
An ONIOM-Based High-Level Thermochemistry Study on Hydrogen Abstraction Reactions of Large Straight-Chain Alkanes by Hydrogen, Hydroxyl, and Hydroperoxyl Radicals

Abstract
:1. Introduction
2. Computational Methods
2.1. Potential Energy Surface
2.2. QCISD(T)/CBS and ONIOM Energies
2.2.1. QCISD(T)/CBS Single-Point Energies
2.2.2. ONIOM[QCISD(T)/CBS] Single-Point Energies
3. Results and Discussion
3.1. Hydrogen Abstraction Reactions of n-CnH2n+2 + H (n = 1–16)
3.1.1. Validation and Comparison of Two [QCISD(T)/CBS] Methods
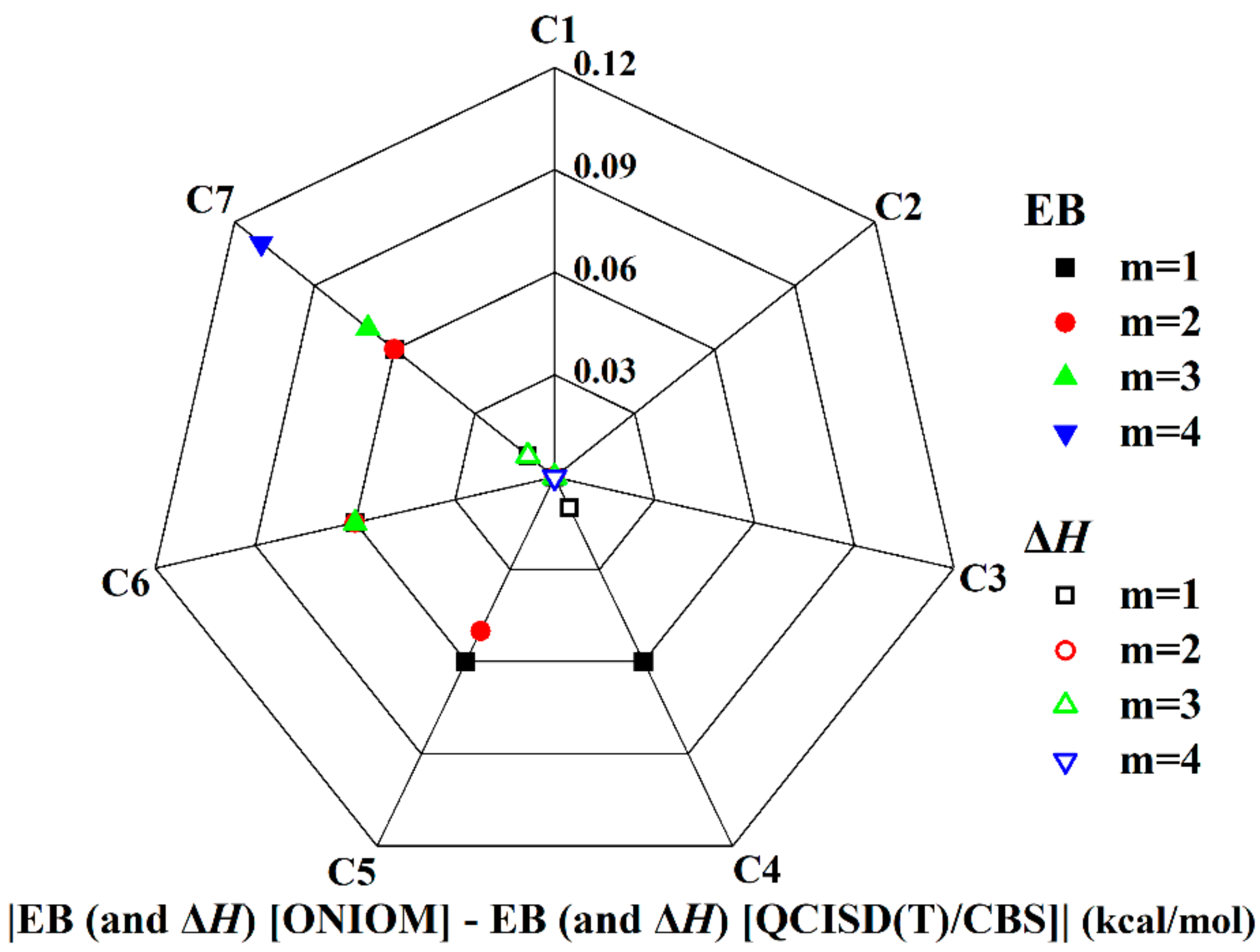
3.1.2. Validation of ONIOM Energies of n-CnH2n+2 + H (n = 1–9)
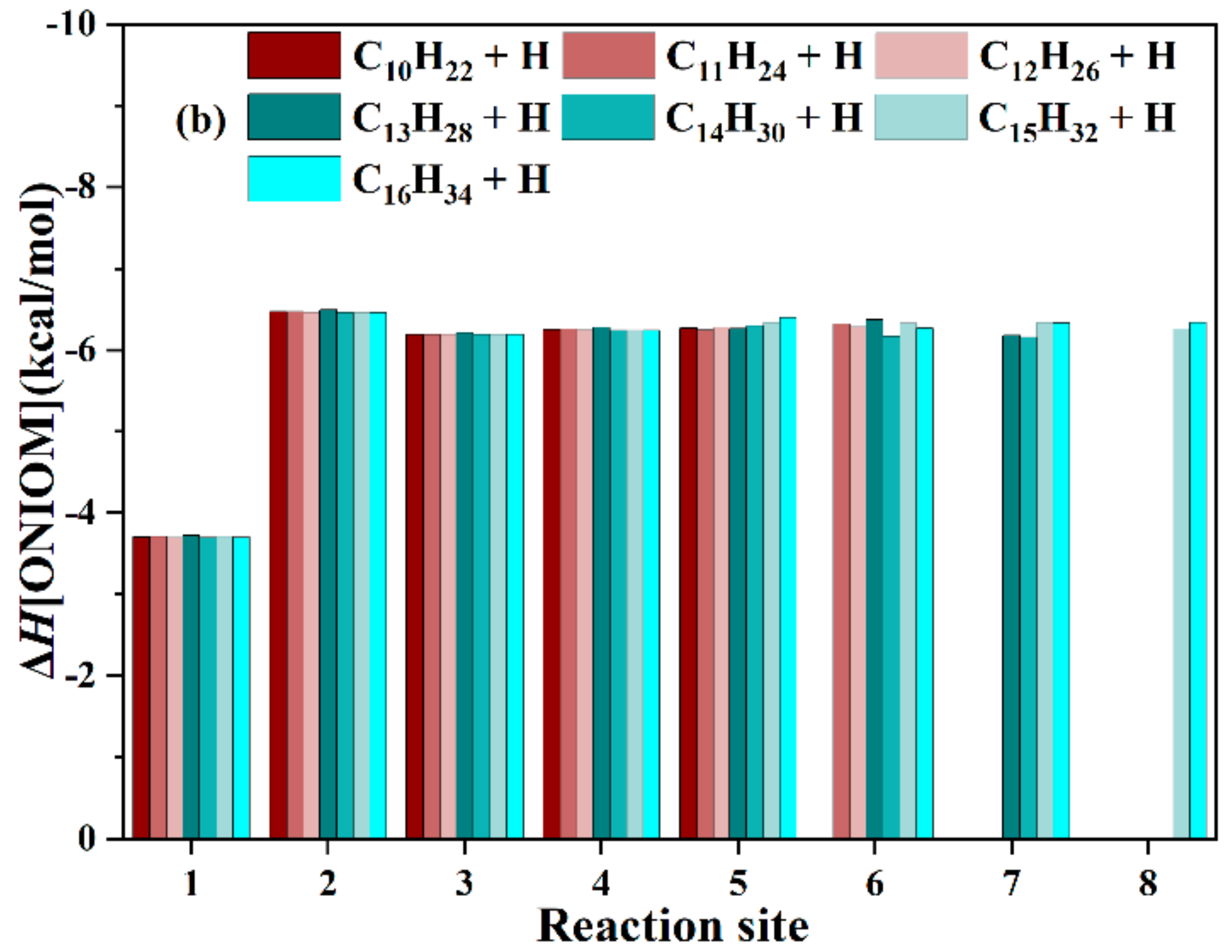
3.1.3. ONIOM Energies of n-CnH2n+2 + H (n = 10–16)
3.2. Hydrogen Abstraction Reactions of n-CnH2n+2 + OH (n = 1–16)
3.2.1. Validation and Comparison of Two [QCISD(T)/CBS] Methods
3.2.2. Validation of ONIOM Energies of n-CnH2n+2 + OH (n = 1–8)
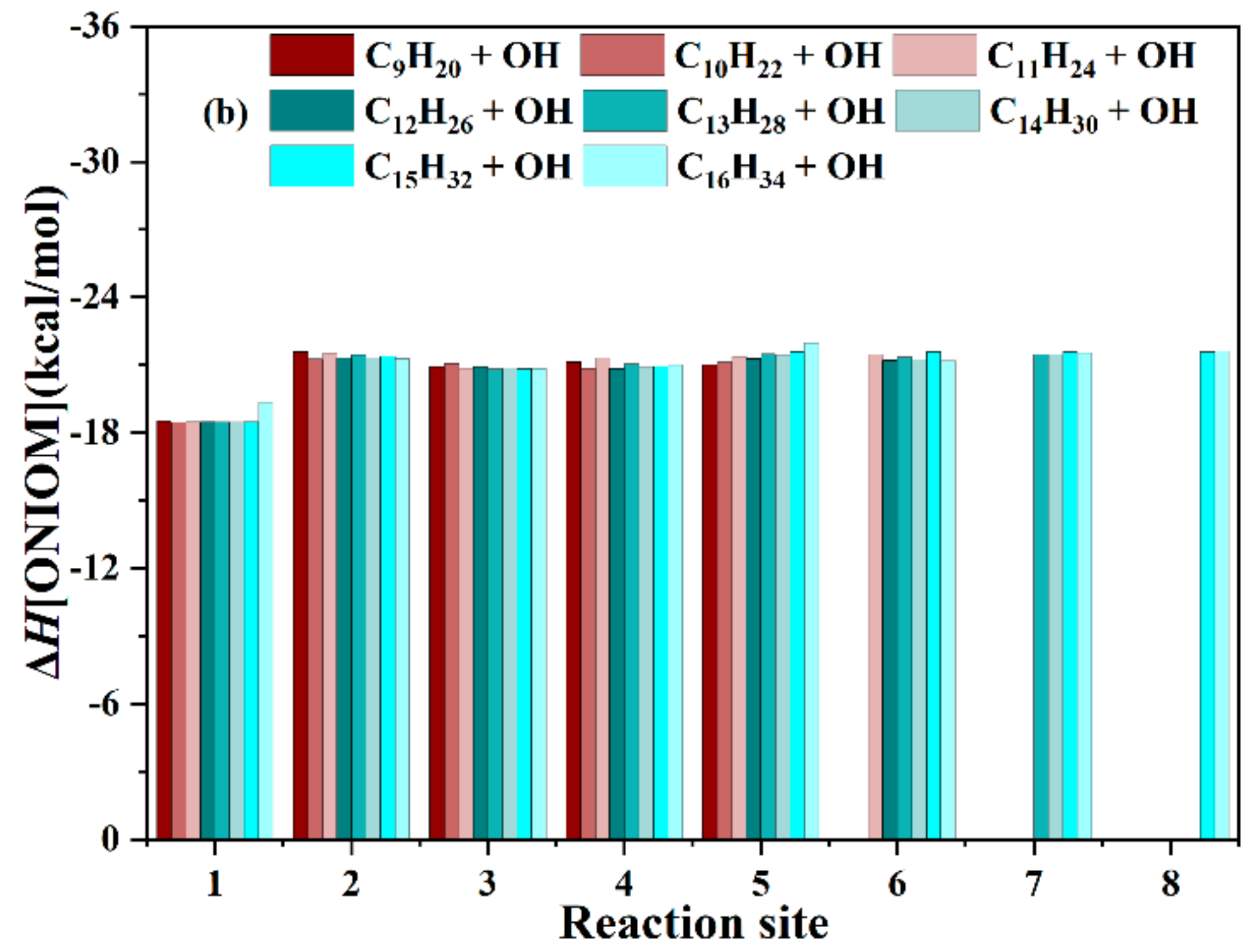
3.2.3. ONIOM Energies of n-CnH2n+2 + OH (n = 9–16)
3.3. Hydrogen Abstraction Reactions of n-CnH2n+2 + HO2 (n = 1–16)
3.3.1. Validation and Comparison of Two [QCISD(T)/CBS] Methods
3.3.2. Validation of ONIOM Energies of CnH2n+2 + HO2 (n = 1–7)
3.3.3. ONIOM Energies of CnH2n+2 + HO2 (n = 8–16)
3.4. Comparision with Literature Data
3.4.1. Comparison with Data from Kinetic Modelling
3.4.2. Comparison with Data from Ab Initio Calculations
3.4.3. Comparison with Saturated Methyl Ester
3.4.4. Comparison to Unsaturated Methyl Ester
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| CAP | Chemically active portion |
| CBS | Complete basis set |
| CCSD(T) | Coupled cluster with single, double, and perturbative triple excitations |
| DFT | Density functional theory |
| DZ | Double Zeta (referring to the quality of a basis set) |
| EB | Energy barrier |
| ΔH | Heat of reaction |
| IRC | Intrinsic reaction coordinate |
| MP2 | Second-order Møller–Plesset perturbation theory correction |
| ONIOM | Our own n-layered integrated molecular orbital and molecular mechanics |
| pV | Polarized valence (referring to the type of functions in a basis set) |
| QCISD(T) | Quadratic configuration interaction with single and double excitations and a perturbative estimate of triple excitations |
| QZ | Quadruple Zeta (referring to the quality of a basis set) |
| TZ | Triple Zeta (referring to the quality of a basis set) |
| ZPE | Zero-point energy |
| cc | Correlation-consistent (referring to the type of basis set) |
References
- Westbrook, C.K.; Dryer, F.L. Chemical kinetics and modeling of combustion processes. Symp. Int. Combust. 1981, 18, 749–767. [Google Scholar] [CrossRef]
- Dagaut, P.; Cathonnet, M. The ignition, oxidation, and combustion of kerosene: A review of experimental and kinetic modeling. Prog. Energy Combust. Sci. 2006, 32, 48–92. [Google Scholar] [CrossRef]
- Edwards, T.; Colket, M.; Cernansky, N.; Dryer, F.; Egolfopoulos, F.; Friend, D.; Law, E.; Lenhert, D.; Lindstedt, P.; Pitsch, H.; et al. Development of an Experimental Database and Kinetic Models for Surrogate Jet Fuels. In Proceedings of the 45th AIAA Aerospace Sciences Meeting and Exhibit, AIAA-2007-0770, Reno, NV, USA, 8–11 January 2007. [Google Scholar]
- Farrell, J.T.; Cernansky, N.P.; Dryer, F.L.; Law, C.K.; Friend, D.G.; Hergart, C.A.; McDavid, R.M.; Patel, A.K.; Mueller, C.J.; Pitsch, H. Development of an Experimental Database and Kinetic Models for Surrogate Diesel Fuels; SAE Technical Paper 2007-01-0201; SAE: Warrendale, PA, USA, 2007. [Google Scholar] [CrossRef]
- Pitz, W.J.; Cernansky, N.P.; Dryer, F.L.; Egolfopoulos, F.N.; Farrell, J.T.; Friend, D.G.; Pitsch, H. Development of an Experimental Database and Chemical Kinetic Models for Surrogate Gasoline Fuels; SAE Technical Paper 2007-01-0175; SAE: Warrendale, PA, USA, 2007. [Google Scholar]
