Physicochemical and Additive Controls on the Multistep Precipitation Pathway of Gypsum

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Turbidity Experiments
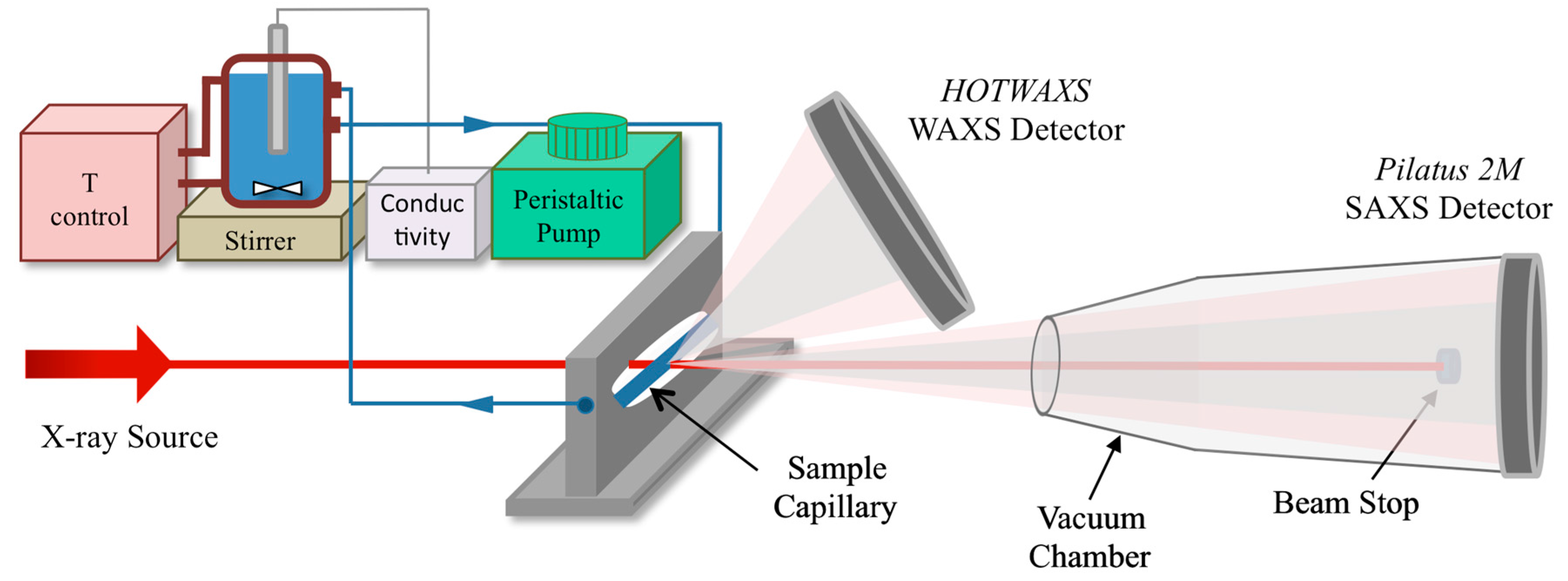
