
Asphaltene Behavior during Thermal Recovery: A Molecular Study Based on Realistic Structures

College of Petroleum Engineering & Geosciences, King Fahd University of Petroleum and Minerals (KFUPM), Dhahran 31261, Saudi Arabia
Minerals 2022, 12(10), 1315; https://doi.org/10.3390/min12101315
Submission received: 3 September 2022
/
Revised: 13 October 2022
/
Accepted: 17 October 2022
/
Published: 18 October 2022
(This article belongs to the Special Issue Mineralogical, Petrophysical and Hydromechanical Properties of Reservoirs and Caprocks)
Abstract
:Asphaltene precipitation and deposition can occur at both the surface and subsurface levels, leading to the formation of organic-based scales. Asphaltene precipitation can also lead to changes in petrophysical properties such as wettability, which affects the ultimate recovery. Asphaltene precipitation is linked to changes in fluid composition driven by pressure drawdown and temperature variation across the reservoir. Thus, asphaltene deposition can adversely influence the ultimate recovery. Thermal recovery methods are invoked to mitigate the adverse effects of asphaltene precipitation. The behavior of asphaltene under thermal recovery along with the link between the asphaltene molecular structure and its response to the increase in temperature during thermal recovery are not fully understood. In this paper, realistic asphaltene structures based on actual crude samples were recreated on a computational platform, and several characteristics of the asphaltene structures (density, viscosity, and interfacial tension) were evaluated during the heating process. The density of asphaltene was correlated with the percentage of aromatic carbon in its structure. The viscosity and interfacial tension decreased substantially as the temperature increased. The IFT reduced by approximately 30 mN/m as the temperature was increased from 300 K to 450 K. Moreover, the mechanical stability of asphaltene was found to be highly influenced by heating. The findings provide nanoscale insights into the behavior of asphaltene during thermal recovery, which can be used to improve the design of thermal recovery processes.
1. Introduction
Petroleum typically includes four components: aliphatic hydrocarbon saturates (e.g., n-alkanes), polycyclic aromatics and their alkylated derivatives, resins, and asphaltenes, which are collectively known as SARA (saturates, aromatics, resins, and asphaltenes). During field production, along with secondary and tertiary recovery phases, crude oil is subject to continuous changes in pressure and temperature, which in turn trigger asphaltene deposition [1]. Asphaltene precipitation leads to severe plugging formation and alters the wettability, which consequently reduces the ultimate recovery. Moreover, asphaltene deposition causes enormous increases in operational expenses and difficulties in field maintenance. Asphaltene deposition can be mitigated through chemical soaking, mechanical scraping, microbial, and dissolution approaches. Physical cleaning includes processes such as scratching and pigging, which are costly. A proactive approach involves the addition of crystal modifiers and asphaltene stabilizers such as sodium dodecyl sulfate, triton X-100, and benzoic acid to inhibit aggregation [2,3]. These treatments are commonly used in surface facilities but are rarely applied to the reservoir itself because of technical difficulties.
The above methods rely on either shifting the thermodynamic equilibrium to reverse precipitation or inhibiting aggregation by chemically altering the bonding between asphaltene units. However, the formation of asphaltene is inevitable, especially under subsurface conditions where rock–oil interactions, along with the temperature and pressure, are difficult to control. Hence, enhanced recovery methods such as thermal recovery are invoked to mitigate the adverse effect of asphaltene deposition on the ultimate recovery [4]. The primary aim of thermal recovery is to reach a temperature that provides favorable asphaltene transport and interfacial properties.
Asphaltenes are high-molecular-weight compounds containing nitrogen (N), sulfur (S), and oxygen (O) heteroatoms as minor skeletal components. Asphaltenes possess island–ocean, rosary, or archipelago molecular structures [5,6]. The presence of N, S, O, and other non-hydrocarbon ingredients such as iron, nickel, manganese, and copper affect the chemical and physical behaviors of asphaltene. Generally, asphaltenes consist of carbon (80%–86%), hydrogen (6%–8%), sulfur (2%–3%), nitrogen (0.5%–2%), oxygen (0.5%–2%), and trace metals (0.1%–0.2%) [7]. Asphaltenes are soluble in aromatic solvents such as benzene, toluene, and xylene, while they are insoluble in liquefied gases such as ethane, methane, and propane [7]. The solid parts of asphaltenes are soluble in toluene and insoluble in n-heptane.
The precipitation of asphaltenes is initiated by the shifting of the thermodynamic equilibrium of the crude oil. Disturbed asphaltenes regain their equilibrium state by associating with other surrounding hydrocarbon molecules. Association with other asphaltene molecules leads to aggregation and eventually precipitation from the oil phase onto the rock surface [8,9]. The asphaltene deposition envelope (ADE) highlights the effects of pressure and temperature changes on asphaltene formation (as shown in Figure 1). The typical ADE diagram shows that asphaltene precipitation increases as the pressure decreases to the bubble point, with the most precipitation occurring at the bubble point [10]. When the pressure drops below the bubble point, precipitation decreases [7]. The rate of precipitation is also affected by changes in temperature. For heavy crude oils, asphaltene precipitation increases with increasing temperature. In contrast, for light oil, precipitation decreases with increasing temperature. Many studies have focused on the nonlinear relationships between asphaltene precipitation and changes in pressure and temperature [3]. Typically, asphaltene precipitation is correlated with the presence of heavy hydrocarbons, and reducing the density of the crude oil will reduce precipitation [11,12]. The deposition of asphaltene on rock grains, which is induced by the polarity of asphaltenes, is linked to alterations in wettability and early water breakthrough during secondary recovery [13,14,15,16,17,18]. The interactions between rock and crude oil are augmented by the polarity of asphaltene and acid/base reactions. The asphaltene content was found to be a key factor in such interactions [19,20]. The experimental findings are consistent with microscopic investigations based on molecular simulation [21,22,23,24,25,26,27,28,29,30]. Molecular simulations have linked the asphaltene aggregation rate to the structure of the asphaltene model [31,32,33,34,35,36] and the type of organic solvent [37]. In general, molecular simulations complement the experimental results and reveal asphaltene aggregation in an organic solvent followed by thin-film formation [38,39,40]. However, a comprehensive description of asphaltene film dynamics throughout a typical production process and different enhanced oil recovery schemes has not been reported. Thermal recovery processes are applied to heavy oil reservoirs [41]. Despite the differences among these thermal methods, they are all based on increasing the temperature of the heavy oil to improve its flow parameters and interfacial properties [42]. Asphaltene is a primary component of heavy oil, and a thorough assessment of its behavior during thermal processes is crucial for understanding the overall effect of heating on heavy oil [43,44]. In this article, the behavior of asphaltene and asphaltene–water interactions were microscopically studied under a range of temperatures reflective of typical heating profiles encountered in real reservoirs. Realistic asphaltene structures derived from an actual oil sample were used to generate the asphaltene phase for comprehensive molecular simulations. The remainder of the paper is organized as follows: Section 2 outlines the methodology, including the recreation of asphaltene within a computational platform, the governing potential function, the characteristics of the asphaltene film, and asphaltene–water interactions under a range of temperatures. Section 3 then presents the results and discussion, and concluding remarks are provided in Section 4.
2. Methodology
Representative asphaltene molecular units served as the building blocks in the simulations. Alqam et al. [5] derived realistic asphaltene structures based on the detailed characterization of oil samples collected from a carbonate formation; from these asphaltene structures, three models were selected (Table 1). The selected models vary in the percentage of aromatic carbon, molecular weight, and carbon-hydrogen ratio. Moreover, the third model has two island-like fused benzene rings connected by a sulfur atom [45].
The variation in the aforementioned characteristics provides an opportunity to elucidate the effects of aromaticity, polarity, and the presence of heteroatoms on the thermodynamics and volumetric properties of asphaltene. The interactions within the asphaltene molecules and between asphaltene and other molecules such as water (i.e., in the case of hot water/steam injection) were captured through the non-bonding 9-6 potential function given by:
Equation (1) models the interactions between two atomic centers with charges and and separated by a distance . The potential exhibits a minimum at a distance greater than , and the maximum attraction is given by . In the second term of the right side of Equation (1), is the dielectric constant.
For unalike centers, a proper mixing rule is needed. The sixth-power averaging rule has been shown to be sufficient for approximating and , as given in Equations (2) and (3) [46]:
The and values for different atomic centers were parametrized using a predefined forcefield. Forcefields such as COMPASS and CFF93 have been extensively used for organic-based materials [47]. These forcefields have been complimented by the launch of the polymer-consistent forcefield PCFF+ [32], which was used to configure the parameters appearing in Equations (1)–(3). The same molecular modeling approach has been applied in similar systems and achieved decent results that reasonably match the experimental results [48,49,50,51,52,53,54,55].