- Pitz, W.J.; Mueller, C.J. Recent progress in the development of diesel surrogate fuels. Prog. Energy Combust. Sci. 2011, 37, 330–350. [Google Scholar] [CrossRef]
- Sarathy, S.; Farooq, A.; Kalghatgi, G.T. Recent progress in gasoline surrogate fuels. Prog. Energy Combust. Sci. 2018, 65, 67–108. [Google Scholar] [CrossRef]
- Mehl, M.; Pitz, W.J.; Westbrook, C.K.; Curran, H. Kinetic modeling of gasoline surrogate components and mixtures under engine conditions. Proc. Combust. Inst. 2011, 33, 193–200. [Google Scholar] [CrossRef]
- Jerzembeck, S.; Peters, N.; Pepiotdesjardins, P.; Pitsch, H. Laminar burning velocities at high pressure for primary reference fuels and gasoline: Experimental and numerical investigation. Combust. Flame 2009, 156, 292–301. [Google Scholar] [CrossRef]
- Sileghem, L.; Alekseev, V.; Vancoillie, J.; Van Geem, K.; Nilsson, E.; Verhelst, S.; Konnov, A. Laminar burning velocity of gasoline and the gasoline surrogate components iso-octane, n-heptane and toluene. Fuel 2013, 112, 355–365. [Google Scholar] [CrossRef]
- Lemaire, R.; Faccinetto, A.; Therssen, E.; Ziskind, M.; Focsa, C.; Desgroux, P. Experimental comparison of soot formation in turbulent flames of Diesel and surrogate Diesel fuels. Proc. Combust. Inst. 2009, 32, 737–744. [Google Scholar] [CrossRef]
- Mati, K.; Ristori, A.; Gaïl, S.; Pengloan, G.; Dagaut, P. The oxidation of a diesel fuel at 1–10 atm: Experimental study in a JSR and detailed chemical kinetic modeling. Proc. Combust. Inst. 2007, 31, 2939–2946. [Google Scholar] [CrossRef]
- Honnet, S.; Seshadri, K.; Niemann, U.; Peters, N. A surrogate fuel for kerosene. Proc. Combust. Inst. 2009, 32, 485–492. [Google Scholar] [CrossRef]
- Dagaut, P.; El Bakali, A.; Ristori, A. The combustion of kerosene: Experimental results and kinetic modelling using 1- to 3-component surrogate model fuels. Fuel 2006, 85, 944–956. [Google Scholar] [CrossRef]
- Strelkova, M.I.; Kirillov, I.A.; Potapkin, B.V.; Safonov, A.A.; Sukhanov, L.P.; Umanskiy, S.Y.; Deminsky, M.A.; Dean, A.J.; Varatharajan, B.; Tentner, A.M. Detailed and reduced mechanisms of jet a combustion at high temperatures. Combust. Sci. Technol. 2008, 180, 1788–1802. [Google Scholar] [CrossRef]
- Mawid, M.; Sekar, B. Development of a detailed JP-8/Jet-A chemical kinetic mechanism for high pressure conditions in gas turbine combustors. In Proceedings of the ASME Turbo Expo 2006: Power for Land, Sea, and Air, Barcelona, Spain, 8–11 May 2006; pp. 369–388. [Google Scholar]
- Dahm, K.; Virk, P.; Bounaceur, R.; Battin-Leclerc, F.; Marquaire, P.; Fournet, R.; Daniau, E.; Bouchez, M. Experimental and modelling investigation of the thermal decomposition of n-dodecane. J. Anal. Appl. Pyrolysis 2004, 71, 865–881. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Dryer, F.L. Chemical kinetic modeling of hydrocarbon combustion. Prog. Energy Combust. Sci. 1984, 10, 1–57. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. Prog. Energy Combust. Sci. 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Curran, H.; Gaffuri, P.; Pitz, W.; Westbrook, C. A Comprehensive modeling study of n-heptane oxidation. Combust. Flame 1998, 114, 149–177. [Google Scholar] [CrossRef]
- Curran, H.; Gaffuri, P.; Pitz, W.; Westbrook, C. A comprehensive modeling study of iso-octane oxidation. Combust. Flame 2002, 129, 253–280. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Pitz, W.J.; Herbinet, O.; Curran, H.J.; Silke, E.J. A comprehensive detailed chemical kinetic reaction mechanism for combustion of n-alkane hydrocarbons from n-octane to n-hexadecane. Combust. Flame 2009, 156, 181–199. [Google Scholar] [CrossRef]