2.2. Synchrotron In Situ Small- and Wide-Angle X-ray Scattering
SAXS and WAXS Data Processing
2.3. AFM Experiments
3. Results
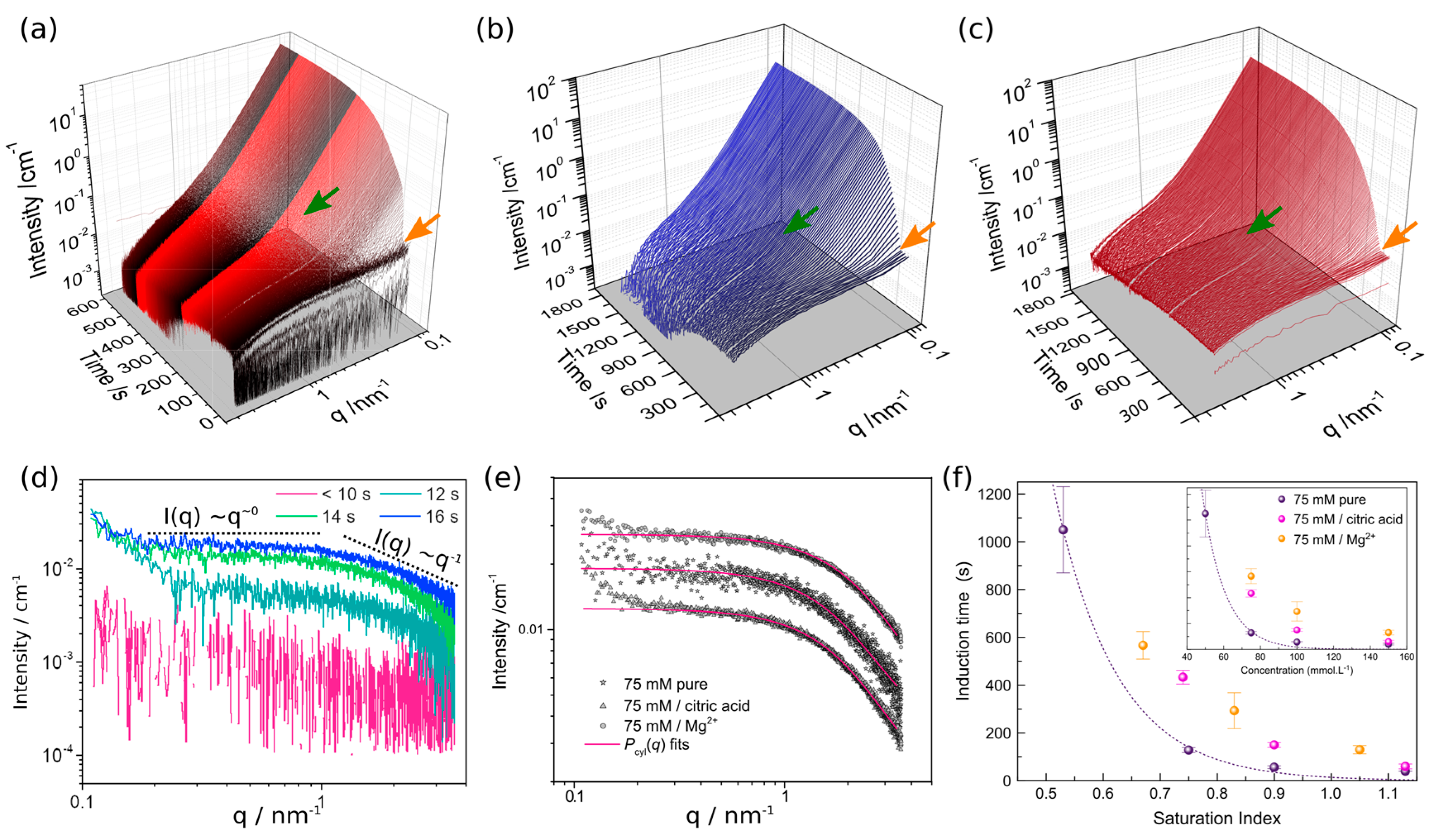
3.1. The Initial Stages of Gypsum Formation