2.1. Asphaltene Film
The asphaltene films were constructed from the respective asphaltene macromolecules given in Table 1 by starting from some number of macromolecular units and condensing them to yield the final structures. For this purpose, molecular dynamics simulations were run using LAMMPS. First, 100 units of asphaltene were initialized in a low-density three-dimensional periodic boundary cell (i.e., asphaltene was initialized at a low density by placing the molecular units in a relatively large cell to avoid any molecular instability). Subsequently, the energy was minimized, the molecular velocity was initialized, and a simulation run was conducted in an isochoric–isothermal NVT ensemble at 900 K. The high temperature helped to speed up the molecular relaxation (i.e., 250 ps in this study). A 200 ps run was then conducted in an isobaric–isothermal ensemble at 20.67 MPa and the same temperature. The temperature was then gradually decreased to 700 K, 500 K, and finally to 350 K through three consecutive runs in the NPT ensemble. In this study, a final temperature of 350 K and a final pressure of 20.67 MPa were selected to represent typical conditions encountered in the reservoir. The final density of the asphaltene 1, asphaltene 2, and asphaltene 3 films were 1.14, 1.02, and 1.13 g/cm3, respectively, and the three cells had respective sizes of 41.08 × 41.08 × 41.08 Å, 40.55 × 40.55 × 40.55 Å, and 42.52 × 42.52 × 42.52 Å. These cell sizes were sufficient to yield representative volumes, as demonstrated in previous studies [49,54]. The final structures are shown in Figure 2. The structures were further analyzed in terms of their ability to host molecules by characterizing the intermolecular spaces. This was carried out by the continuous insertion of nonoverlapping spheres with a predefined threshold [56]. The distributions of intermolecular space in the asphaltene structures are shown in Figure 3.
2.2. Viscosity Calculations
The viscosity, which is a crucial parameter for assessing the effect of heating on mobility, was calculated from the velocity and molecular trajectories of the equilibrated asphaltene films during a simulation run under the NVT ensemble with an extended simulation time of 5 ns [57,58]. The viscosity package of MedeA software was used to complete the calculations. Since one of the primary objectives of this study was to model the viscosity of asphaltene as a function of temperature, the viscosity calculations were repeated under increasing temperature. An equilibration molecular dynamic simulation consisting of runs under the NPT and NVT ensembles was conducted prior to the viscosity calculations for the considered range of temperatures.
2.3. Water–Asphaltene Interactions and Interfacial Tension Calculations
The interactions between water at elevated temperatures and asphaltene were quantified using two approaches. In the first approach, the water molecules hosted by asphaltene at a given temperature were assessed by running equilibrium Gibbs ensemble Monte Carlo simulations. These simulations mimicked the water molecules crossing the interface from the aqueous phase to the asphaltene film. The Gibbs ensemble Monte Carlo simulations were run using the water fugacity as an input for the calculations (see Table 2).
To calculate the interfacial tension (IFT), the boundary of asphaltene was adjusted in the z-direction to allow for the stacking of asphaltene by water. The water layer consisted of 600 water molecules with a density of 1.0 g/cm3. The simulation workflow consisted of an equilibration stage for the changes in temperature and an extended NVT simulation, with the last 300 ps used to calculate IFT. This simulation time was sufficient for the IFT value to converge, as shown in Figure 4. The calculations were carried out using the IFT package in MedeA software.
2.4. Mechanical Stability of the Asphaltene Film
The asphaltene film is subject to forces during primary and enhanced recovery. During thermal recovery, the increase in temperature may also affect film stability. In this study, the mechanical properties of the asphaltene films were assessed under continuous strain in concert with energy quantification. The relationship between the strain and change in energy is governed by Equation (4):
where U is defined as the change in energy (Etot − E0) resulting from the applied strain e divided by the initial volume V0. The subscripts i and j yield a tensor with 36 elements across the structure, and C is a stiffness matrix component. The number of elements is reduced to three as the asphaltene film forms a cubic cell. The energy was quantified by running equilibration molecular dynamics simulations before and after deformation. The simulation workflow consisted of a minimization stage followed by an extended NVT run. This approach has been applied extensively to investigate the mechanical stability of different materials [59].
3. Results and Discussion
3.1. Asphaltene Physical Properties
Asphaltene density is used to macroscopically distinguish asphaltene based on type. In this study, three asphaltene models with different chemical structures were constructed from single units through molecular dynamics simulation. The density of each structure was obtained as a function of temperature based on equilibration molecular dynamics simulations (Figure 5).
The density of the first and third structures (asphaltene 1 and 3) was higher than that of the second structure (asphaltene 2). This could be attributed to the higher fraction of sheet-like carbon (i.e., aromatic carbon) in the molecular structure of asphaltene 1 and 3, which allowed for more condensed stacking compared to asphaltene 2. For all structures, the density decreased as temperature increased. Interestingly, the density of asphaltene 2 decreased to slightly below 1 g/cm3 at high temperatures.
Next, the viscosity was analyzed for the same range of temperatures, as shown in Figure 6. At a low temperature (i.e., 300 K), the viscosities of the asphaltene 1, 2, and 3 structures were 963, 390, and 149 cP, respectively. These values are within the range anticipated for waxy and heavy organic-based materials. Asphaltene 1 had a higher viscosity than the other two, which can be attributed to its structure. Asphaltene 1 has eight fused benzene rings while asphaltene 2 has five. Asphaltene 3 has two separate three and four fused benzene rings. Interestingly, the viscosity at lower temperatures followed the order of the size of fused benzene rings.
As the temperature increased from 300 to 350 K, the viscosity reduced drastically to 60.3 and 19.6, and 57.0 cP, respectively, for the three structures. These changes suggest that the pour point (i.e., the temperature above which a material is considered to behave like a liquid) was in the range of 300–350 K. As the temperature continued to increase to 450 K, the viscosity continued to reduce to 19.4 cP for asphaltene 1 and 1.3 cP for asphaltene 2. Asphaltene 3, however, showed less sensitivity to a further decrease in the temperature as the viscosity was maintained almost the same with increasing the temperature. This suggests that the intermolecular forces induced by the polarity of sulfur restrict further reduction in the viscosity.
3.2. Interactions with Water and Interfacial Behavior
Thermal recovery often involves the injection of hot water/steam. Hence, it is vital to understand the interactions between water and asphaltene as a function of temperature. For this purpose, the asphaltene films were molecularly stacked by a water layer, and IFT calculations were performed for the three structures (Figure 7).
The initial IFT values for asphaltene 1, 2, and 3 were 75.7, 72.0, and 67.0 mN/m, respectively. Similar to the density and viscosity, the IFT decreased with increasing temperature, reaching 35.4, 27.0, and 24.0 mN/m for the three asphaltenes, respectively, without a noticeable influence of sulfur. The visual representations of the stacked asphaltene–water systems (Figure 8) show that the dispersion effect of water molecules increased as the temperature increased, suggesting that heating can instigate emulsion. Notably, the effect of temperature was more pronounced in asphaltene 2 and 3 compared to asphaltene 1, consistent with the reduction in IFT.
To provide more insight into the effect of temperature on asphaltene–water interactions, Gibbs ensemble Monte Carlo simulations were employed for the three structures. These simulations aimed to quantify the capacity of asphaltene to disperse the water layer by hosting water molecules. As outlined in the methodology section, water fugacity was evaluated as a function of temperature, and the number of water molecules entrapped by asphaltene was determined (Figure 9). Asphaltene became able to host water molecules when the temperature exceeded 350 K, and the capacity to host water increased drastically as the temperature increased further. Compared to asphaltene 1, asphaltene 2 had a stronger capacity for hosting water molecules. These results are consistent with the IFT calculations and the visual representations of the asphaltene–water systems are given in Figure 7 and Figure 8.
3.3. Mechanical Integrity
The molecular simulation framework provides an opportunity to assess the mechanical stability of the asphaltene films using the approach outlined in Section 2.4. The asphaltene films were continuously deformed to a final strain of 0.4 with a strain increment of 0.02. The stress–strain relationships of the three films were then obtained and plotted at temperatures of 300 and 450 K (Figure 10, Figure 11 and Figure 12).
The stress–strain relationships for the asphaltene structures revealed solid-like characteristics at 300 K with elastic deformation followed by mechanical failure via strain-softening behavior. The asphaltene 3 structure had a slightly higher yield strength than the other two structures. This could be attributed to the structural difference between the three models. Upon heating to 450 K, the behavior completely shifted, and the three films deformed plastically without a clear elastic region. Notably, the toughness (i.e., the area enclosed by the stress–strain curve) decreased from 0.0457 to 0.0312 GPa for the asphaltene 1 model. The toughness of the asphaltene 2 model decreased from 0.0390 to 0.0256 GPa. Similarly, the toughness of asphaltene 3 decreased from 0.0498 to 0.0331 GPa. The reduction in the toughness of asphaltene 3 was lesser than the other two structures, which was also observed in the viscosity trend of the same model. Again, resistance to heating might be attributed to the presence of sulfur.