- Ritter, E.R.; Bozzelli, J.W. THERM: Thermodynamic property estimation for gas phase radicals and molecules. Int. J. Chem. Kinet. 1991, 23, 767–778. [Google Scholar] [CrossRef]
- Lay, T.H.; Bozzelli, J.W.; Dean, A.M.; Ritter, E.R. Hydrogen atom bond increments for calculation of thermodynamic properties of hydrocarbon radical species. J. Phys. Chem. 1995, 99, 14514–14527. [Google Scholar] [CrossRef]
- Benson, S.W. Thermochemical Kinetics; Wiley: Hoboken, NJ, USA, 1976. [Google Scholar]
- Papajak, E.; Truhlar, D.G. What are the most efficient basis set strategies for correlated wave function calculations of reaction energies and barrier heights? J. Chem. Phys. 2012, 137, 064110. [Google Scholar] [CrossRef] [PubMed]
- Zádor, J.; Taatjes, C.A.; Fernandes, R.X. Kinetics of elementary reactions in low-temperature autoignition chemistry. Prog. Energy Combust. Sci. 2011, 37, 371–421. [Google Scholar] [CrossRef]
- Goldsmith, C.F.; Magoon, G.R.; Green, W.H. Database of Small Molecule Thermochemistry for Combustion. J. Phys. Chem. A 2012, 116, 9033–9057. [Google Scholar] [CrossRef] [PubMed]
- El-Nahas, A.M.; Navarro, M.V.; Simmie, J.M.; Bozzelli, J.W.; Curran, H.J.; Dooley, S.; Metcalfe, W. Enthalpies of formation, bond dissociation energies and reaction paths for the decomposition of model biofuels: Ethyl propanoate and methyl butanoate. J. Phys. Chem. A 2007, 111, 3727–3739. [Google Scholar] [CrossRef]
- Osmont, A.; Catoire, L.; Dagaut, P. Thermodynamic data for the modeling of the thermal decomposition of biodiesel. 1. Saturated and monounsaturated FAMEs. J. Phys. Chem. A 2009, 114, 3788–3795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, P. Towards high-level theoretical studies of large biodiesel molecules: An ONIOM [QCISD(T)/CBS:DFT] study of hydrogen abstraction reactions of CnH2n+1COOCmH2m+1 + H. Phys. Chem. Chem. Phys. 2015, 17, 200–208. [Google Scholar] [CrossRef]
- Vreven, T.; Byun, K.S.; Komáromi, I.; Dapprich, S.; Montgomery, J.A., Jr.; Morokuma, K.; Frisch, M.J. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput. 2006, 2, 815–826. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, Q.; Chi, Y.; Zhang, P. Toward high-level theoretical studies of large biodiesel molecules: An ONIOM [QCISD(T)/CBS:DFT] study of the reactions between unsaturated methyl esters (CnH2n−1COOCH3) and hydrogen radical. J. Phys. Chem. A 2018, 122, 4882–4893. [Google Scholar] [CrossRef]
- He, C.; Chi, Y.; Zhang, P. Approximate reconstruction of torsional potential energy surface based on voronoi tessellation. Proc. Combust. Inst. 2020, 38, 757–766. [Google Scholar] [CrossRef]
- Meng, Q.; Zhang, L.; Chen, Q.; Chi, Y.; Zhang, P. Influence of torsional anharmonicity on the reactions of methyl butanoate with hydroperoxyl radical. J. Phys. Chem. A 2020, 124, 8643–8652. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Iparraguirre, J.; Curran, H.J.; Klopper, W.; Simmie, J.M. Accurate benchmark calculation of the reaction barrier height for hydrogen abstraction by the hydroperoxyl radical from methane. implications for CnH2n+2 where n = 2 → 4. J. Phys. Chem. A 2008, 112, 7047–7054. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N. Are reaction rate coefficients additive? Revised transition state theory calculations for OH + alkane reactions. Int. J. Chem. Kinet. 1991, 23, 397–417. [Google Scholar] [CrossRef]
- Cohen, N. The use of transition-state theory to extrapolate rate coefficients for reactions of H atoms with alkanes. Int. J. Chem. Kinet. 1991, 23, 683–700. [Google Scholar] [CrossRef]
