.2 Hz, 1H, ArH), 5.52 (s, 1H, ArCH), 4.74 (s, 1H, H-1), 4.32 (d, J = 12.5 Hz, 1H, H-6a), 4.08 (dd, J = 12.6, 2.0 Hz, 1H, H-6b), 3.99–3.96 (m, 3H, H-2, H-3, H-5), 3.86–3.76 (m, 8H, 2 × OCH2, OCH3, H-4), 3.71–3.65 (m, 8H, 4 × OCH2), 3.53 (s, 3H, OCH3), 3.45–2.73 (m, 8H, 3 × NCH2, NCH2CH2Ar).
13C NMR (126 MHz, CDCl3) δ 157.32 (ArCCH), 138.18 (ArCCH), 130.86 (ArC), 129.06 (ArC), 128.15 (ArC), 126.63 (ArC), 120.97 (ArC), 110.48 (ArCH), 101.28 (C-1OMe), 78.08 (CH), 74.78 (CH), 74.72 (CH) 71.88 (OCH2) 70.86 (OCH2), 69.87 (OCH2), 69.11 (C-6), 65.83 (CH), 55.90 (OCH3), 55.43 (NCH2), 29.37 (NCH2CH2Ar).
2.2.9. Methyl-4,6-O-benzylidene-α-d-galactopyranoside-2,3-ditosylate (10)
The synthesis of the α analogue was performed similarly to that of the aforementioned β analogue. Thus, methyl 4,6-
O-benzylidene-α-
d-galactopyranoside (
9) (12.83 g, 45 mmol) was dissolved in 33 mL of dry pyridine, and to this solution 24.99 g (131 mmol) tosyl chloride in 50 mL of dry chloroform was added dropwise, while the internal temperature was kept under 40 °C. The reaction mixture was then stirred at 40 °C for 30 h. Work-up procedures were identical as described in
Section 2.2.1. The crude product was recrystallized from CHCl
3–hexane to afford the pure product.
Yield: 75% (20.11 g), white solid. Mp.: 177–179 °C
1H NMR (300 MHz, CDCl3) δ 7.75–7.65 (m, 4H, ArH), 7.49–7.32 (m, 5H, ArH), 7.26 (m, 4H, ArH), 5.36 (s, 1H, ArCH), 5.00 (dd, J = 8.1, 4.5 Hz, 2H, H-3, H-4), 4.83 (dd, J = 10.3, 3.4 Hz, 1H, H-2), 4.45 (d, J = 3.6 Hz, 1H, H-1), 4.24 (d, J = 12.7 Hz, 1H, H-6a), 4.02 (d, J = 12.6 Hz, 1H, H-6b), 3.70 (s, 1H, H-5), 3.37 (s, 3H, OCH3), 2.44 (s, 3H, ArCH3), 2.41 (s, 3H, ArCH3).
13C NMR (75 MHz, CDCl3) δ 145.02 (ArC), 144.92 (ArC), 137.15 (ArC), 133.75 (ArC), 132.88 (ArC), 129.71 (ArC), 129.68 (ArC), 129.04 (ArC), 128.26 (ArC), 128.09 (ArC), 127.80 (ArC), 126.06 (ArC), 100.53 (ArCH), 98.45 (C-1OMe), 74.86 (CH), 74.39 (CH), 73.44 (CH), 68.75 (CH), 62.09 (CH), 56.01 (OCH3), 21.71 (ArCH3).
2.2.10. Methyl-4,6-O-benzylidene-α-d-idopyranoside (12)
Ditosylate 10 (20.11 g, 34 mmol) was dissolved in dry dimethyl sulfoxide (320 mL), and then 60 mL of freshly prepared 3 M NaOMe solution in MeOH was added by stirring. The reaction mixture was allowed to react for 5 h, and then it was poured into 1 L of ice water while stirring. After some time, a beige solid precipitated which was filtered. This was a mixture of methyl 4,6-O-benzylidene-2,3-anhydro-d-gulopyranoside and methyl 4,6-O-benzylidene-2,3-anhydro-d-talopyranodside and was used in the next step without purification.
The crude mixture of anhydro sugars was suspended in 250 mL of 5 M potassium hydroxide solution and was kept at reflux until complete conversion was achieved. The solution was then neutralized to phenolphthalein with 50% acetic acid. The product precipitated at this point and was filtered and washed with water. Recrystallization was performed from CHCl3–hexane.
Yield: 80% (7.71 g), white solid. Mp.: 145–149 °C
1H NMR (500 MHz, CD3OD) δ 7.50–7.44 (m, 2H, ArH), 7.37–7.31 (m, 3H, ArH), 5.59 (s, 1H, ArCH), 4.71 (s, 1H, H-1), 4.61 (s, 2H, OH), 4.24–4.14 (m, 2H, H-6a, H-6b), 4.06 (s, 1H, H-4), 3.88 (s, 1H, H-3), 3.81–3.78 (m, 1H, H-5), 3.56–3.52 (m, 1H, H-2), 3.42 (s, 3H, OCH3).