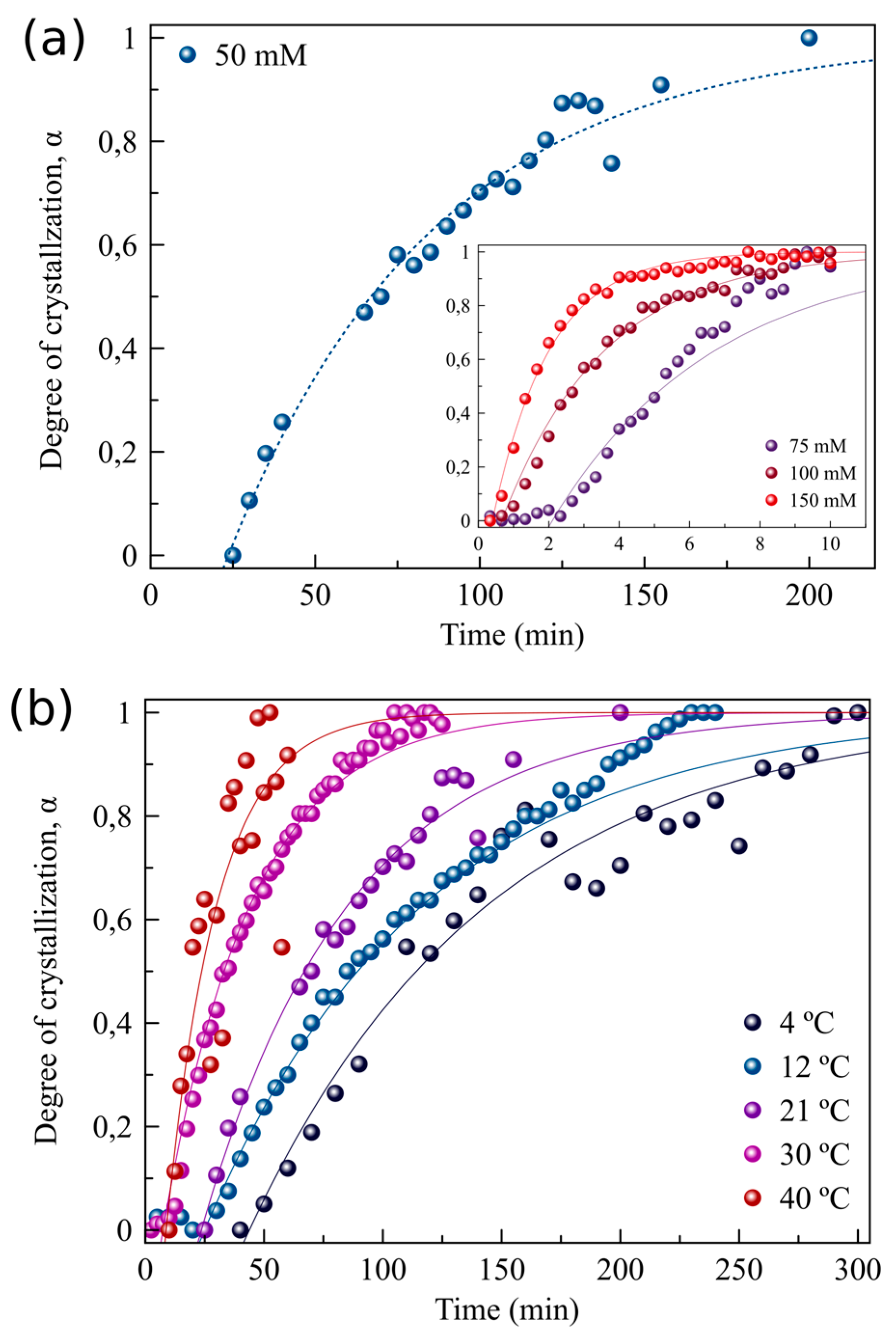
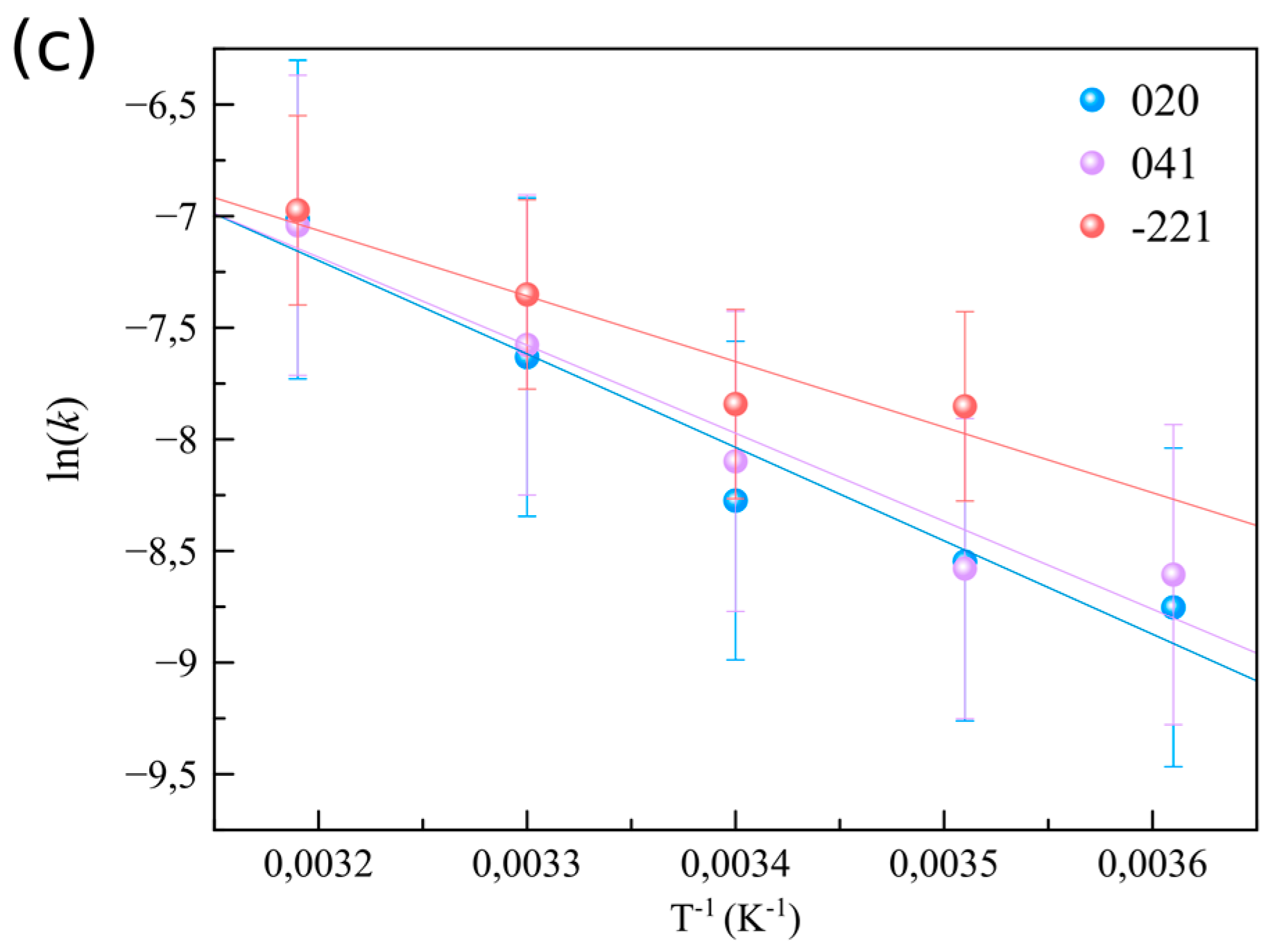
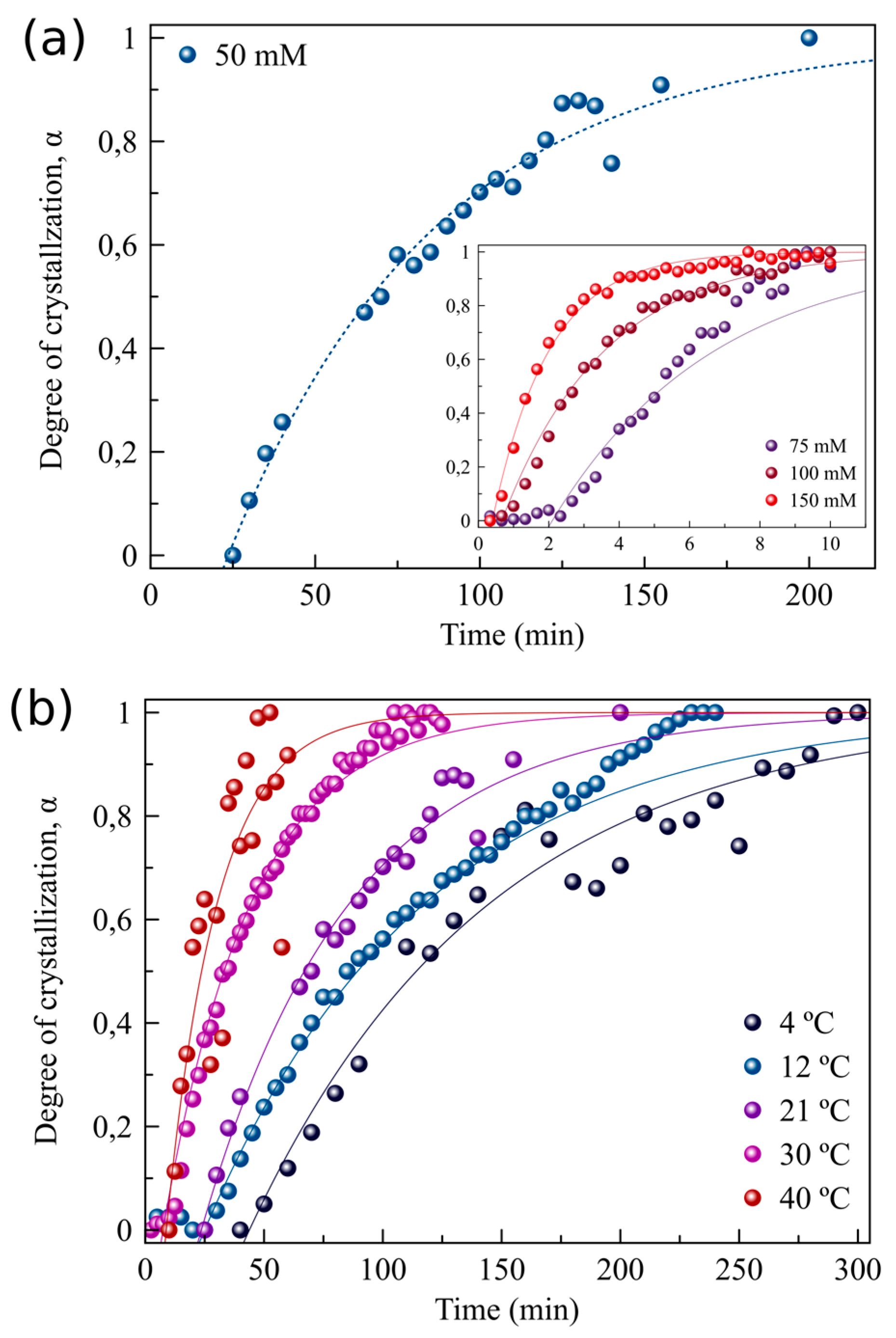
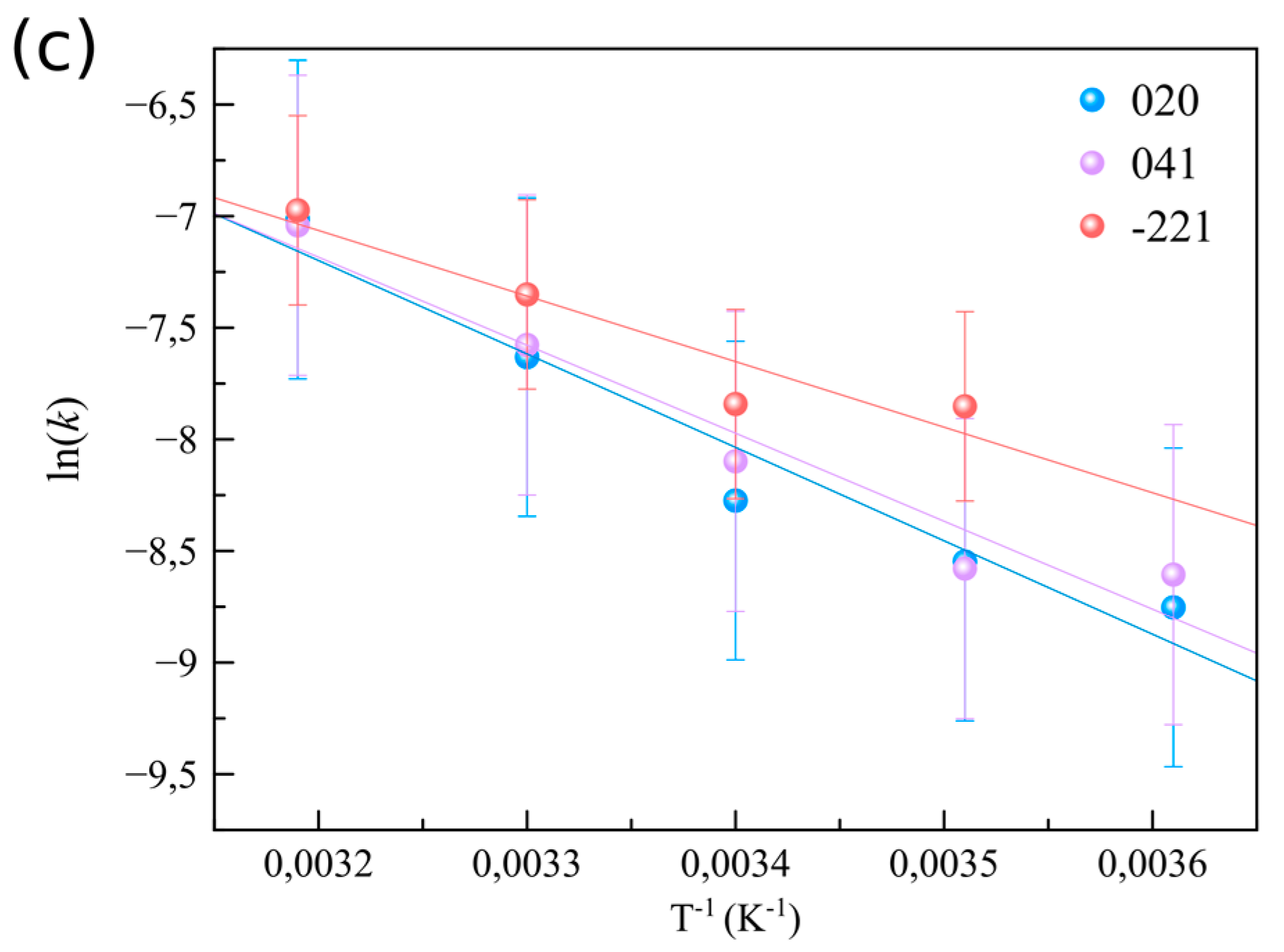
3.2. The Effect of Supersaturation