3.4. Wettability Alteration
The water/asphaltene interface showed high sensitivity to the increase in temperature, as discussed in Section 3.2. The overall impact of thermal recovery, however, is also governed by the rock/fluid interactions (i.e., wettability). In general, it is well-documented that maximum recovery is anticipated at intermediate to strong water-wetness conditions. Upon deposition, the thin layer of asphaltene coating the rock surface acts to shift wettability toward oil-wet characteristics [19,54]. The deposited asphaltene film is, however, subject to deterioration during thermal recovery (see Figure 8, Figure 9, Figure 10, Figure 11 and Figure 12). To cast more light on wettability alteration, asphaltene, water, and substrate (calcite) were stacked and equilibrated at two temperatures to elucidate the wettability mode. The calcite structure is recreated in a similar manner as in other molecular simulation studies [60,61]. The structures were constructed as calcite coated with a thin layer of asphaltene interacting with water. Water and asphaltene layers of equal thickness were selected (i.e., 1 nm) while 4 nm of calcite was used. Snapshots of the system were taken during an extended NVT stage (i.e., 5 ns) to qualitatively assess the wettability, as shown in Figure 13.
It can be seen that the system at the higher temperature showed a drastic change where water molecules caused the detachment and replacement of the asphaltene film with time. These observations are consistent with the results of IFT and mechanical integrity analyses.
3.5. Consistency with Experimental Observations
This study provided in-depth nanoscale investigations of the changes in the physical and interfacial properties of asphaltene during a thermal recovery scheme. Some efforts have been done experimentally to unfold the underlying mechanisms of thermal recovery enhancement. These studies vary in their scale from simple core scale to full reservoir scale. Table 3 provides a summary of some studies highlighting the primary property under investigation.
The molecular simulation investigation reported in this study showed that increasing the temperature could reverse the wettability by washing out the precipitated asphaltene layer (see Figure 13) which is consistent with the experimental observations reported by Rao [62] and Roosta et al. [65]. Moreover, the increase in the temperature reduces the interfacial tension significantly for all asphaltene types considered in this study which is also consistent with the experimentally reported studies [64,66].
4. Conclusions
Thermal recovery methods have been applied to enhance recovery from heavy oil reservoirs. The effects of heating during thermal recovery on the characteristics of crude oil have been actively investigated. However, the behavior of asphaltene and its interactions with the aqueous phase is not well understood. In this study, realistic asphaltene models were created in a computational platform to assess the characteristics of asphaltene upon heating. The following conclusions can be drawn:
- The density of asphaltene was influenced by the chemical structure and temperature. Asphaltene containing a higher percentage of aromatic carbon had a higher density. The density was inversely correlated with temperature.
- Similar to the density, the viscosity of asphaltene increased with increasing temperature. The asphaltene structure containing the largest share of fused benzene rings had the highest viscosity at lower temperatures. At higher temperatures, however, the presence of sulfur seemed to restrict further reduction in the viscosity. The pour points of the asphaltene models were between 300 and 350 K.
- The temperature affected the interfacial tension, which decreased from more than 70 mN/m at 300 K to approximately 24 mN/m at 450 K. This can be explained by the enhanced ability of asphaltene to disperse the water phase under increasing temperature.
- Mechanical analysis revealed the solid-like behavior of asphaltene with both elastic and plastic deformation at 300 K. The mechanical behavior changed drastically upon heating, and the toughness of the asphaltene films decreased by approximately 60%.
- The findings provide in-depth insights into asphaltene behavior as a function of temperature. These insights can be used to optimize the design of thermal recovery processes. The findings of this study should be experimentally validated and repeated by extending this study to other asphaltene models.
Funding
This research received no external funding.
Data Availability Statement
The molecular simulation data, including the constructed asphaltene structures used to support the findings of this study, are available from the corresponding author upon request.
Acknowledgments
The author acknowledges the support provided by the College of Petroleum Engineering and Geosciences at King Fahd University of Petroleum and Minerals in Saudi Arabia. The results reported in this study were obtained using MedeA software.
Conflicts of Interest
The authors declare no conflict of interest.
Nomenclature
| Temperature, K | |
| Presssure, psi, MPa, or Pa | |
| Pore volume, m3 | |
| Young’s modulus, GPa | |
| Bulk modulus, GPa | |
| Shear modulus, GPa | |
| Poisson’s ratio, dimensionless parameter | |
| Separation distance between two center forces | |
| Atomic charge | |
| Dielectric constant | |
| Distance at which the interaction potential is zero | |
| Maximum amplitude of the potential well | |
| E0 | Energy of the structure, J |
| Etot | Energy after deformation, J |
References
- Mullins, O.C.; Sabbah, H.; Eyssautier, J.; Pomerantz, A.E.; Barré, L.; Andrews, A.B.; Ruiz-Morales, Y.; Mostowfi, F.; McFarlane, R.; Goual, L.; et al. Advances in asphaltene science and the Yen-Mullins model. Energy Fuels 2012, 26, 3986–4003. [Google Scholar] [CrossRef]
- Farooq, U.; Patil, A.; Panjwani, B.; Simonsen, G. Review on application of nanotechnology for asphaltene adsorption, crude oil demulsification, and produced water treatment. Energy Fuels 2021, 35, 19191–19210. [Google Scholar] [CrossRef]
- Adebiyi, F.M. An insight into asphalene precipitation, deposition and management stratagems in petroleum industry. J. Pipeline Sci. Eng. 2021, 1, 419–427. [Google Scholar] [CrossRef]
- Mukhametshina, A.; Hascakir, B. Bitumen extraction by expanding solvent-steam assisted gravity drainage (ES-SAGD) with asphaltene solvents and non-solvents. In Proceedings of the SPE Heavy Oil Conference Canada, Calgary, AB, Canada, 10–12 June 2014. [Google Scholar]