13C NMR (126 MHz, CD3OD) δ 142.05 (ArCCH), 132.61 (ArC), 131.76 (ArC), 129.81 (ArC), 106.72 (ArCH), 104.79 (C-1OMe), 80.66 (CH), 73.62 (CH), 73.26 (C-6), 72.49 (CH), 64.06 (CH), 58.48 (OCH3).
2.2.11. Methyl-4,6-O-benzylidene-2,3-bis-O-[(2-chloroethoxy)ethyl]-α-d-idopyranoside (13)
A two-necked, round-bottom flask equipped with a mechanical stirrer was charged with methyl-4,6-O-benzylidene-α-d-idopyranoside (12) (7.71 g, 27 mmol) and bis(2-chloroethyl) ether (96.0 mL, 820 mmol). To the resulting suspension, tetrabutylammonium hydrogensulfate (9.27 g, 27 mmol) and 50 m/m% NaOH solution (96 mL) were added. The resulting mixture was stirred vigorously for 10 h, after which it was diluted with water (250 mL) and dichloromethane (250 mL). The phases were separated, and the organic phase was washed with water (3 × 100 mL), dried over Na2SO4, and concentrated. The excess bis(2-chloroethyl) ether was then removed by vacuum distillation. The crude product (10.86 g) solidified after cooling, so attempts were made to purify it by recrystallization. The use of EtOH-hexane and diisopropyl ether were both unsuccessful, the product that crystallized was still impure according to TLC and NMR analysis. Thus, the material was purified by column chromatography on silica gel (300 g) with gradient elution—dichloromethane-MeOH 100:1 → 100:3.
Yield: 30% (4.03 g), off-white semi-solid.
1H NMR (500 MHz, CDCl3) δ 7.53–7.47 (m, 2H, ArH), 7.38–7.31 (m, 3H, ArH), 5.53 (s, 1H, ArCH), 4.73 (d, J = 4.7 Hz, 1H, H-1), 4.27 (d, J = 12.8 Hz, 1H, H-6a), 4.16–4.07 (m, 2H, H-6b, H-2), 3.97–3.59 (m, 17H, 6 × OCH2, H-3, H-4, H-5, CH2Cl), 3.59–3.55 (m, 2H, CH2Cl), 3.42 (s, 3H, OCH3).
13C NMR (126 MHz, CDCl3) δ 137.93 (ArCCH), 128.94 (ArC), 128.14 (ArC), 126.21 (ArC), 102.78 (ArCH), 100.24 (C-1OMe), 80.95 (CH), 79.07 (CH), 78.20 (CH), 71.35 (OCH2), 71.34 (OCH2), 71.28 (OCH2), 70.73 (OCH2), 70.69 (OCH2), 70.67 (OCH2), 69.26 (C-6), 61.97 (CH), 55.43 (OCH3), 42.92 (CH2Cl), 42.84 (CH2Cl).
2.2.12. Methyl-4,6-O-benzylidene-2,3-bis-O-[(2-iodoethoxy) ethyl]-α-d-idopyranoside (14)
Bis-chloro podant 13 (4.03 g, 8 mmol) was dissolved in 40 mL of dry acetone along with 4.88 g (33 mmol) of dry sodium iodide and a small amount of dry sodium carbonate. The reaction mixture was refluxed for 40 h while simultaneously, a white precipitate appeared. This was then filtered from the mixture, and acetone was distilled off. The crude product was dissolved in a mixture of 50 mL dichloromethane and 50 mL of water, and the phases were separated. The aqueous phase was extracted with 20 mL of dichloromethane, and then the combined organic phase was washed with water (3 × 30 mL), dried over sodium sulfate, filtered, and concentrated.
Yield: 96% (5.27 g), light-brown solid. Mp.: 77–80 °C.
1H NMR (500 MHz, CDCl3) δ 7.53–7.47 (m, 2H, ArH), 7.38–7.31 (m, 3H, ArH), 5.54 (s, 1H, ArCH), 4.73 (d, J = 4.8 Hz, 1H, H-1), 4.28 (d, J = 12.9 Hz, 1H, H-6a), 4.17–4.10 (m, 2H, H-6b, H-2), 3.88–3.76 (m, 5H, OCH2, H-3, H-4, H-5), 3.76–3.71 (m, 4H, 2-OCH2), 3.71–3.60 (m, 6H, 3-OCH2), 3.42 (s, 3H OCH3), 3.23 (t, J = 5.6 Hz, 2H, CH2I), 3.20 (t, J = 5.6 Hz, 2H, CH2I).
13C NMR (126 MHz, CDCl3) δ 137.91 (ArCCH), 128.95 (ArC), 128.16 (ArC), 126.22 (ArC), 102.81 (ArCH), 100.22 (C-1OMe), 81.03 (CH), 79.15 (CH), 78.28 (CH), 71.94 (OCH2), 71.90 (OCH2), 71.35 (OCH2), 70.79 (OCH2), 70.31 (OCH2), 70.27 (OCH2), 69.24 (C-6), 62.02 (CH), 55.47 (OCH3), 3.29 (CH2I), 3.13 (CH2I).