- Liu, D.; Khaled, F.; Giri, B.R.; Assaf, E.; Fittschen, C.; Farooq, A. H-abstraction by OH from large branched alkanes: Overall rate measurements and site-specific tertiary rate calculations. J. Phys. Chem. A 2017, 121, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Badra, J.; Elwardany, A.; Farooq, A. Shock tube measurements of the rate constants for seven large alkanes + OH. Proc. Combust. Inst. 2015, 35, 189–196. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Michael, J.V. Rate constants for OH with selected large alkanes: Shock-tube measurements and an improved group scheme. J. Phys. Chem. A 2009, 113, 5047–5060. [Google Scholar] [CrossRef]
- Hashemi, H.; Christensen, J.M.; Glarborg, P.; Gersen, S.; Essen, M.; Wand, Z.; Ju, Y. High-pressure oxidation of n-butane. Int. J. Chem. Kinet. 2023, 55, 688–706. [Google Scholar] [CrossRef]
- Hashemi, H.; Christensen, J.M.; Harding, L.B.; Klippenstein, S.J.; Glarborg, P. High-pressure oxidation of propane. Proc. Combust. Inst. 2018, 37, 461–468. [Google Scholar] [CrossRef]
- Hashemi, H.; Jacobsen, J.G.; Rasmussen, C.T.; Christensen, J.M.; Glarborg, P.; Gersen, S.; van Essen, M.; Levinsky, H.B.; Klippenstein, S.J. High-pressure oxidation of ethane. Combust. Flame 2017, 182, 150–166. [Google Scholar] [CrossRef]
- Hashemi, H.; Christensen, J.M.; Gersen, S.; Levinsky, H.; Klippenstein, S.J.; Glarborg, P. High-pressure oxidation of methane. Combust. Flame 2016, 172, 349–364. [Google Scholar] [CrossRef]
- Peukert, S.L.; Sivaramakrishnan, R.; Michael, J.V. High temperature rate constants for H/D+ n-C4H10 and i-C4H10. Proc. Combust. Inst. 2015, 35, 171–179. [Google Scholar] [CrossRef]
- Khaled, F.; Giri, B.R.; Szőri, M.; Viskolcz, B.; Farooq, A. An experimental and theoretical study on the kinetic isotope effect of C2H6 and C2D6 reaction with OH. Chem. Phys. Lett. 2015, 641, 158–162. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Michael, J.; Ruscict, B. High-temperature rate constants for H/D+ C2H6 and C3H8. Int. J. Chem. Kinet. 2012, 44, 194–205. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Srinivasan, N.; Su, M.-C.; Michael, J. High temperature rate constants for OH+ alkanes. Proc. Combust. Inst. 2009, 32, 107–114. [Google Scholar] [CrossRef]
- Krasnoperov, L.N.; Michael, J. Shock tube studies using a novel multipass absorption cell: Rate constant results for OH + H2 and OH + C2H6. J. Phys. Chem. A 2004, 108, 5643–5648. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J. New analytical potential energy surface for the CH4+H hydrogen abstraction reaction: Thermal rate constants and kinetic isotope effects. J. Chem. Phys. 2002, 116, 10664–10673. [Google Scholar] [CrossRef]
- Wu, J.; Ning, H.; Xu, X.; Ren, W. Accurate entropy calculation for large flexible hydrocarbons using a multi-structural 2-dimensional torsion method. Phys. Chem. Chem. Phys. 2019, 21, 10003–10010. [Google Scholar] [CrossRef]
- Chi, Y.; Meng, Q.; He, C.; Zhang, P. Metric-based assessment method for MS-T formalism with small subsets of torsional conformers. J. Phys. Chem. A 2022, 126, 8305–8314. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, X.; Zhai, Y.; Zhang, L.; Zhang, P.; Sheng, L. A theoretical investigation on Bell-Evans-Polanyi correlations for hydrogen abstraction reactions of large biodiesel molecules by H and OH radicals. Combust. Flame 2020, 214, 394–406. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Thomson, M.J.; Pitz, W.J.; Lu, T. An experimental and kinetic modeling study of methyl decanoate combustion. Proc. Combust. Inst. 2011, 33, 399–405. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Martin, J.M.; Uzan, O. Basis set convergence in second-row compounds. The importance of core polarization functions. Chem. Phys. Lett. 1998, 282, 16–24. [Google Scholar] [CrossRef]