- Alqam, M.H.; Abu-Khamsin, S.A.; Sultan, A.S.; Al-Afnan, S.F.; Alawani, N.A. An investigation of factors influencing carbonate rock wettability. Energy Rep. 2021, 7, 1125–1132. [Google Scholar] [CrossRef]
- Schuler, B.; Meyer, G.; Peña, D.; Mullins, O.C.; Gross, L. Unraveling the molecular structures of asphaltenes by atomic force microscopy. J. Am. Chem. Soc. 2015, 137, 9870–9876. [Google Scholar] [CrossRef]
- Hasanvand, M.Z.; Montazeri, M.; Salehzadeh, M.; Amiri, M.; Fathinasab, M. A literature review of asphaltene entity, precipitation, and deposition: Introducing recent models of deposition in the well column. J. Oil Gas Petrochem. Sci. 2018, 1, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Mullins, O. Review of the molecular structure and aggregation of asphaltenes and petroleomics. SPE J. 2008, 13, 48–57. [Google Scholar] [CrossRef]
- Andersen, S.I.; Birdi, K.S. Aggregation of asphaltenes as determined by calorimetry. J. Colloid Interface Sci. 1991, 142, 497–502. [Google Scholar] [CrossRef]
- Alizadeh, A.; Nakhli, H.; Kharrat, R.; Ghazanfari, M.H. An experimental investigation of asphaltene precipitation during natural production of heavy and light oil reservoirs: The role of pressure and temperature. Pet. Sci. Technol. 2011, 29, 1054–1065. [Google Scholar] [CrossRef]
- Mansoori, G.A. Modeling of asphaltene and other heavy organic depositions. J. Pet. Sci. Eng. 1997, 17, 101–111. [Google Scholar] [CrossRef]
- Speight, J.G. Petroleum asphaltenes—Part 1: Asphaltenes, resins and the structure of petroleum. Oil Gas Sci. Technol. 2004, 59, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.G. Wettability literature survey—Part 6: The effects of wettability on waterflooding. J. Pet. Technol. 1987, 39, 1605–1622. [Google Scholar] [CrossRef]
- Berezin, V.M.; Yarygina, V.S.; Dubrovina, N.A. Adsorption of asphaltenes and tar from petroleum by sandstone. Neftepromysl. Delo 1982, 5, 15–17. [Google Scholar]
- Buckley, J.S.; Liu, Y.; Monsterleet, S. Mechanisms of wetting alteration by crude oils. SPE J. 1998, 3, 54–61. [Google Scholar] [CrossRef]
- Collins, S.H.; Melrose, J.C. Adsorption of asphaltenes and water on reservoir rock minerals. In Proceedings of the SPE Oilfield and Geothermal Chemistry Symposium, Denver, CO, USA, 1–3 June 1983. [Google Scholar]
- Basu, S.; Sharma, M.M. Investigating the role of crude oil components on wettability alteration using atomic force microscopy. SPE J. 1999, 4, 235–241. [Google Scholar] [CrossRef]
- Crocker, M.E.; Marchin, L.M. Wettability and adsorption characteristics of crude-oil. J. Pet. Technol. 1998, 40, 470–479. [Google Scholar] [CrossRef]
- Amin, J.S.; Nikooee, E.; Ghatee, M.H.; Ayatollahi, S.; Alamdari, A.; Sedghamiz, T. Investigating the effect of different asphaltene structures on surface topography and wettability alteration. Appl. Surf. Sci. 2011, 257, 8341–8349. [Google Scholar] [CrossRef]
- Standal, S.; Haavik, J.; Blokhus, A.M.; Skauge, A. Effect of polar organic components on wettability as studied by adsorption and contact angles. J. Pet. Sci. Eng. 1999, 24, 131–144. [Google Scholar] [CrossRef]
- Zhang, L.; Greenfield, M.L. Analyzing properties of model asphalts using molecular simulation. Energy Fuels 2007, 21, 1712–1716. [Google Scholar] [CrossRef]
- Fenistein, D.; Barré, L.; Broseta, D.; Espinat, D.; Livet, A.; Roux, J.N.; Scarsella, M. Viscosimetric and neutron scattering study of asphaltene aggregates in mixed toluene/heptane solvents. Langmuir 1998, 14, 1013–1020. [Google Scholar] [CrossRef]
- Gao, F.; Xu, Z.; Liu, G.; Yuan, S. Molecular dynamics simulation: The behavior of asphaltene in crude oil and at the oil/water interface. Energy Fuels 2014, 28, 7368–7376. [Google Scholar] [CrossRef]
- Headen, T.F.; Boek, E.S.; Skipper, N.T. Evidence for asphaltene nanoaggregation in toluene and heptane from molecular dynamics simulations. Energy Fuels 2009, 23, 1220–1229. [Google Scholar] [CrossRef]
- Hu, C.; Garcia, N.C.; Xu, R.; Cao, T.; Yen, A.; Garner, S.A.; Macias, J.M.; Joshi, N.; Hartman, R.L. Interfacial properties of asphaltenes at the heptol–brine interface. Energy Fuels 2016, 30, 80–87. [Google Scholar] [CrossRef]
- Jian, C.; Tang, T. Molecular dynamics simulations reveal inhomogeneity-enhanced stacking of violanthrone-78-based polyaromatic compounds in n-heptane−toluene mixtures. J. Phys. Chem. B 2015, 119, 8660–8668. [Google Scholar] [CrossRef]
- Jian, C.; Tang, T. One-dimensional self-assembly of polyaromatic compounds revealed by molecular dynamics simulations. J. Phys. Chem. 2014, 118, 12772–12780. [Google Scholar] [CrossRef]
- Jian, C.; Tang, T.; Bhattacharjee, S. Molecular dynamics investigation on the aggregation of Violanthrone78-based model asphaltenes in toluene. Energy Fuels 2014, 28, 3604–3613. [Google Scholar] [CrossRef]
- Jian, C.; Tang, T.; Bhattacharjee, S. Probing the effect of side-chain length on the aggregation of a model asphaltene using molecular dynamics simulations. Energy Fuels 2017, 27, 2057–2067. [Google Scholar] [CrossRef]
- Kuznicki, T.; Masliyah, J.H.; Bhattacharjee, S. Aggregation and partitioning of model asphaltenes at toluene—water interfaces: Molecular dynamics simulations. Energy Fuels 2009, 23, 5027–5035. [Google Scholar] [CrossRef]
- Sedghi, M.; Goual, L.; Welch, W.; Kubelka, J. Effect of asphaltene structure on association and aggregation using molecular dynamics. J. Phys. Chem. 2013, 117, 5765–5776. [Google Scholar] [CrossRef]
- Ungerer, P.; Rigby, D.; Leblanc, B.; Yiannourakou, M. Sensitivity of the aggregation behaviour of asphaltenes to molecular weight and structure using molecular dynamics. Mol. Simul. 2013, 40, 115–122. [Google Scholar] [CrossRef]
- Teklebrhan, R.B.; Ge, L.; Bhattacharjee, S.; Xu, Z.; Sjöblom, J. Probing structure–nanoaggregation relations of polyaromatic surfactants: A molecular dynamics simulation and dynamic light scattering study. J. Phys. Chem. B 2012, 116, 5907–5918. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Molinari, D. A multiscale approach to the simulation of asphaltenes. Comput. Theor. Chem. 2011, 975, 76–82. [Google Scholar] [CrossRef]
- Stukan, M.R.; Ligneul, P.; Boek, E.S. Molecular dynamics simulation of spontaneous imbibition in nanopores and recovery of asphaltenic crude oils using surfactants for EOR applications. Oil Gas Sci. Technol. 2012, 67, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Porte, G.; Zhou, H.; Lazzeri, V. Reversible description of asphaltene colloidal association and precipitation. Langmuir 2003, 19, 40–47. [Google Scholar] [CrossRef]