2.2.13. Methyl-4,6-O-benzylidene-2,3-dideoxy-α-d-idopyranosido[2,3-h]-N-[3-hydroxypropyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (2a)
Bis-iodo podant 14 (2.60 g, 4 mmol) was dissolved in dry acetonitrile (38 mL) under Ar. To this solution, 2.44 g (23 mmol) of dry sodium carbonate, then 295 µL (4 mmol) of 3-aminopropanol were added, and the mixture was refluxed for 35 h. It was then filtered, and the solvent was distilled off. The residue was dissolved in dichloromethane (50 mL) and washed with water (3 × 30 mL). The aqueous phase was extracted once with dichloromethane and the combined organic phase was dried over sodium sulfate, filtered, and concentrated. The crude product (2.54 g) was purified by column chromatography on aluminium oxide (50 g) using isocratic elution with dichloromethane–MeOH 100:2.
Yield: 68% (1.31 g), orange–white solid. Mp.: 97–101 °C
1H NMR (500 MHz, CDCl3) δ 7.51–7.46 (m, 2H, ArH), 7.37–7.30 (m, 3H, ArH), 5.52 (s, 1H, ArCH), 4.69 (d, J = 5.4 Hz, 1H, H-1), 4.27 (d, J = 12.9 Hz, 1H, H-6a), 4.17–4.08 (m, 2H, H-2, H-3), 3.95–3.51 (m, 17H, H-6b, H-4, H-5, 7 × OCH2), 3.41 (s, 3H, OCH3), 2.87–2.73 (m, 2H, NCH2), 2.71–2.63 (m, 4H, 2 × NCH2), 1.72–1.58 (m, 2H, CH2CH2CH2OH).
13C NMR (126 MHz, CDCl3) δ 137.90 (ArCCH), 128.87 (ArC), 128.11 (ArC), 126.17 (ArC), 103.49 (ArCH), 99.92 (C-1OMe), 81.24 (CH), 79.49 (CH), 79.38 (CH), 71.39 (OCH2), 70.38 (OCH2), 70.34 (OCH2), 70.26 (OCH2), 69.14 (C-6), 69.04 (OCH2), 64.27 (OCH2), 62.47 (CH), 56.95 (NCH2), 55.31 (OCH3), 54.57 (NCH2), 54.44 (NCH2), 28.46 (CH2CH2CH2).
2.2.14. Methyl-4,6-O-benzylidene-2,3-dideoxy-α-d-idopyranosido[2,3-h]-N-[2-(2-methoxyphenyl)ethyl]-1,4,7,10-tetraoxa-13-azacyclopentadecane (2b)
Synthesis was carried out according to the previous procedure (
Section 2.2.13) with the same quantities, except for the amine; 560 µL (4 mmol) of 2-(2-methoxyphenyl)ethylamine was used.
After work-up, the crude product (2.47 g) was purified by column chromatography (50 g of silica gel) with isocratic elution (dichloromethane-MeOH 100:5).
Yield: 62% (1.35 g), light-brown solid. Mp.: 118–121 °C
1H NMR (500 MHz, CDCl3) δ 7.55–7.50 (m, 2H, ArH), 7.41–7.34 (m, 3H, ArH), 7.23–7.12 (m, 2H, ArH), 6.92–6.82 (m, 2H, ArH), 5.56 (s, 1H, ArCH), 4.75 (d, J = 5.4 Hz, 1H, H-1), 4.32 (d, J = 12.9 Hz, 1H, H-6a), 4.21–4.11 (m, 2H, H-6b, H-5), 3.98–3.91 (m, 2H, H-3, H-4), 3.90–3.84 (m, 2H, OCH2), 3.83 (s, 3H, ArOCH3), 3.78–3.58 (m, 10H, 5 × OCH2), 3.50 (dd, J = 10.1, 5.4 Hz, 1H, H-2), 3.46 (s, 3H, OCH3), 2.99–2.73 (m, 8H, 3 × NCH2, ArCH2).
13C NMR (126 MHz, CDCl3) δ 157.51 (ArCOMe), 137.89 (ArC), 130.38 (ArC), 128.89 (ArC), 128.13 (ArC), 127.27 (ArC), 126.17 (ArC), 120.43 (ArC), 110.24 (ArC), 103.57 (ArCH), 99.94 (C-1OMe), 81.35 (CH), 79.54 (CH), 79.47 (CH), 71.38 (OCH2), 70.53 (OCH2), 70.43 (OCH2), 70.32 (OCH2), 69.14 (C-6), 62.49 (CH), 56.83 (NCH2), 55.31 (OCH3), 55.23 (OCH3), 54.22 (NCH2), 54.15 (NCH2), 28.08 (ArCH2).