- Zhang, P.; Klippenstein, S.J.; Law, C.K. Ab initio kinetics for the decomposition of hydroxybutyl and butoxy radicals of n-butanol. J. Phys. Chem. A 2013, 117, 1890–1906. [Google Scholar] [CrossRef]
- Meng, Q.; Chi, Y.; Zhang, L.; Zhang, P.; Sheng, L. Towards high-level theoretical studies of large biodiesel molecules: An ONIOM/RRKM/Master-equation approach to the isomerization and dissociation kinetics of methyl decanoate radicals. Phys. Chem. Chem. Phys. 2019, 21, 5232–5242. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Battin-Leclerc, F.; Blurock, E.; Bounaceur, R.; Fournet, R.; Glaude, P.-A.; Herbinet, O.; Sirjean, B.; Warth, V. Towards cleaner combustion engines through groundbreaking detailed chemical kinetic models. Chem. Soc. Rev. 2011, 40, 4762–4782. [Google Scholar] [CrossRef]
- Nehse, M.; Warnat, J.; Chevalier, C. Kinetic modeling of the oxidation of large aliphatic hydrocarbons. Symp. Int. Combust. 1996, 26, 773–780. [Google Scholar] [CrossRef]
- Petrova, M.; Williams, F. A small detailed chemical-kinetic mechanism for hydrocarbon combustion. Combust. Flame 2006, 144, 526–544. [Google Scholar] [CrossRef]
- Warnatz, J. Resolution of gas phase and surface combustion chemistry into elementary reactions. Symp. Int. Combust. 1992, 24, 553–579. [Google Scholar] [CrossRef]
- Cohen, N. The use of transition-state theory to extrapolate rate coefficients for reactions of oh with alkanes. Int. J. Chem. Kinet. 1982, 14, 1339–1362. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11–24. [Google Scholar] [CrossRef]
- Rossi, I.; Truhlar, D.G. Improved general scaling factors and systematic tests of the SAC method for estimating correlation energies of molecules. Chem. Phys. Lett. 1995, 234, 64–70. [Google Scholar] [CrossRef]
- Wang, P.; Yan, J.; Yan, T.; Ao, C.; Zhang, L.; Lei, L. Kinetic study of H-abstraction and preliminary pyrolysis of n-decane in post-injection fuels. Combust. Flame 2024, 262, 113367. [Google Scholar] [CrossRef]
- Chi, Y.; You, X. Kinetics of hydrogen abstraction reactions of methyl palmitate and octadecane by hydrogen atoms. J. Phys. Chem. A 2019, 123, 3058–3067. [Google Scholar] [CrossRef]
- Gruber, B.; Czakó, G. Benchmark ab initio characterization of the abstraction and substitution pathways of the OH+ CH4/C2H6 reactions. Phys. Chem. Chem. Phys. 2020, 22, 14560–14569. [Google Scholar] [CrossRef]
- Seal, P.; Oyedepo, G.; Truhlar, D.G. Kinetics of the hydrogen atom abstraction reactions from 1-butanol by hydroxyl radical: Theory matches experiment and more. J. Phys. Chem. A 2013, 117, 275–282. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/ CBS]1 | [QCISD(T)/ CBS]2 | Difference | [QCISD(T)/ CBS]1 | [QCISD(T)/CBS]2 | Difference | |||
| C1 | H-R1 | H + CH4 → H2 + CH3 | 13.45 | 13.34 | −0.11 | −0.15 | −0.30 | −0.15 |
| C2 | H-R2 | H + CH3CH3 → H2 + CH2CH3 | 10.27 | 10.17 | −0.10 | −3.96 | −4.05 | −0.09 |
| C3 | H-R3 | H + CH3CH2CH3 → H2 + CH2CH2CH3 | 10.21 | 10.11 | −0.10 | −3.58 | −3.66 | −0.08 |
| H-R4 | H + CH3CH2CH3 → H2 + CH3CHCH3 | 7.58 | 7.49 | −0.09 | −6.72 | −6.74 | −0.02 | |
| C4 | H-R5 | H + CH3(CH2)2CH3 → H2 + CH2(CH2)2CH3 | 10.11 | 10.01 | −0.10 | −3.64 | −3.73 | −0.09 |
| H-R6 | H + CH3(CH2)2CH3 → H2 + CH3CHCH2CH3 | 7.47 | 7.39 | −0.08 | −6.43 | −6.45 | −0.02 | |
| C5 | H-R7 | H + CH3(CH2)3CH3 → H2 + CH2(CH2)3CH3 | 10.04 | 9.95 | −0.09 | −3.61 | −3.69 | −0.08 |
| H-R8 | H + CH3(CH2)3CH3 → H2 + CH3CH(CH2)2CH3 | 7.46 | 7.38 | −0.08 | −6.43 | −6.45 | −0.02 | |
| H-R9 | H + CH3(CH2)3CH3 → H2 + CH3CH2CHCH2CH3 | 7.46 | 7.38 | −0.08 | −6.11 | −6.13 | −0.02 | |
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/CBS]2 | ONIOM | Difference | [QCISD(T)/CBS]2 | ONIOM | Difference | |||
| C1 | H-R1 | H + CH4 → H2 + CH3 | 13.34 | 13.34 | 0.00 | −0.30 | −0.30 | 0.00 |