- Kuznicki, T.; Masliyah, J.H.; Bhattacharjee, S. Molecular dynamics study of model molecules resembling asphaltene-like structures in aqueous organic solvent systems. Energy Fuels 2008, 22, 2379–2389. [Google Scholar] [CrossRef]
- Mikami, Y.; Liang, Y.; Matsuoka, T.; Boek, E.S. Molecular dynamics simulations of asphaltenes at the oil–water interface: From nanoaggregation to thin-film formation. Energy Fuels 2013, 27, 1838–1845. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Y.; Ren, S. Molecular dynamics simulation of self-aggregation of asphaltenes at an oil/water interface: Formation and destruction of the asphaltene protective film. Energy Fuels 2015, 29, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.; Mansoori, G.A. Effect of CO2 on the interfacial and transport properties of water/binary and asphaltenic oils: Insights from molecular dynamics. Energy Fuels 2018, 32, 5409–5417. [Google Scholar] [CrossRef]
- Green, D.W.; Willhite, G.P. Enhanced Oil Recovery (6); Henry L. Doherty Memorial Fund of AIME, Society of Petroleum Engineers: Richardson, TX, USA, 1998. [Google Scholar]
- Aljawad, M.S.; Alafnan, S.; Abu-Khamsin, S. Artificial lift and mobility enhancement of heavy oil reservoirs utilizing a renewable energy-powered heating element. ACS Omega 2019, 4, 20048–20058. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, K.; Hammami, A.; Kharrat, A.; Zhang, D.; Allenson, S.; Creek, J.; Kabir, S.; Jamaluddin, A.; Marshall, A.G.; Rodgers, R.P.; et al. Asphaltenes—Problematic but rich in potential. Oilfield Rev. 2007, 19, 24–43. [Google Scholar]
- Gray, M.R.; Assenheimer, G.; Boddez, L.; McCaffrey, W.C. Melting and fluid behavior of asphaltene films at 200−500 °C. Energy Fuels 2004, 18, 1419–1423. [Google Scholar] [CrossRef]
- Alafnan, S. Utilization of depleted heavy oil reservoirs for carbon dioxide storage and sequestration: A molecular level assessment. Int. J. Greenh. Gas Control 2022, 119, 103741. [Google Scholar] [CrossRef]
- Waldmann, M.; Hagler, A.T. New combining rules for rare gas van der Waals parameters. J. Comput. Chem. 1993, 14, 1077–1084. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Afagwu, C.; Al-Afnan, S.; Patil, S.; Aljaberi, J.; Mahmoud, M.A.; Li, J. The impact of pore structure and adsorption behavior on kerogen tortuosity. Fuel 2021, 303, 121261. [Google Scholar] [CrossRef]
- Alafnan, S. Petrophysics of kerogens based on realistic structures. ACS Omega 2021, 6, 9549–9558. [Google Scholar] [CrossRef]
- Alafnan, S.; Solling, T.; Mahmoud, M. Effect of kerogen thermal maturity on methane adsorption capacity: A molecular modeling approach. Molecules 2020, 25, 3764. [Google Scholar] [CrossRef]
- Alafnan, S.; Sultan, A.S.; Aljaberi, J. Molecular fractionation in the organic materials of source rocks. ACS Omega 2020, 5, 18968–18974. [Google Scholar] [CrossRef]
- Alafnan, S.; Falola, Y.; Al Mansour, O.; AlSamadony, K.; Awotunde, A.; Aljawad, M. Enhanced recovery from organic-rich shales through carbon dioxide injection: Molecular-level investigation. Energy Fuels 2020, 34, 16089–16098. [Google Scholar] [CrossRef]
- Aljaberi, J.; Alafnan, S.; Glatz, G.; Sultan, A.S.; Afagwu, C. The impact of kerogen tortuosity on shale permeability. SPE J. 2020, 26, 765–779. [Google Scholar] [CrossRef]
- Alqam, M.H.; Abu-Khamsin, S.A.; Alafnan, S.F.; Sultan, A.S.; Al-Majed, A.; Okasha, T. The impact of carbonated water on wettability: Combined experimental and molecular simulation approach. SPE J. 2021, 27, 945–957. [Google Scholar] [CrossRef]
- Alafnan, S. Utilization of supercritical carbon dioxide for mechanical degradation of organic matters contained in shales. Fuel 2022, 319, 123427. [Google Scholar] [CrossRef]
- Barber, C.B.; Dobkin, D.P.; Huhdanpaa, H. The quickhull algorithm for convex hulls. ACM Trans. Math. Softw. 1996, 22, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Greenfield, M.L. Relaxation time, diffusion, and viscosity analysis of model asphalt systems using molecular simulation. J. Chem. Phys. 2007, 127, 194502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dysthe, D.K.; Fuchs, A.H.; Rousseau, B. Fluid transport properties by equilibrium molecular dynamics. I. Methodology at extreme fluid states. J. Chem. Phys. 1999, 110, 4047–4059. [Google Scholar] [CrossRef] [Green Version]
- Alafnan, S. The impact of pore structure on kerogen geomechanics. Geofluids 2021, 2021, 4093895. [Google Scholar] [CrossRef]
- Yang, M.; Rodger, P.M.; Harding, J.H.; Stipp, S.L.S. Molecular dynamics simulations of peptides on calcite surface. Mol. Simul. 2009, 35, 547–553. [Google Scholar] [CrossRef]
- Mutisya, S.M.; Kirch, A.; de Almeida, J.M.; Sánchez, V.M.; Miranda, C.R. Molecular Dynamics Simulations of Water Confined in Calcite Slit Pores: An NMR Spin Relaxation and Hydrogen Bond Analysis. J. Phys. Chem. C 2017, 121, 6674–6684. [Google Scholar] [CrossRef]
- Rao, D.N. Wettability effects in thermal recovery operations. SPE Reserv. Eval. Eng. 1999, 2, 420–430. [Google Scholar] [CrossRef]
- Tang, G.-Q.; Lowry, D.; Lee, V. Recovery mechanism of steam injection in heavy oil carbonate reservoir. In Proceedings of the SPE Western North American Region Meeting, Anchorage, Alaska, USA, 7–11 May 2011. [Google Scholar]
- Al-Hadhrami, H.S.; Blunt, M.J. Thermally induced wettability alteration to improve oil recovery in fractured reservoirs. In Proceedings of the SPE/DOE Improved Oil Recovery Symposium, Tulsa, OK, USA, 3–5 April 2001; p. SPE 71866. [Google Scholar]
- Roosta, A.B.; Escrochi, M.F.; Khatibi, V.J.; Ayatollahi, V.J.S.; Schafiee, M. Investigating the mechanism of thermally induced wettability alteration. In Proceedings of the SPE Middle East Oil and Gas Show and Conference, Manama, Bahrain, 15–18 March 2009. [Google Scholar]
- Bardon, C.; Longeron, D.G. Influence of very low interfacial tensions on relative permeability. Soc. Pet. Eng. J. 1980, 20, 391–401. [Google Scholar] [CrossRef]
Figure 1.
Typical asphaltene deposition envelope (ADE).

Figure 2.
Visual representations of the final asphaltene structures.

Figure 3.
Intermolecular space (in radius) for the asphaltene films, revealing similar pore size distributions.
Figure 3.
Intermolecular space (in radius) for the asphaltene films, revealing similar pore size distributions.

Figure 4.
Example of an IFT calculation showing the convergence of the IFT value to the final calculated value.
Figure 4.
Example of an IFT calculation showing the convergence of the IFT value to the final calculated value.
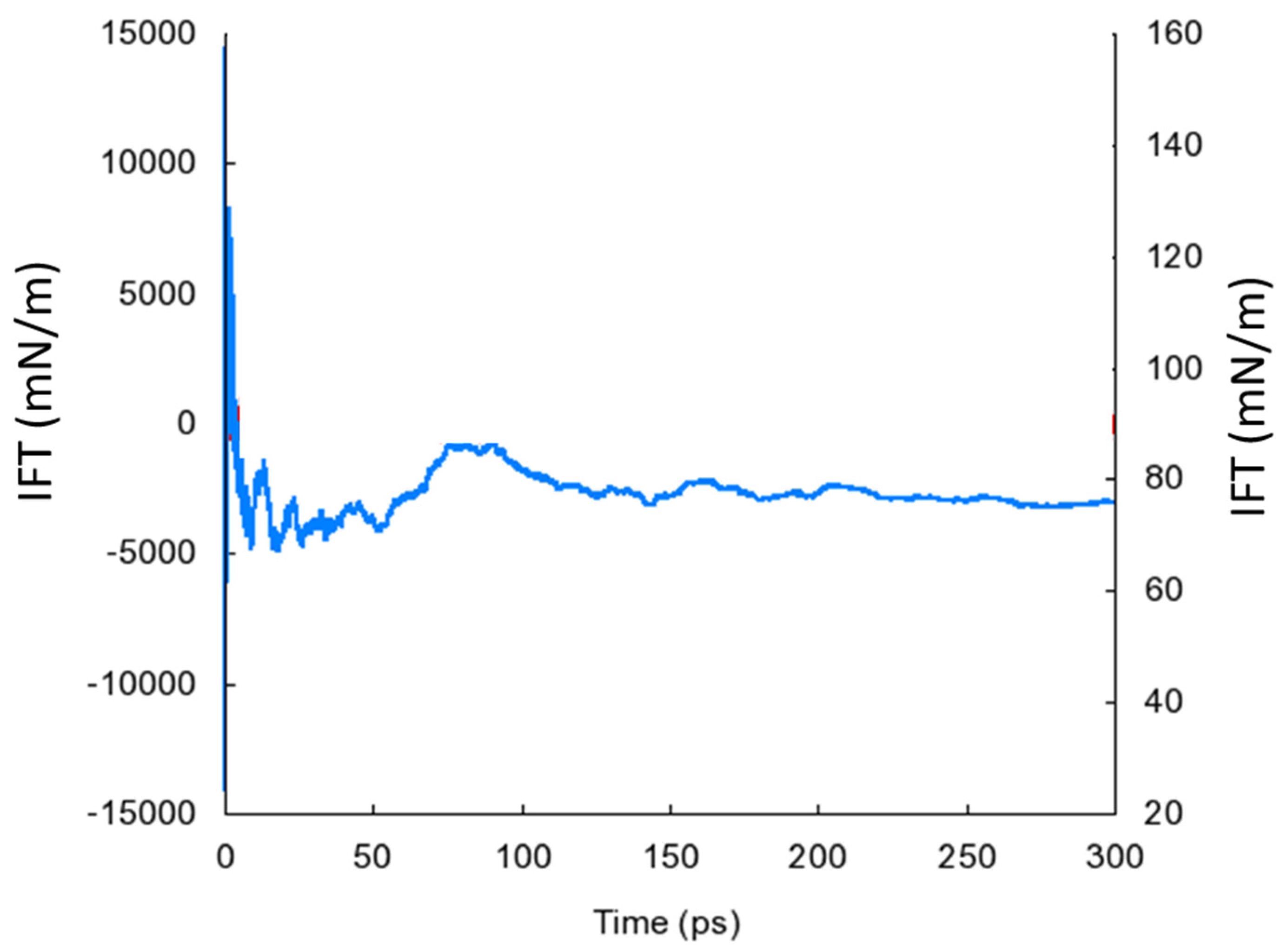
Figure 5.
Density profiles of the asphaltene structures as a function of temperature.