2.3. Asymmetric Syntheses
2.3.1. Diethyl 2-acetamido-2-(2-nitro-1-phenylethyl) Malonate (17)
Diethyl acetamidomalonate (
16) (0.163 g, 0.75 mmol), β-nitrostyrene (
15) (0.075 g, 0.5 mmol) and the appropriate crown catalyst (10 mol%) were dissolved in a 2.5 mL, 4:1 mixture of dry Et
2O and dry THF. The THF was added first and then the mixture was diluted with the appropriate amount of Et
2O. After a short period of stirring, anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added, and the mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–ethyl-acetate 4:1). After completion, the mixture was concentrated, and the residue was dissolved in dichloromethane and filtered. The filtrate was washed with 10% aqueous HCl (3 × 5 mL), and then dried (Na
2CO
3 and Na
2SO
4). The crude product obtained after evaporating the solvent was purified by preparative TLC (hexane –ethyl-acetate 3:1) to give an off-white solid with an Mp of 135–136 °C. Yields and ee values can be observed in
Table 1.
1H NMR (500 MHz, CDCl3), δ 7.31–7.28 (m, 3H, ArH), 7.22–7.18 (m, 2H, ArH), 6.89 (br s, NH), 5.54–5.48 (m, 1H, PhCH), 4.73–4.66 (m, 2H, OCH2), 4.34–4.23 (m, 2H, OCH2), 4.20–4.13 (m, 1H, CH2NO2), 4.08–4.01 (m, 1H, CH2NO2), 2.12 (s, 3H, COCH3), 1.27 (t, J = 7 Hz, 3H, CH3CH2), 1.25 (t, J = 7 Hz, 3H, CH3CH2)
13C NMR (75 MHz, CDCl3), δ 170.10 (COCH3), 166.43 (C(O)O), 165.71 (C(O)O), 133.78 (ArC), 128.75 (ArC), 128.70 (ArC), 128.69 (ArC), 76.83 (HNCCO), 67.21 (CNO2), 63.56 (CH2CH3), 62.75 (CH2CH3), 48.30 (PhCCNO2), 22.97 (COCH3), 13.84 (CH2CH3), 13.76 (CH2CH3).
2.3.2. Diethyl 2,2-dicyano-3-phenylcyclopropane-1,1-dicarboxylate (20)
Benzylidenemalononitrile (
18) (0.077 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol) and the appropriate crown ether (10 mol%) were dissolved in a 2.5 mL, 4:1 mixture of dry Et
2O and dry THF. The THF was added first, and then the mixture was diluted with the appropriate amount of Et
2O. Then, dry Na
2CO
3 (0.11 g, 1 mmol) was added. The reaction mixture was stirred at room temperature. After completion of the reaction, the mixture was concentrated, and the residue was dissolved in dichloromethane and filtered. The filtrate was washed with 10% aqueous HCl (3 × 5 mL) and then dried (Na
2CO
3 and Na
2SO
4) and concentrated. The crude product was purified by preparative TLC using hexane–ethyl-acetate (5:1) as the eluent to give a yellow oil. Yields and ee values can be observed in
Table 2.
1H NMR (300 MHz, CDCl3), δ 7.45–7.35 (m, 5H, ArH), 4.43 (q, J = 7.2 Hz, 2H, OCH2), 4.30–4.18 (m, 2H, OCH2), 3.96 (s, 1H, ArCH), 1.39 (t, J = 7.2 Hz, 3H, CH2CH3), 1.19 (t, J = 7.2 Hz, 3H, CH2CH3)
13C NMR (75 MHz, CDCl3), δ 163.05 (COOC2H5), 161.06 (COOC2H5), 129.67 (ArC), 129.10 (ArC), 128.76 (ArC), 127.31 (ArC), 111.86 (CN), 109.71 (CN), 64.50 (CH2CH3), 63.62 (CH2CH3), 46.39 (OCCCO), 40.08 (PhCH), 16.32 (NCCCN), 13.97 (CH2CH3), 13.60 (CH2CH3).
2.3.3. Triethyl 2-cyano-3-phenylcyclopropane-1,1,2-tricarboxylate (22a)
Benzylidenecyanoacetic acid ethyl ester (
21a) (0.101 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in dry dichloromethane (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, giving a yellow oil. Yields and ee values can be observed in
Table 3.
1H NMR (500 MHz, CDCl3) δ 7.41–7.31 (m, 5H, ArH), 4.42–4.21 (m, 4H, OCH2CH3), 4.14 (qd, J = 7.1, 2.0 Hz, 2H, OCH2CH3), 3.94 (s, 1H, ArCH), 1.39 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.31 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.10 (t, J = 7.1 11 Hz, 3H, OCH2CH3)
13C NMR (75 MHz, CDCl3) δ 164.52 (COOCH2), 164.05 (COOCH2), 162.74 (COOCH2), 129.90 (ArC), 128.93 (ArC), 128.84 (ArC), 128.77(ArC), 112.94 (CN), 64.33 (OCH2CH3), 63.17 (OCH2CH3), 63.02 (OCH2CH3), 48.07 (OCCCO), 39.37 (ArCH), 30.91 (NCCCO), 14.18 (OCH2CH3), 14.08 (OCH2CH3), 13.77 (OCH2CH3).
2.3.4. Triethyl 3-(2-chlorophenyl)-2-cyanocyclopropane-1,1,2-tricarboxylate (22b)
Ethyl 3-(2-chlorophenyl)-2-cyanoacrylate (
21b) (0.118 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in dry dichloromethane (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, giving a yellow oil. Yields and ee values can be observed in
Table 3.