| C2 | H-R2 | H + CH3CH3 → H2 + CH2CH3 | 10.17 | 10.17 | 0.00 | −4.05 | −4.05 | 0.00 |
| C3 | H-R3 | H + CH3CH2CH3 → H2 + CH2CH2CH3 | 10.11 | 10.11 | 0.00 | −3.66 | −3.66 | 0.00 |
| H-R4 | H + CH3CH2CH3 → H2 + CH3CHCH3 | 7.49 | 7.49 | 0.00 | −6.74 | −6.74 | 0.00 | |
| C4 | H-R5 | H + CH3(CH2)2CH3 → H2 + CH2(CH2)2CH3 | 10.01 | 10.05 | 0.04 | −3.73 | −3.72 | 0.01 |
| H-R6 | H + CH3(CH2)2CH3 → H2 + CH3CHCH2CH3 | 7.39 | 7.39 | 0.00 | −6.45 | −6.45 | 0.00 | |
| C5 | H-R7 | H + CH3(CH2)3CH3 → H2 + CH2(CH2)3CH3 | 9.95 | 10.00 | 0.05 | −3.69 | −3.69 | 0.00 |
| H-R8 | H + CH3(CH2)3CH3 → H2 + CH3CH(CH2)2CH3 | 7.38 | 7.42 | 0.04 | −6.45 | −6.45 | 0.00 | |
| H-R9 | H + CH3(CH2)3CH3 → H2 + CH3CH2CHCH2CH3 | 7.38 | 7.38 | 0.00 | −6.13 | −6.13 | 0.00 | |
| C6 | H-R10 | H + CH3(CH2)4CH3 → H2 + CH2(CH2)4CH3 | 9.91 | 9.98 | 0.07 | −3.72 | −3.72 | 0.00 |
| H-R11 | H + CH3(CH2)4CH3 → H2 + CH3CH(CH2)3CH3 | 7.39 | 7.44 | 0.05 | −6.49 | −6.49 | 0.00 | |
| H-R12 | H + CH3(CH2)4CH3 → H2 + CH3CH2CH(CH2)2CH3 | 7.28 | 7.32 | 0.04 | −6.19 | −6.19 | 0.00 | |
| C7 | H-R13 | H + CH3(CH2)5CH3 → H2 + CH2(CH2)5CH3 | 9.92 | 9.99 | 0.07 | −3.71 | −3.70 | 0.01 |
| H-R14 | H + CH3(CH2)5CH3 → H2 + CH3CH(CH2)4CH3 | 7.33 | 7.38 | 0.05 | −6.47 | −6.47 | 0.00 | |
| H-R15 | H + CH3(CH2)5CH3 → H2 + CH3CH2CH(CH2)3CH3 | 7.22 | 7.28 | 0.06 | −6.17 | −6.18 | −0.01 | |
| H-R16 | H + CH3(CH2)5CH3 → H2 + CH3(CH2)2CH(CH2)2CH3 | 7.23 | 7.30 | 0.07 | −6.21 | −6.21 | 0.00 | |
| C8 | H-R17 | H + CH3(CH2)6CH3 → H2 + CH2(CH2)6CH3 | 9.86 | 9.94 | 0.08 | −3.71 | −3.71 | 0.00 |
| H-R18 | H + CH3(CH2)6CH3 → H2 + CH3CH(CH2)5CH3 | 7.28 | 7.34 | 0.06 | −6.48 | −6.48 | 0.00 | |
| H-R19 | H + CH3(CH2)6CH3 → H2 + CH3CH2CH(CH2)4CH3 | 7.15 | 7.21 | 0.06 | −6.17 | −6.18 | −0.01 | |
| H-R20 | H + CH3(CH2)6CH3 → H2 + CH3(CH2)2CH(CH2)3CH3 | 7.13 | 7.22 | 0.09 | −6.24 | −6.24 | 0.00 | |
| C9 | H-R21 | H + CH3(CH2)7CH3 → H2 + CH2(CH2)7CH3 | 9.77 | 9.86 | 0.09 | −3.70 | −3.70 | 0.00 |
| H-R22 | H + CH3(CH2)7CH3 → H2 + CH3CH(CH2)6CH3 | 7.13 | 7.20 | 0.07 | −6.46 | −6.47 | −0.01 | |
| H-R23 | H + CH3(CH2)7CH3 → H2 + CH3CH2CH(CH2)5CH3 | 7.02 | 7.09 | 0.07 | −6.17 | −6.18 | −0.01 | |
| H-R24 | H + CH3(CH2)7CH3 → H2 + CH3(CH2)2CH(CH2)4CH3 | 7.01 | 7.10 | 0.09 | −6.23 | −6.25 | −0.02 | |
| H-R25 | H + CH3(CH2)7CH3 → H2 + CH3(CH2)3CH(CH2)3CH3 | 6.92 | 7.04 | 0.12 | −6.20 | −6.22 | −0.02 | |
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/CBS]1 | [QCISD(T)/CBS]2 | Difference | [QCISD(T)/CBS]1 | [QCISD(T)/CBS]2 | Difference | |||
| C1 | OH-R1 | OH + CH4 → H2O + CH3 | 4.83 | 5.02 | 0.19 | −14.84 | −14.97 | −0.13 |
| C2 | OH-R2 | OH + CH3CH3 → H2O + CH2CH3 | 2.43 | 2.63 | 0.20 | −18.42 | −18.48 | −0.06 |
| C3 | OH-R3 | OH + CH3CH2CH3 → H2O + CH2CH2CH3 | 1.63 | 1.81 | 0.18 | −18.40 | −18.46 | −0.06 |
| OH-R4 | OH + CH3CH2CH3 → H2O + CH3CHCH3 | 0.29 | 0.49 | 0.20 | −21.47 | −21.47 | 0.00 | |
| C4 | OH-R5 | OH + CH3(CH2)2CH3 → H2O + CH2(CH2)2CH3 | 2.72 | 2.88 | 0.16 | −17.29 | −17.36 | −0.07 |
| OH-R6 | OH + CH3(CH2)2CH3 → H2O + CH3CHCH2CH3 | 0.81 | 0.99 | 0.18 | −20.27 | −20.27 | 0.00 | |
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/CBS]2 | ONIOM | Difference | [QCISD(T)/CBS]2 | ONIOM | Difference | |||
| C1 | OH-R1 | OH + CH4 → H2O + CH3 | 5.02 | 5.02 | 0.00 | −14.97 | −14.97 | 0.00 |
| C2 | OH-R2 | OH + CH3CH3 → H2O + CH2CH3 | 2.63 | 2.63 | 0.00 | −18.48 | −18.48 | 0.00 |
| C3 | OH-R3 | OH + CH3CH2CH3 → H2O + CH2CH2CH3 | 1.81 | 1.81 | 0.00 | −18.46 | −18.46 | 0.00 |
| OH-R4 | OH + CH3CH2CH3 → H2O + CH3CHCH3 | 0.49 | 0.49 | 0.00 | −21.47 | −21.47 | 0.00 | |
| C4 | OH-R5 | OH + CH3(CH2)2CH3 → H2O + CH2(CH2)2CH3 | 2.88 | 2.89 | 0.01 | −17.36 | −17.39 | −0.03 |