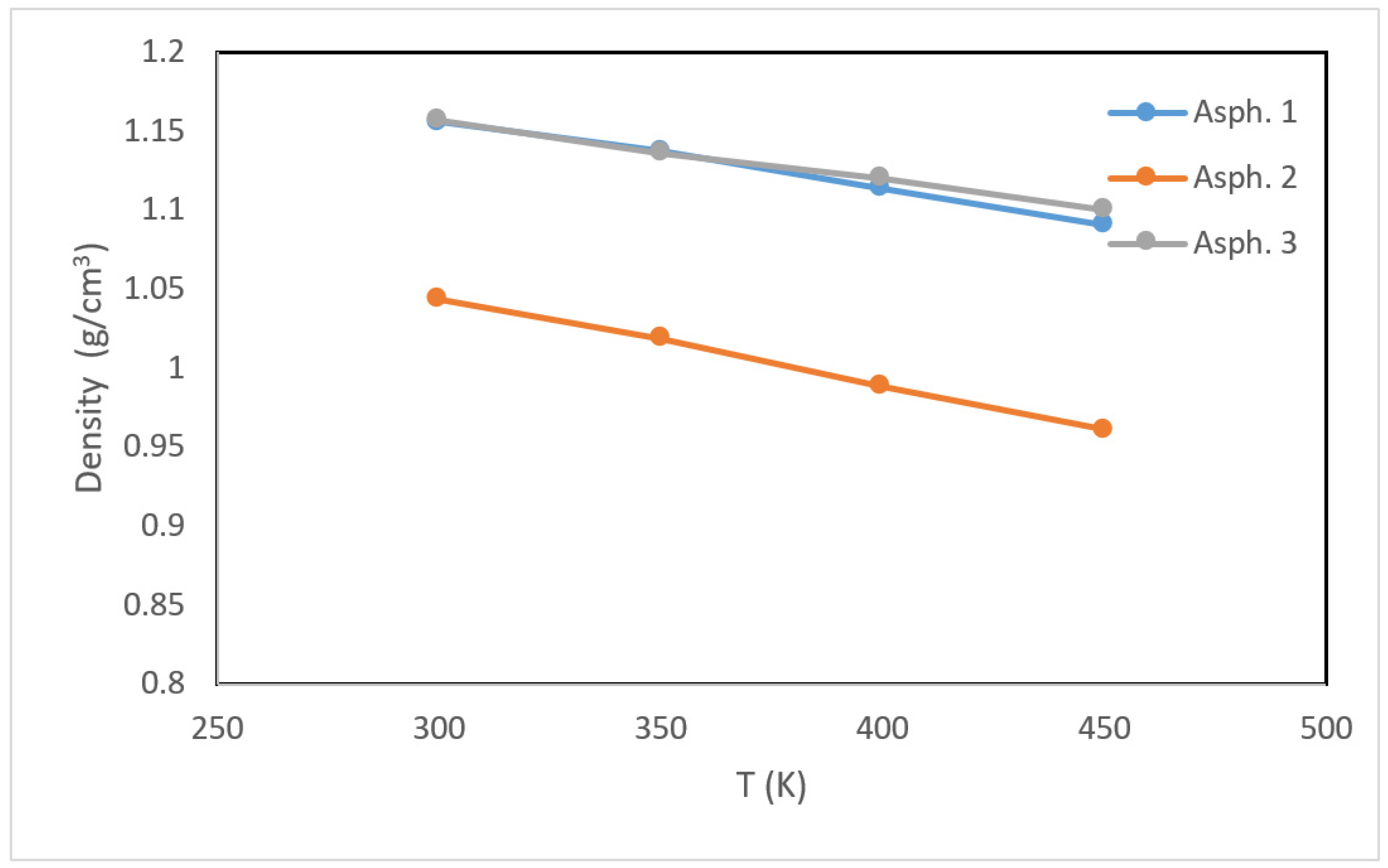
Figure 6.
Viscosity profiles for the asphaltene structures as a function of temperature. In all structures, the viscosity decreases drastically as the temperature increases.
Figure 6.
Viscosity profiles for the asphaltene structures as a function of temperature. In all structures, the viscosity decreases drastically as the temperature increases.
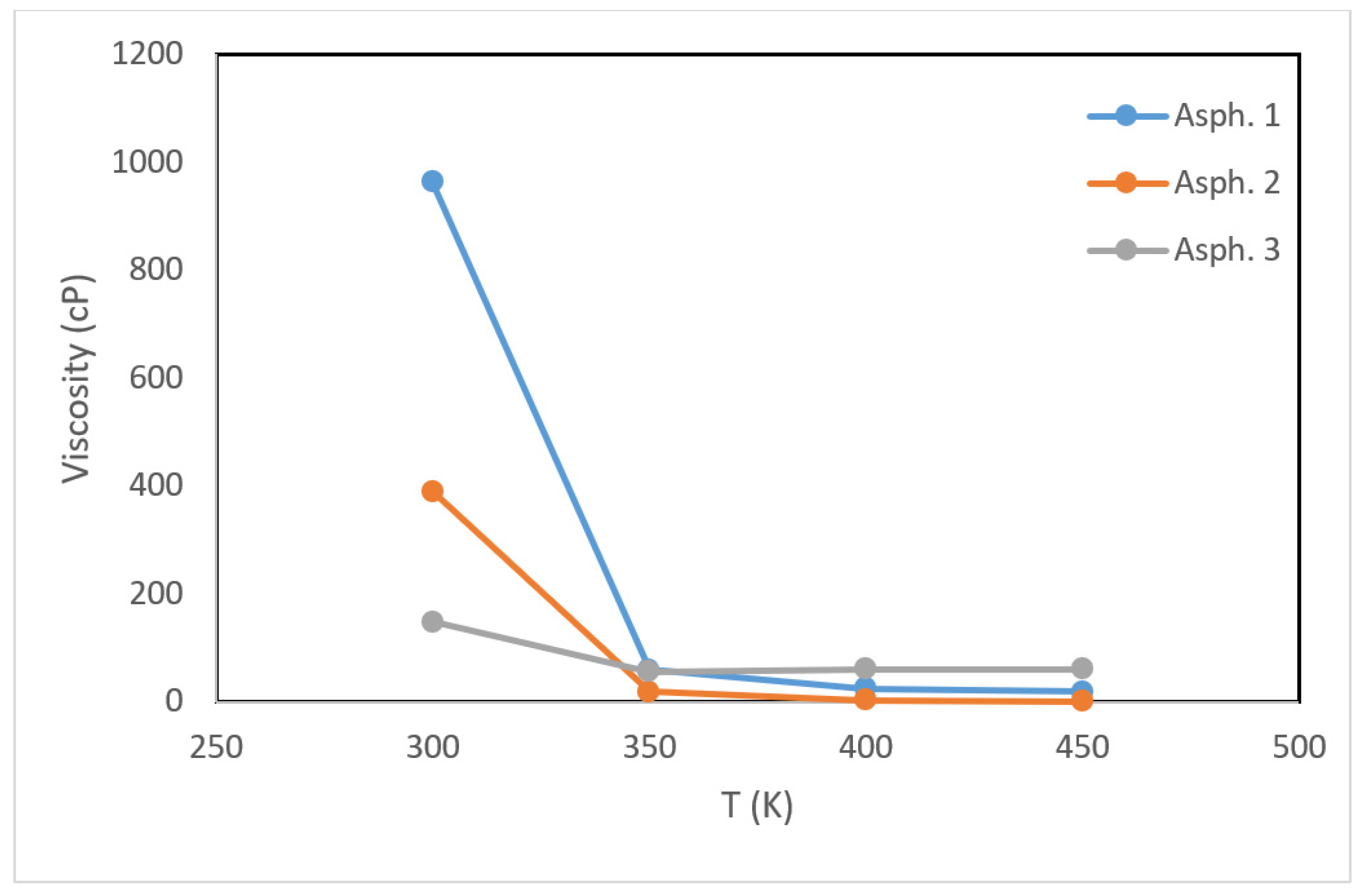
Figure 7.
Calculated IFT values of the asphaltene structures as a function of temperature.