1H NMR (500 MHz, CDCl3) δ 7.45–7.37 (m, 2H, ArH), 7.33–7.25 (m, 2H, ArH), 4.45–4.23 (m, 4H, OCH2CH3), 4.19 (q, J = 7.3 Hz, 2H, OCH2CH3), 3.86 (s, 1H, ArCH), 1.39 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.30 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.16 (t, J = 7.2 Hz, 3H, OCH2CH3).
13C NMR (126 MHz, CDCl3) δ 164.22 (COOCH2), 163.62 (COOCH2), 162.71 (COOCH2), 135.17 (ArCCl), 130.20 (ArC), 129.95 (ArC), 129.90 (ArC), 128.04 (ArC), 126.88 (ArC), 112.57 (CN), 64.25 (OCH2CH3), 63.15 (OCH2CH3), 62.75 (OCH2CH3), 47.61 (OCCCO), 38.30 (ArCH), 31.49 (NCCCO), 14.04 (OCH2CH3), 13.89 (OCH2CH3), 13.60 (OCH2CH3).
2.3.5. Triethyl 3-(3-chlorophenyl)-2-cyanocyclopropane-1,1,2-tricarboxylate (22c)
Ethyl 3-(3-chlorophenyl)-2-cyanoacrylate (
21c) (0.118 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in dry dichloromethane (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1. mmol) was added. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, giving a yellow oil. Yields and ee values can be observed in
Table 3.
1H NMR (500 MHz, CDCl3) δ 7.35 (s, 1H, ArH), 7.33–7.28 (m, 3H, ArH), 4.42–4.21 (m, 4H, OCH2CH3), 4.21–4.12 (m, 2H, OCH2CH3), 3.87 (s, 1H, ArCH), 1.38 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.14 (t, J = 7.1 Hz, 3H, OCH2CH3).13C NMR (126 MHz, CDCl3) δ 164.07 (COOCH2), 163.61 (COOCH2), 162.32 (COOCH2), 134.65 (ArCCl), 131.64 (ArC), 130.11 (ArC), 128.97 (ArC), 128.92 (ArC), 126.89 (ArC), 112.42, (CN) 64.34 (OCH2CH3), 63.26 (OCH2CH3), 63.00 (OCH2CH3), 47.66 (OCCCO), 38.31 (ArCH), 30.64 (NCCCO), 13.99 (OCH2CH3), 13.89 (OCH2CH3), 13.70 (OCH2CH3).
2.3.6. Triethyl 3-(4-chlorophenyl)-2-cyanocyclopropane-1,1,2-tricarboxylate (22d)
Ethyl 3-(4-chlorophenyl)-2-cyanoacrylate (
21d) (0.118 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in dry dichloromethane (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, giving a yellow oil. Yields and ee values can be observed in
Table 3.
1H NMR (500 MHz, CDCl3) δ 7.35–7.31 (m, 4H, ArH), 4.42–4.19 (m, 4H, OCH2CH3), 4.15 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.86 (s, 1H, ArCH), 1.38 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.13 (t, J = 7.0 Hz, 3H, OCH2CH3).
13C NMR (126 MHz, CDCl3) δ 164.11 (COOCH2), 163.67 (COOCH2), 162.36 (COOCH2), 134.75 (ArCCl), 130.09 (2 × ArC), 129.05 (2 × ArC), 128.22 (ArC), 112.56 (CN), 64.31 (OCH2CH3), 63.20 (OCH2CH3), 62.96 (OCH2CH3), 47.76 (OCCCO), 38.39 (ArCH), 30.73 (NCCCO), 13.99 (OCH2CH3), 13.89 (OCH2CH3), 13.69 (OCH2CH3).
2.3.7. Diethyl 1′,3′-dioxo-3-phenyl-1′,3′-dihydrospiro[cyclopropane-1,2′-indene]-2,2-dicarboxylate (24a)
Benzylidene-1,3-indanedione (
23a) (0.117 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in a mixture of dry diethyl ether–tetrahydrofurane 4:1 (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The THF was added first, and then the mixture was diluted with the appropriate amount of Et
2O. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, resulting in a pale-yellow oil. Yields and ee values can be observed in
Table 4.
1H NMR (500 MHz, CDCl3) δ 8.04–7.93 (m, 2H, ArH), 7.88–7.81 (m, 2H, ArH), 7.35–7.25 (m, 5H, ArH), 4.40–4.25 (m, 2H, OCH2), 4.25–4.15 (m, 2H, OCH2), 4.16 (s, 1H, PhCH), 1.31 (t, J = 7.2 Hz, 3H, CH3), 1.18 (t, J = 7.2 Hz, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ 194.69 (ArCO), 191.41 (ArCO), 164.37 (COOC2H5), 162.53 (COOC2H5), 143.10 (ArC), 141.24 (ArC), 135.58 (ArC), 135.25 (ArC), 130.17 (ArC), 127.86 (ArC), 123.18 (ArC), 62.89 (CH2CH3), 62.39 (CH2CH3), 51.74 (ArOCCCOAr), 45.23(OOCCCOO), 42.35 (PhCH), 13.98 (CH2CH3), 13.71 (CH2CH3).