| OH-R6 | OH + CH3(CH2)2CH3 → H2O + CH3CHCH2CH3 | 0.99 | 0.99 | 0.00 | −20.27 | −20.27 | 0.00 | |
| C5 | OH-R7 | OH + CH3(CH2)3CH3 → H2O + CH2(CH2)3CH3 | 1.95 | 1.98 | 0.03 | −18.59 | −18.59 | 0.00 |
| OH-R8 | OH + CH3(CH2)3CH3 → H2O + CH3CH(CH2)2CH3 | −0.29 | −0.27 | 0.02 | −21.29 | −21.30 | −0.01 | |
| OH-R9 | OH + CH3(CH2)3CH3 → H2O + CH3CH2CHCH2CH3 | −0.13 | −0.13 | 0.00 | −20.93 | −20.93 | 0.00 | |
| C6 | OH-R10 | OH + CH3(CH2)4CH3 → H2O + CH2(CH2)4CH3 | 1.92 | 1.95 | 0.03 | −18.12 | −18.13 | −0.01 |
| OH-R11 | OH + CH3(CH2)4CH3 → H2O + CH3CH(CH2)3CH3 | 0.37 | 0.41 | 0.04 | −20.81 | −20.84 | −0.03 | |
| OH-R12 | OH + CH3(CH2)4CH3 → H2O + CH3CH2CH(CH2)2CH3 | 0.26 | 0.28 | 0.02 | −20.52 | −20.54 | −0.02 | |
| C7 | OH-R13 | OH + CH3(CH2)5CH3 → H2O + CH2(CH2)5CH3 | 1.64 | 1.68 | 0.03 | −18.55 | −18.55 | 0.00 |
| OH-R14 | OH + CH3(CH2)5CH3 → H2O + CH3CH(CH2)4CH3 | −0.17 | −0.13 | 0.04 | −21.67 | −21.68 | −0.01 | |
| OH-R15 | OH + CH3(CH2)5CH3 → H2O + CH3CH2CH(CH2)3CH3 | −0.79 | −0.74 | 0.05 | −20.94 | −20.96 | −0.02 | |
| OH-R16 | OH + CH3(CH2)5CH3 → H2O + CH3(CH2)2CH(CH2)2CH3 | −0.94 | −0.86 | 0.08 | −21.40 | −21.43 | −0.03 | |
| C8 | OH-R17 | OH + CH3(CH2)6CH3 → H2O + CH2(CH2)6CH3 | 2.29 | 2.31 | 0.02 | −18.39 | −18.40 | −0.01 |
| OH-R18 | OH + CH3(CH2)6CH3 → H2O + CH3CH(CH2)5CH3 | −0.14 | −0.08 | 0.06 | −21.14 | −21.16 | −0.02 | |
| OH-R19 | OH + CH3(CH2)6CH3 → H2O + CH3CH2CH(CH2)4CH3 | −0.24 | −0.20 | 0.04 | −20.94 | −20.97 | −0.03 | |
| OH-R20 | OH + CH3(CH2)6CH3 → H2O + CH3(CH2)2CH(CH2)3CH3 | −0.86 | −0.76 | 0.10 | −20.89 | −20.93 | −0.04 | |
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/CBS]1 | [QCISD(T)/CBS]2 | Difference | [QCISD(T)/CBS]1 | [QCISD(T)/CBS]2 | Difference | |||
| C1 | HO2-R1 | HO2 + CH4 → H2O2 + CH3 | 24.29 | 24.11 | −0.18 | 17.21 | 17.04 | −0.17 |
| C2 | HO2-R2 | HO2 + CH3CH3 → H2O2 + CH2CH3 | 20.01 | 19.90 | −0.11 | 13.40 | 13.29 | −0.11 |
| C3 | HO2-R3 | HO2 + CH3CH2CH3 → H2O2 + CH2CH2CH3 | 19.51 | 19.40 | −0.09 | 13.78 | 13.68 | −0.10 |
| HO2-R4 | HO2 + CH3CH2CH3 → H2O2 + CH3CHCH3 | 16.92 | 16.86 | −0.06 | 10.64 | 10.60 | −0.04 | |
| No. | Reactions | EB | ΔH | |||||
|---|---|---|---|---|---|---|---|---|
| [QCISD(T)/CBS]2 | ONIOM | Difference | [QCISD(T)/CBS]2 | ONIOM | Difference | |||
| C1 | HO2-R1 | HO2 + CH4 → H2O2 + CH3 | 24.11 | 24.11 | 0.00 | 17.04 | 17.04 | 0.00 |
| C2 | HO2-R2 | HO2 + CH3CH3 → H2O2 + CH2CH3 | 19.90 | 19.90 | 0.00 | 13.29 | 13.29 | 0.00 |
| C3 | HO2-R3 | HO2 + CH3CH2CH3 → H2O2 + CH2CH2CH3 | 19.40 | 19.40 | 0.00 | 13.68 | 13.68 | 0.00 |
| HO2-R4 | HO2 + CH3CH2CH3 → H2O2 + CH3CHCH3 | 16.86 | 16.86 | 0.00 | 10.60 | 10.60 | 0.00 | |
| C4 | HO2-R5 | HO2 + CH3(CH2)2CH3 → H2O2 + CH2(CH2)2CH3 | 19.25 | 19.31 | 0.06 | 13.61 | 13.62 | 0.01 |
| HO2-R6 | HO2 + CH3(CH2)2CH3 → H2O2 + CH3CHCH2CH3 | 16.37 | 16.37 | 0.00 | 10.89 | 10.89 | 0.00 | |
| C5 | HO2-R7 | HO2 + CH3(CH2)3CH3 → H2O2 + CH2(CH2)3CH3 | 19.58 | 19.64 | 0.06 | 13.65 | 13.65 | 0.00 |
| HO2-R8 | HO2 + CH3(CH2)3CH3 → H2O2 + CH3CH(CH2)2CH3 | 16.24 | 16.29 | 0.05 | 10.89 | 10.89 | 0.00 | |
| HO2-R9 | HO2 + CH3(CH2)3CH3 → H2O2 + CH3CH2CHCH2CH3 | 16.10 | 16.10 | 0.00 | 11.21 | 11.21 | 0.00 | |
| C6 | HO2-R10 | HO2 + CH3(CH2)4CH3 → H2O2 + CH2(CH2)4CH3 | 19.52 | 19.58 | 0.06 | 13.62 | 13.62 | 0.00 |
| HO2-R11 | HO2 + CH3(CH2)4CH3 → H2O2 + CH3CH(CH2)3CH3 | 16.38 | 16.44 | 0.06 | 10.85 | 10.85 | 0.00 | |
| HO2-R12 | HO2 + CH3(CH2)4CH3 → H2O2 + CH3CH2CH(CH2)2CH3 | 15.99 | 16.05 | 0.06 | 11.15 | 11.15 | 0.00 | |
| C7 | HO2-R13 | HO2 + CH3(CH2)5CH3 → H2O2 + CH2(CH2)5CH3 | 19.45 | 19.51 | 0.06 | 13.63 | 13.64 | 0.01 |
| HO2-R14 | HO2 + CH3(CH2)5CH3 → H2O2 + CH3CH(CH2)4CH3 | 16.16 | 16.22 | 0.06 | 10.87 | 10.87 | 0.00 | |
| HO2-R15 | HO2 + CH3(CH2)5CH3 → H2O2 + CH3CH2CH(CH2)3CH3 | 16.39 | 16.46 | 0.07 | 11.17 | 11.16 | −0.01 | |