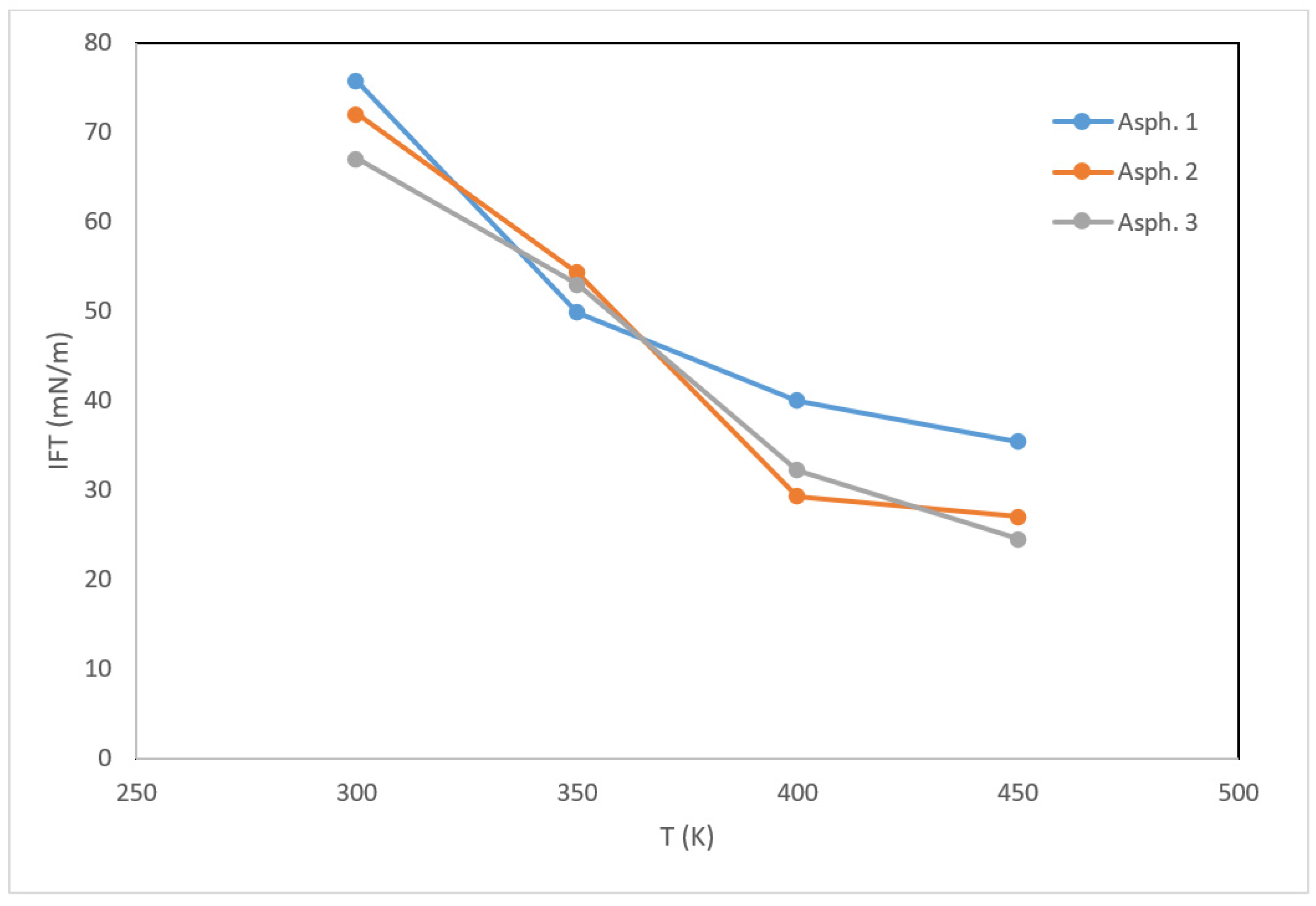
Figure 8.
Visual representations of the water–asphaltene stacked systems upon the completion of the IFT calculations.
Figure 8.
Visual representations of the water–asphaltene stacked systems upon the completion of the IFT calculations.

Figure 9.
Asphaltene–water interactions (presented as the millimoles of water hosted per kilogram of asphaltene) as a function of temperature.
Figure 9.
Asphaltene–water interactions (presented as the millimoles of water hosted per kilogram of asphaltene) as a function of temperature.
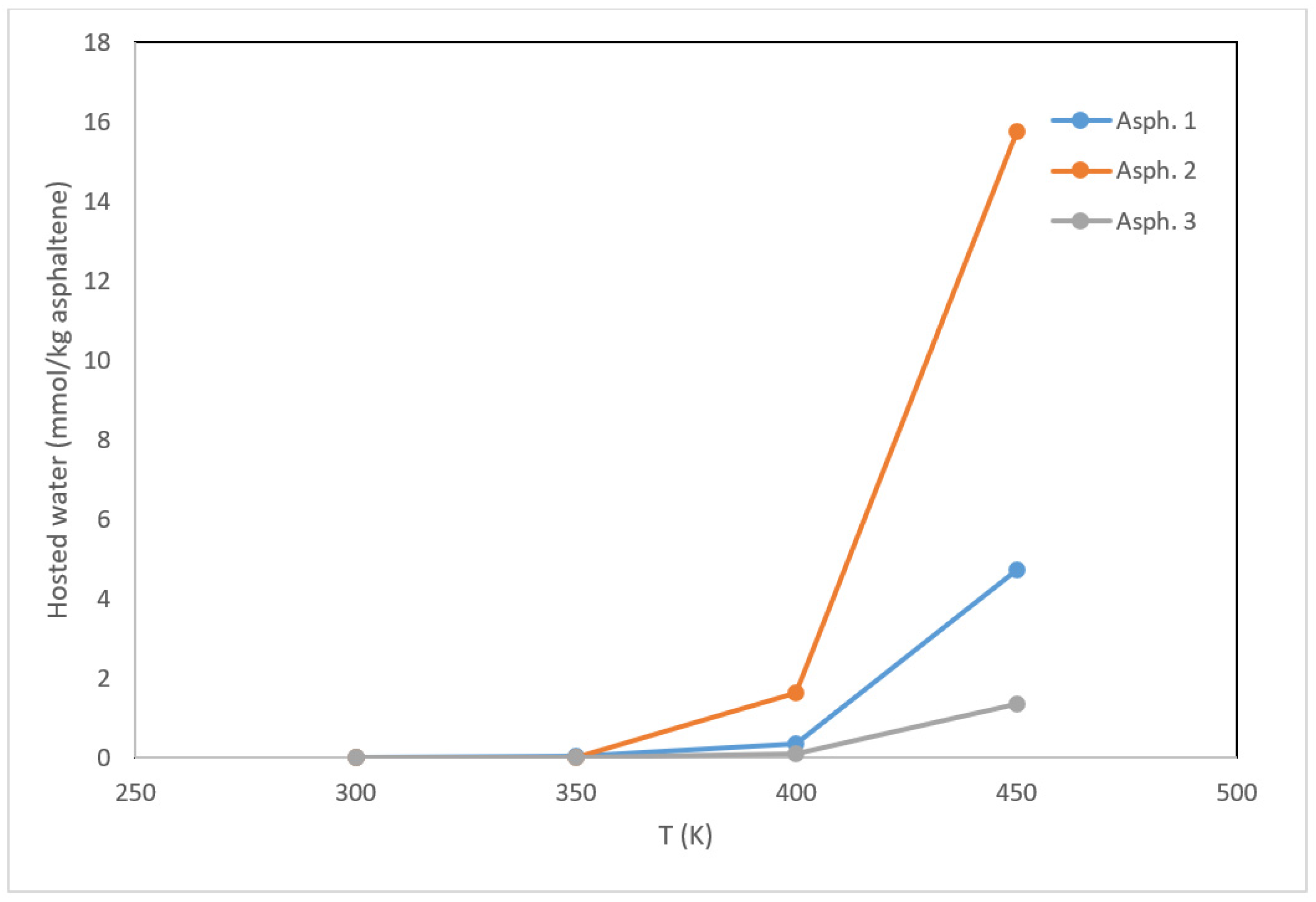
Figure 10.
Stress–strain relationships for the asphaltene 1 structure at 300 and 450 K.
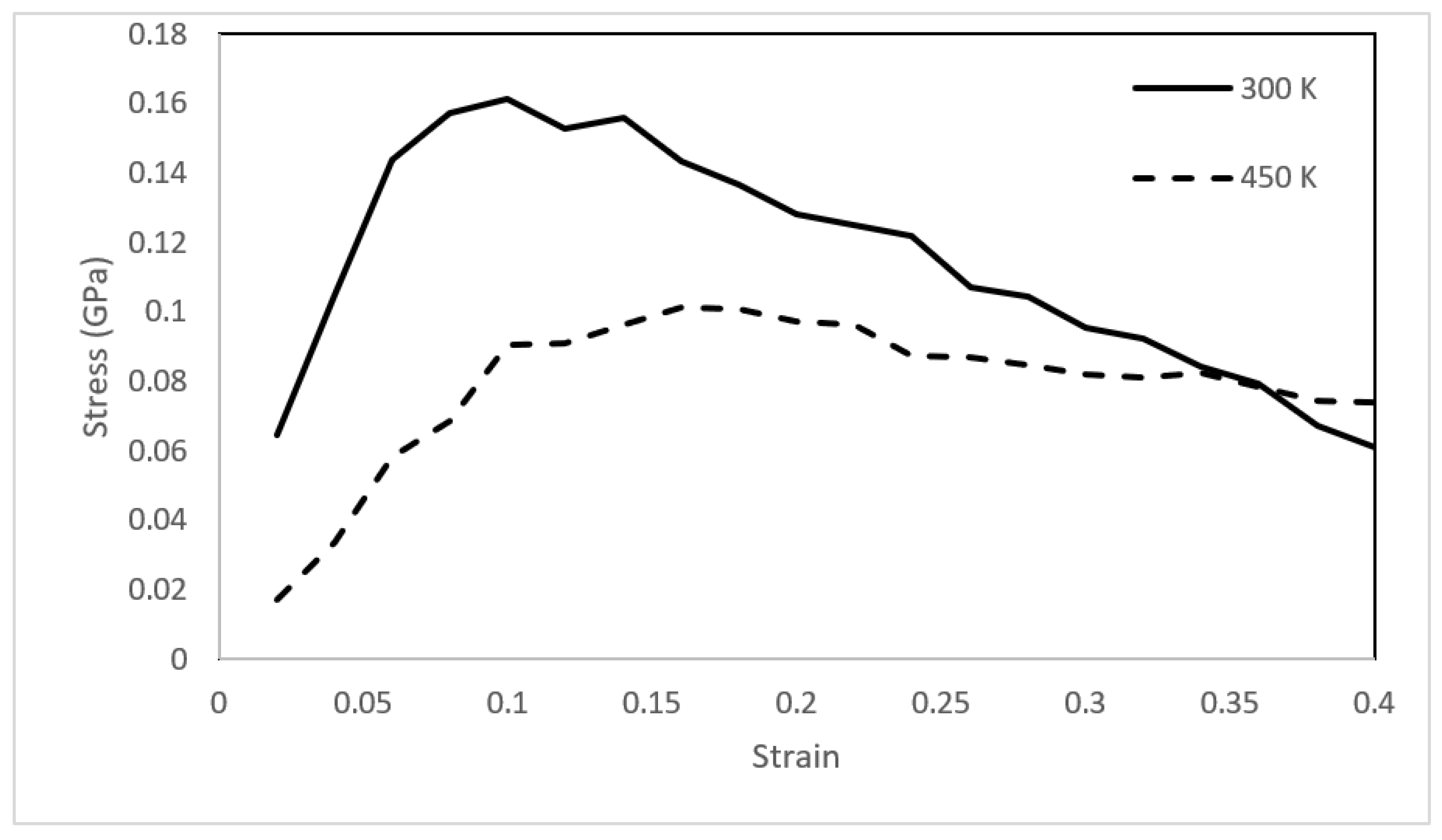
Figure 11.