2.3.8. Diethyl 3-(4-nitrophenyl)-1′,3′-dioxo-1′,3′-dihydrospiro[cyclopropane-1,2′-indene]-2,2-dicarboxylate (24b)
2-(4-Nitrobenzylidene)-1
H-indene-1,3(2
H)-dione (
23b) (0.140 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in a mixture of dry diethyl ether–tetrahydrofurane 4:1 (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The THF was added first and then the mixture was diluted with the appropriate amount of Et
2O. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, resulting in an orange oil. Yields and ee values can be observed in
Table 4.
1H NMR (500 MHz, CDCl3), δ 8.15 (d, J = 8.5 Hz, 2H, ArH), 8.07–8.02 (m, 1H, ArH), 8.02–7.97 (m, 1H, ArH), 7.93–7.88 (m, 2H, ArH), 7.52 (d, J = 8.5 Hz, 2H, ArH), 4.37–4.28 (m, 2H, OCH2), 4.23–4.14 (m, 2H, OCH2), 4.17 (s, 1H, ArCH), 1.32 (t, J = 7 Hz, 3H, CH2CH3), 1.17 (t, J = 7 Hz, 3H, CH2CH3).
13C NMR (75 MHz, CDCl3), δ 193.71 (ArCO), 191.57 (ArCO), 163.74 (COOC2H5), 162.16 (COOC2H5), 147.38 (ArC), 142.89 (ArC), 141.44 (ArC), 137.87 (ArC), 135.78 (ArC), 131.37 (ArC), 123.38 (ArC), 123.03 (ArC), 63.24 (CH2CH3), 62.73 (CH2CH3), 51.33 (ArOCCCOAr), 44.94 (OOCCCOO), 40.47 (ArCH), 14.00 (CH2CH3), 13.73 (CH2CH3).
2.3.9. Diethyl 3-(4-methoxyphenyl)-1′,3′-dioxo-1′,3′-dihydrospiro[cyclopropane-1,2′-indene]-2,2-dicarboxylate (24c)
2-(4-Methoxybenzylidene)-1
H-indene-1,3(2
H)-dione (
23c) (0.132 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol %) were dissolved in a mixture of dry diethyl ether–tetrahydrofurane 4:1 (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The THF was added first, and then the mixture was diluted with the appropriate amount of Et
2O. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, resulting in a yellow oil. Yields and ee values can be observed in
Table 4.
1H NMR (500 MHz, CDCl3), δ 8.02–7.98 (m, 1H, ArH), 7.97–7.93 (m, 1H, ArH), 7.86–7.81 (m, 2H, ArH), 7.25 (d, J = 8.5 Hz, 2H, ArH), 6.82 (d, J = 8.5 Hz, 2H, ArH), 4.36–4.26 (m, 2H, OCH2), 4.26–4.16 (m, 2H, OCH2), 4.10 (s, 1H, ArCH), 3.78 (s, 3H, ArOCH3), 1.31 (t, J = 7 Hz, 3H, CH2CH3), 1.21 (t, J = 7 Hz, 3H, CH2CH3).
13C NMR (75 MHz, CDCl3), δ 194.79 (ArCO), 191.57 (ArCO), 164.47 (COOC2H5), 162.75 (COOC2H5), 156.88 (ArC), 142.93 (ArC), 135.88 (ArC), 135.72 (ArC), 125.14 (ArC), 123.11 (ArC), 114.66 (ArC), 62.77 (CH2CH3), 62.38 (CH2CH3), 55.64 (ArOCH3), 51.83 (ArOCCCOAr), 45.20 (OOCCCOO), 41.47 (ArCH), 13.91 (CH2CH3), 13.52 (CH2CH3).
2.3.10. Diethyl 1′,3′-dioxo-3-(4-methylphenyl)-1′,3′-dihydrospiro[cyclopropane-1,2′-indene]-2,2-dicarboxylate (24d)
2-(4-Methylbenzylidene)-1
H-indene-1,3(2
H)-dione (
23d) (0.124 g, 0.5 mmol), diethyl bromomalonate (
19) (130 µL, 0.75 mmol), and the appropriate crown ether (10 mol%) were dissolved in a mixture of dry diethyl ether–tetrahydrofurane 4:1 (3 mL), and anhydrous Na
2CO
3 (0.11 g, 1 mmol) was added. The THF was added first, and then the mixture was diluted with the appropriate amount of Et
2O. The reaction mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–EtOAc 4:1). After completion of the reaction, the mixture was filtered, and the filtrate was concentrated. The crude product was purified by preparative TLC using hexane–EtOAc mixture (5:1) as the eluent, resulting in a pale-yellow oil. Yields and ee values can be observed in
Table 4.