| HO2-R16 | HO2 + CH3(CH2)5CH3 → H2O2 + CH3(CH2)2CH(CH2)2CH3 | 15.77 | 15.88 | 0.11 | 11.13 | 11.13 | 0.00 | |
| Involved Radical | Reaction Sites | ONIOM | Reference |
|---|---|---|---|
| H | Primary sites (CH3 groups) | 9.78 to 10.11 | 7.70 a, 7.70 b, 8.61 c |
| Other sites (CH2 group) | 6.93 to 7.49 | 5.00 a, 5.00 b, 5.93 c | |
| OH | Primary sites (CH3 groups) | 1.68 to 2.89 | 0.45 a, 1.81 b, 2.41 c |
| Other sites (CH2 group) | −1.57 to 0.99 | −0.76 a, 0.70 b, 1.19 c | |
| HO2 | Primary sites (CH3 groups) | 19.00 to 19.69 | 17.00 a, 19.41 b |
| Other sites (CH2 group) | 15.45 to 16.86 | 15.49 a, 17.02 b |
| No. | Reactions | ONIOM | Reference | |
|---|---|---|---|---|
| C1 | H-R1 | H + CH4 → H2 + CH3 | 13.34 | 13.3 a |
| C2 | H-R2 | H + CH3CH3 → H2 + CH2CH3 | 10.17 | 10.30 b |
| C3 | H-R3 | H + CH3CH2CH3 → H2 + CH2CH2CH3 | 10.11 | 10.27 b |
| H-R4 | H + CH3CH2CH3 → H2 + CH3CHCH3 | 7.49 | 7.68 b | |
| C4 | H-R5 | H + CH3(CH2)2CH3 → H2 + CH2(CH2)2CH3 | 10.05 | 10.07 c |
| H-R6 | H + CH3(CH2)2CH3 → H2 + CH3CHCH2CH3 | 7.39 | 7.21 c | |
| C10 | H-R26 | H + CH3(CH2)8CH3 → H2 + CH2(CH2)8CH3 | 9.82 | 10.1 d |
| H-R27 | H + CH3(CH2)8CH3 → H2 + CH3CH(CH2)7CH3 | 7.15 | 7.2 d | |
| H-R28 | H + CH3(CH2)8CH3 → H2 + CH3CH2CH(CH2)6CH3 | 7.05 | 7.1 d | |
| H-R29 | H + CH3(CH2)8CH3 → H2 + CH3(CH2)2CH(CH2)5CH3 | 7.04 | 7.0 d | |
| H-R30 | H + CH3(CH2)8CH3 → H2 + CH3(CH2)3CH(CH2)4CH3 | 7.02 | 7.0 d | |
| C18 | H-R82 | H + CH3(CH2)16CH3 → H2 + CH2(CH2)16CH3 | 9.84 | 10.85 e |
| H-R83 | H + CH3(CH2)16CH3 → H2 + CH3CH(CH2)15CH3 | 7.21 | 7.82 e | |
| H-R84 ~ H-R90 | 6.91~7.10 | 7.7~7.8 e | ||
| No. | Reactions | ONIOM | Reference | |
|---|---|---|---|---|
| C1 | OH-R1 | OH + CH4 → H2O + CH3 | 5.02 | 4.78 a |
| C2 | OH-R2 | OH + CH3CH3 → H2O + CH2CH3 | 2.63 | 2.18 a, 2.22 b |
| C3 | OH-R3 | OH + CH3CH2CH3 → H2O + CH2CH2CH3 | 1.81 | 1.93 c |
| OH-R4 | OH + CH3CH2CH3 → H2O + CH3CHCH3 | 0.49 | 0.93 c | |
| C4 | OH-R5 | OH + CH3(CH2)2CH3 → H2O + CH2(CH2)2CH3 | 2.89 | 2.06 c |
| OH-R6 | OH + CH3(CH2)2CH3 → H2O + CH3CHCH2CH3 | 0.99 | 0.72 c | |
| C5 | OH-R7 | OH + CH3(CH2)3CH3 → H2O + CH2(CH2)3CH3 | 1.98 | 1.98 d |
| OH-R8 | OH + CH3(CH2)3CH3 → H2O + CH3CH(CH2)2CH3 | −0.27 | 0.84 d | |
| OH-R9 | OH + CH3(CH2)3CH3 → H2O + CH3CH2CHCH2CH3 | −0.13 | 0.40 d |
| No. | Reactions | ONIOM | Reference | |
|---|---|---|---|---|
| C1 | HO2-R1 | HO2 + CH4 → H2O2 + CH3 | 24.11 | 24.00 a |
| C2 | HO2-R2 | HO2 + CH3CH3 → H2O2 + CH2CH3 | 19.90 | 19.50 a |
| C3 | HO2-R3 | HO2 + CH3CH2CH3 → H2O2 + CH2CH2CH3 | 19.40 | 19.60 a, 19.30 b |
| HO2-R4 | HO2 + CH3CH2CH3 → H2O2 + CH3CHCH3 | 16.86 | 16.09 a, 16.60 b | |
| C4 | HO2-R5 | HO2 + CH3(CH2)2CH3 → H2O2 + CH2(CH2)2CH3 | 19.31 | 19.46 a |
| HO2-R6 | HO2 + CH3(CH2)2CH3 → H2O2 + CH3CHCH2CH3 | 16.37 | 15.39 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, Y.; Pan, H.; Meng, Q.; Zhang, L.; Zhang, P. An ONIOM-Based High-Level Thermochemistry Study on Hydrogen Abstraction Reactions of Large Straight-Chain Alkanes by Hydrogen, Hydroxyl, and Hydroperoxyl Radicals. Symmetry 2024, 16, 367. https://doi.org/10.3390/sym16030367
Chi Y, Pan H, Meng Q, Zhang L, Zhang P. An ONIOM-Based High-Level Thermochemistry Study on Hydrogen Abstraction Reactions of Large Straight-Chain Alkanes by Hydrogen, Hydroxyl, and Hydroperoxyl Radicals. Symmetry. 2024; 16(3):367. https://doi.org/10.3390/sym16030367
Chicago/Turabian StyleChi, Yicheng, Hao Pan, Qinghui Meng, Lidong Zhang, and Peng Zhang. 2024. "An ONIOM-Based High-Level Thermochemistry Study on Hydrogen Abstraction Reactions of Large Straight-Chain Alkanes by Hydrogen, Hydroxyl, and Hydroperoxyl Radicals" Symmetry 16, no. 3: 367. https://doi.org/10.3390/sym16030367