Stress–strain relationships for the asphaltene 2 structure at 300 and 450 K.
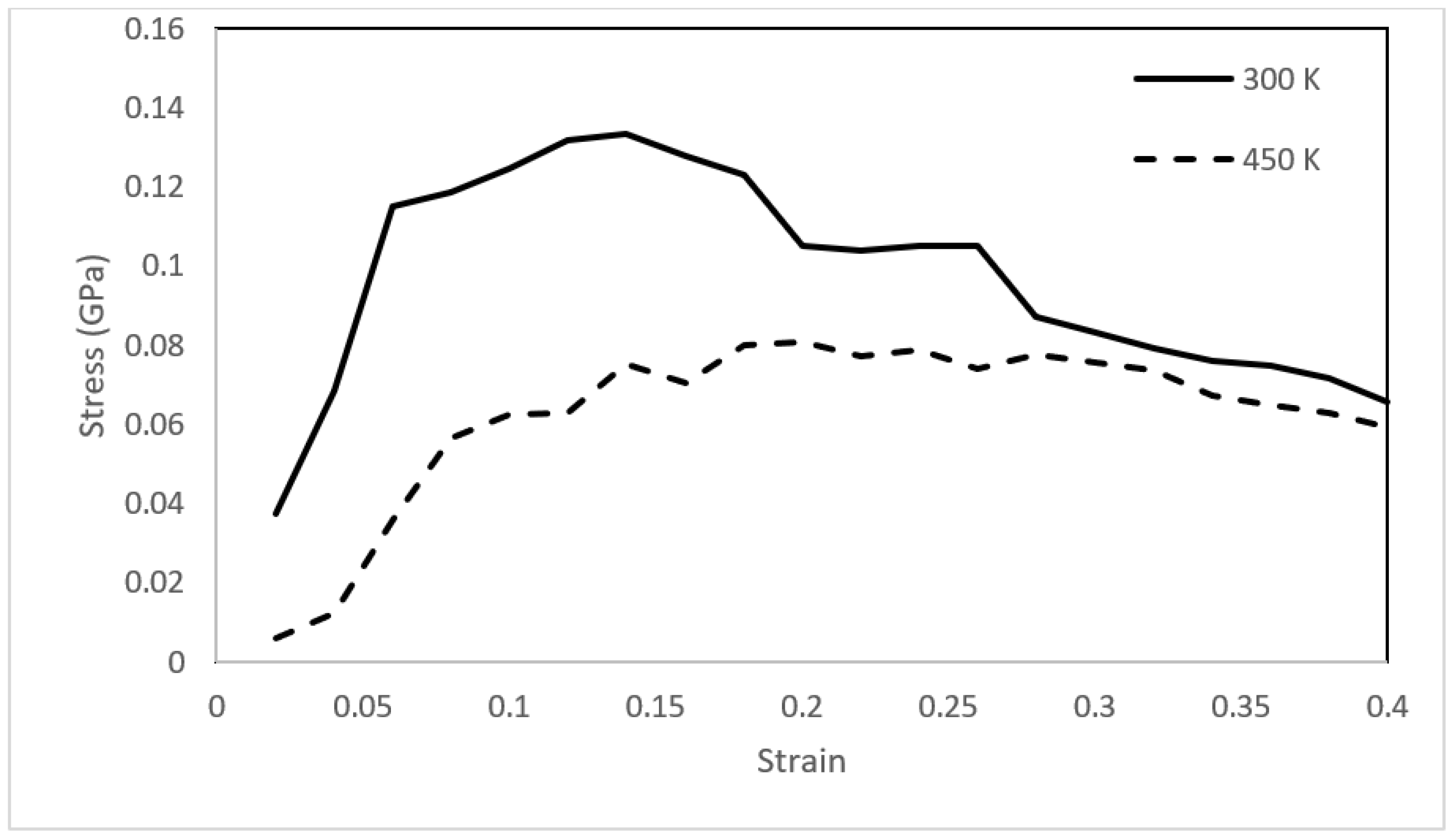
Figure 12.
Stress–strain relationships for the asphaltene 3 structure at 300 and 450 K.

Figure 13.
Stacked system of calcite/asphaltene 3/water showing the time evolution of fluid-fluid, fluid-rock interactions at 300 and 450 K. The red layer represents water, while the black is asphaltene 3 stacked on calcite.
Figure 13.
Stacked system of calcite/asphaltene 3/water showing the time evolution of fluid-fluid, fluid-rock interactions at 300 and 450 K. The red layer represents water, while the black is asphaltene 3 stacked on calcite.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Asphaltene structures with different percentages of aromatic carbon adopted for use in this study.
Table 1.
Asphaltene structures with different percentages of aromatic carbon adopted for use in this study.
| Asphaltene | Molecular Structure | Molecular Formula | Percentage of Aromatic Carbon |
|---|---|---|---|
| 1 | 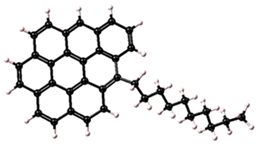 | C37H33 | 73% |
| 2 | 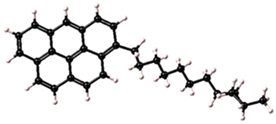 | C31H35 | 58% |
| 3 | 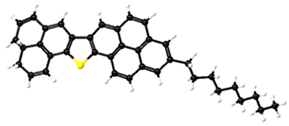 | C38H35S | 73% |
Table 2.
Fugacity of water as a function of temperature calculated using the Peng–Robinson equation of state at 20.67 MPa.
Table 2.
Fugacity of water as a function of temperature calculated using the Peng–Robinson equation of state at 20.67 MPa.
| T (K) | Fugacity (MPa) |
|---|---|
| 300 | 0.004 |
| 350 | 0.052 |
| 375 | 0.137 |
| 400 | 0.311 |
| 450 | 1.149 |
Table 3.
Summary of some experimentally reported investigation of thermal recovery effects on oil physical and interfacial properties.
Table 3.
Summary of some experimentally reported investigation of thermal recovery effects on oil physical and interfacial properties.
| Reference | Type of Formation | Property under Investigation | Observation |
|---|---|---|---|
| Rao [62] | Carbonate | Wettability | Upon hot water injection, the wettability shifted from oil to water wet |
| Tang et al. [63] | Carbonate | Viscosity | The viscosity of the oil reduced to 2 cP as the temperature reached 240 °C |
| Al-Hadrami and Blunt [64] | Carbonate | Interfacial tension | The interfacial tension reduced significantly as the temperature increased which resulted in reduced capillary forces |
| Roosta et al. [65] | Carbonate/Quartz | Wettability | The wettability shifted toward water wet system as the temperature increased |
| Bardon and Longeron [66] | NA | Interfacial tension | The interfacial tension reduced which influenced the relative permeability positively |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alafnan, S. Asphaltene Behavior during Thermal Recovery: A Molecular Study Based on Realistic Structures. Minerals 2022, 12, 1315. https://doi.org/10.3390/min12101315
AMA Style
Alafnan S. Asphaltene Behavior during Thermal Recovery: A Molecular Study Based on Realistic Structures. Minerals. 2022; 12(10):1315. https://doi.org/10.3390/min12101315
Chicago/Turabian StyleAlafnan, Saad. 2022. "Asphaltene Behavior during Thermal Recovery: A Molecular Study Based on Realistic Structures" Minerals 12, no. 10: 1315. https://doi.org/10.3390/min12101315
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.