1H NMR (500 MHz, CDCl3), δ 8.02–7.98 (m, 1H, ArH), 7.97–7.93 (m, 1H, ArH), 7.87–7.81 (m, 2H, ArH), 7.21 (d, J = 7.5 Hz, 2H, ArH), 7.09 (d, J = 7.5 Hz, 2H, ArH), 4.36–4.26 (m, 2H, OCH2), 4.26–4.17 (m, 2H, OCH2), 4.12 (s, 1H, ArCH), 2.31 (s, 3H, ArCH3), 1.31 (t, J = 7 Hz, 3H, CH2CH3), 1.20 (t, J = 7 Hz, 3H, CH2CH3)
13C NMR (75 MHz, CDCl3), δ 194.65 (ArCO), 191.58 (ArCO), 164.52 (COOC2H5), 162.76 (COOC2H5), 142.91 (ArC), 139.78 (ArC), 135.60 (ArC), 133.48 (ArC), 127.65 (ArC), 125.04 (ArC), 123.22 (ArC), 62.76 (CH2CH3), 62.40 (CH2CH3), 51.81 (ArOCCCOAr), 45.20 (OOCCCOO), 41.42 (ArCH), 20.49 (ArCH3), 13.93 (CH2CH3), 13.60 (CH2CH3)
2.3.11. Diethyl 2-cyano-3-phenyl-2-(phenylsulfonyl) cyclopropane-1,1-dicarboxylate (26)
3-Phenyl-2-(phenylsulfonyl) acrylonitrile (
25) (0.134 g, 0.5 mmol) was dissolved in dry dichloromethane (3 mL), and to this solution, 130 µL (0.75 mmol) of diethyl bromomalonate (
19) along with the appropriate crown ether (10 mol%) were added. To start the reaction, 0.11 g (1 mmol) of sodium carbonate was added, and the mixture was stirred at room temperature. The progress of the reaction was monitored by TLC (hexane–EtOAc 4:1). After completion, the mixture was filtered and diluted with dichloromethane. The organic solution was washed with 10% HCl (3 × 10 mL), dried using Na
2SO
4, filtered again and concentrated. Crude product was purified by preparative TLC using hexane–EtOAc 5:1 as the eluent to obtain an off-white solid. Yields and ee values can be observed in
Table 5.
1H NMR (500 MHz, CDCl3) δ 8.13 (dd, J = 8.5, 1.3 Hz, 2H, ArH), 7.81 (tt, J = 7.5, 1.2 Hz, 1H, ArH), 7.68 (t, J = 7.9 Hz, 2H, ArH), 7.31 (dd, J = 4.9, 2.0 Hz, 3H, ArH), 7.17–7.11 (m, 2H, ArH), 4.50–4.37 (m, 2H, OCH2), 4.16 (s, 1H, PhCH), 4.13 (qd, J = 7.1, 2.6 Hz, 2H, OCH2), 1.42 (t, J = 7.1 Hz, 3H, CH3), 1.10 (t, J = 7.1 Hz, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ 162.94 (ArCO), 162.06 (ArCO), 136.12 (ArC), 135.59 (ArC), 130.13 (ArC), 129.59 (ArC), 128.98 (ArC), 128.94 (ArC), 128.65 (ArC), 128.27 (ArC), 111.52 (CN), 63.55 (OCH2CH3), 63.46 (OCH2CH3), 48.24 (NCCS), 47.58 (OCCCO), 36.95 (PhCH), 13.82 (OCH2CH3), 13.60 (OCH2CH3).
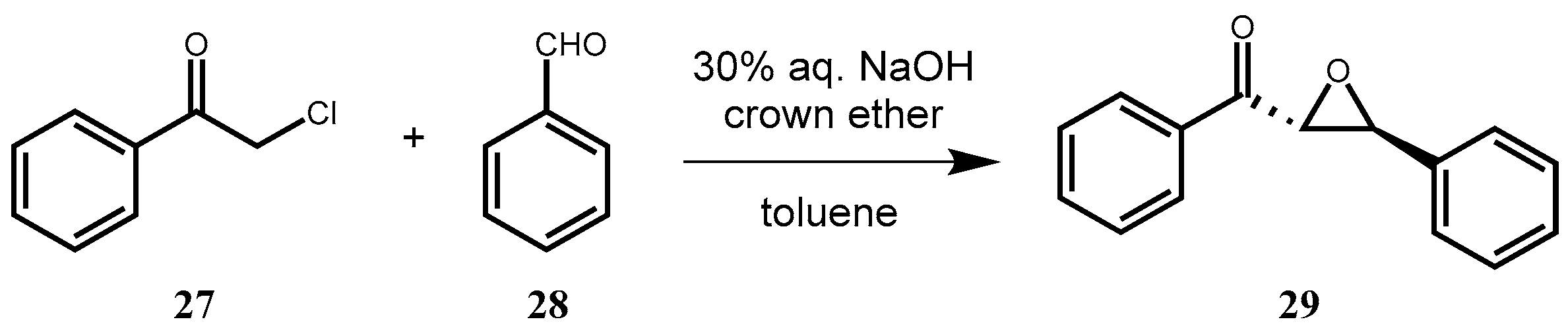
2.3.12. (2R,3S)-Phenyl(3-phenyloxirane-2-yl) Methanone (29) via Darzens-Condensation
Phenacyl chloride (
27) (0.077 g, 0.5 mmol), benzaldehyde (
28) (76 µL, 0.75 mmol) and the appropriate crown catalyst (10 mol%) were dissolved in toluene (1.5 mL). Then, 30% aqueous NaOH solution (0.5 mL) was added, and the mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–ethyl-acetate 10:1). After completion of the reaction, the mixture was diluted with toluene (7 mL) and water (3 mL), and the phases were separated. The organic layer was washed with 10% aqueous HCl solution (3 × 10 mL), dried (Na
2CO
3 and Na
2SO
4), filtered, and concentrated in vacuum. The crude product was purified by preparative TLC (hexane–ethyl-acetate 10:1) to give a yellowish-white powder. Yields and ee values can be observed in
Table 6.
1H NMR (CDCl3, 500 MHz), δ 7.97–7.94 (m, 2H, ArH), 7.60–7.56 (m, 1H, ArH), 7.46–7.44 (m, 2H, ArH), 7.38–7.32 (m, 5H), 4.26 (d, J = 1.9 Hz, 1H, COCH), 4.05 (d, J = 1.9 Hz, 1H, ArCH)
13C NMR (CDCl3, 75 MHz), δ 193.06 (C=O), 135.48 (ArC), 133.97 (ArC), 129.04 (ArC), 128.86 (ArC), 128.76 (ArC), 128.33 (ArC), 125.78 (ArC), 61.00 (OCCO), 59.34 (PhCO).
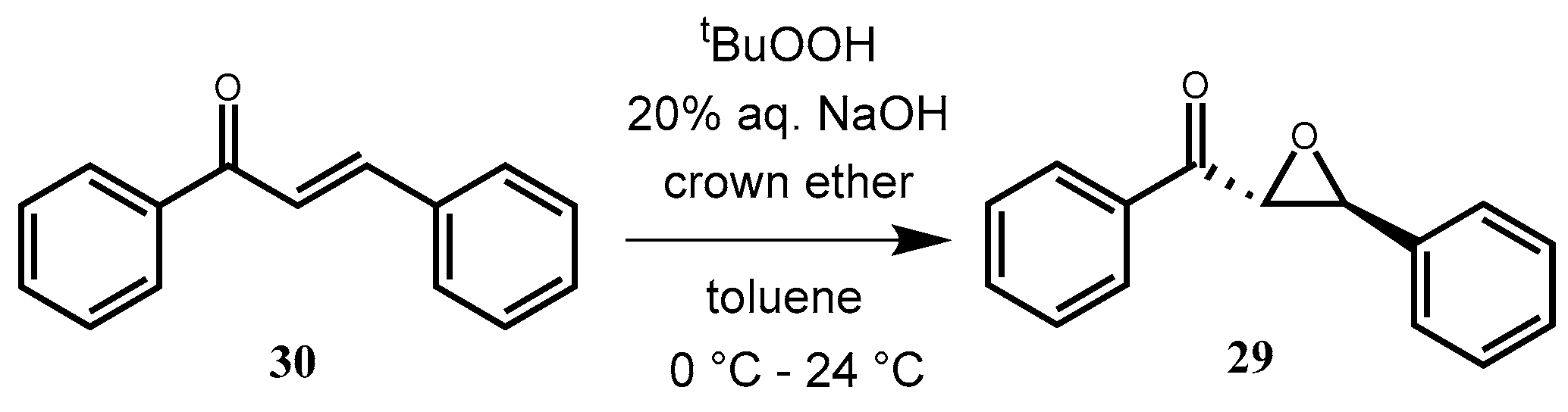
2.3.13. (2R,3S)-Phenyl(3-phenyloxirane-2-yl) Methanone (29) via Epoxidation
trans-Chalcone (
30) (0.104 g, 0.5 mmol) and the appropriate crown catalyst (10 mol%) were dissolved in toluene (3 mL), and then 5.5 M
tert-butylhydroperoxide solution (0.25 mL, in decane) and 20% aqueous NaOH solution (0.5 mL) were added. The mixture was stirred at room temperature. The reaction was monitored by TLC (hexane–ethyl-acetate 10:1). After completion, the reaction mixture was diluted with toluene (7 mL) and water (3 mL), and the phases were separated. The organic layer was washed with 10% aqueous HCl solution (3 × 10 mL), dried (Na
2CO
3 and Na
2SO
4), filtered, and concentrated in vacuum. The crude product was purified by preparative TLC (hexane–ethyl-acetate 10:1) to give a yellowish-white powder. Yields and ee values can be observed in
Table 7.
1H NMR (CDCl3, 500 MHz), δ 7.97–7.94 (m, 2H, ArH), 7.60–7.56 (m, 1H, ArH), 7.46–7.44 (m, 2H, ArH), 7.38–7.32 (m, 5H), 4.26 (d, J = 1.9 Hz, 1H, COCH), 4.05 (d, J = 1.9 Hz, 1H, ArCH)
13C NMR (CDCl3, 75 MHz), δ 193.06 (C=O), 135.48 (ArC), 133.97 (ArC), 129.04 (ArC), 128.86 (ArC), 128.76 (ArC), 128.33 (ArC), 125.78 (ArC), 61.00 (OCCO), 59.34 (PhCO).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}