and Temperature on the Crystallization Kinetics
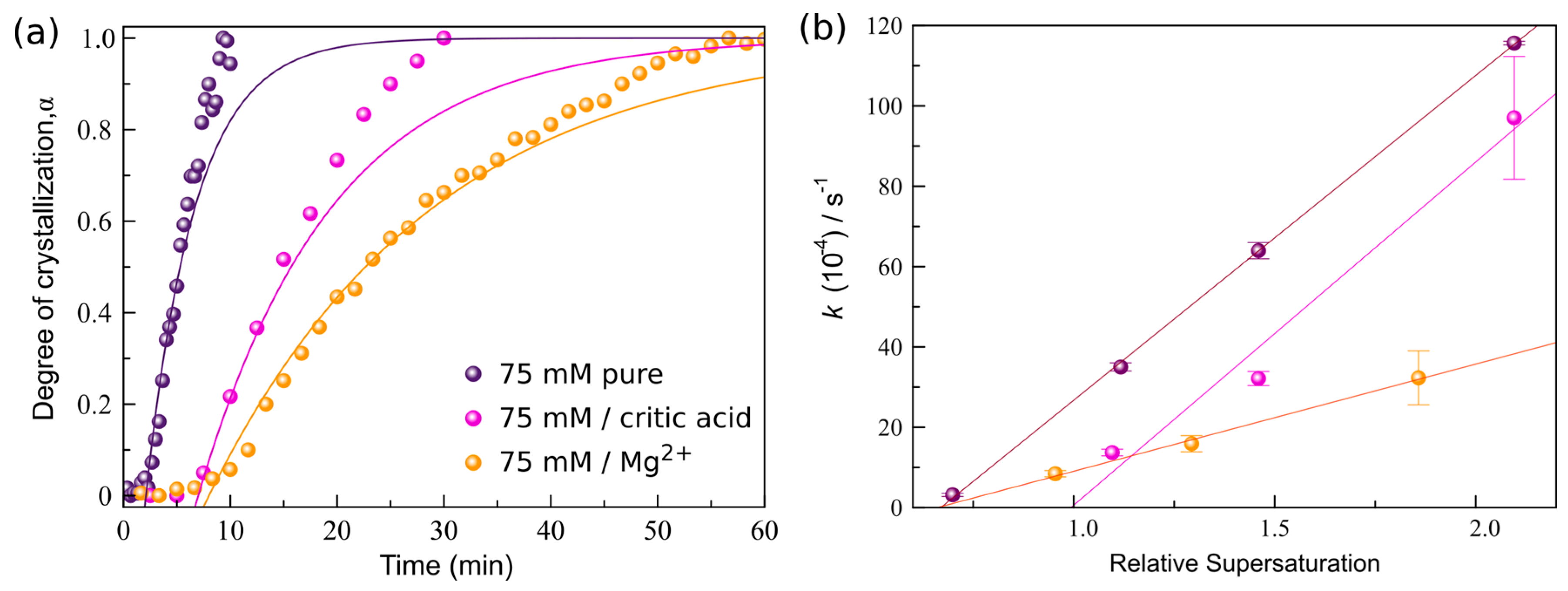
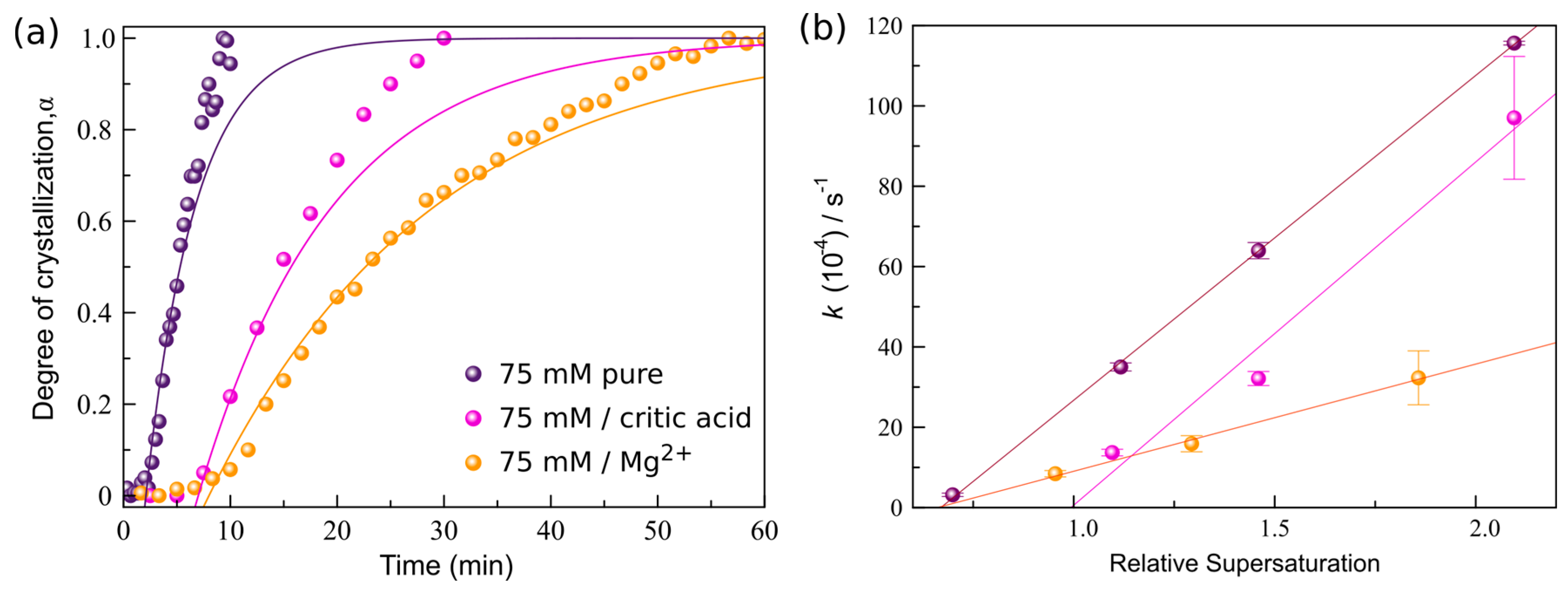
3.3. Crystallization Kinetics in the Presence of Additives
4. Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Warren, J.K. Evaporites: Sediments, Resources and Hydrocarbons; Springer: Berlin, Germany, 2006. [Google Scholar]
- Sharpe, R.; Cork, G. Gypsum and Anhydrite. In Industrial Minerals & Rocks, 7th ed.; Kogel, J.E., Ed.; Society for Mining, Metallurgy and Exploration Inc.: Englewood, CO, USA, 2006; p. 519. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Mi, B.; Elimelech, M. Gypsum scaling and cleaning in forward osmosis: Measurements and mechanisms. Environ. Sci. Technol. 2010, 44, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.E.S.; Stawski, T.M.; Benning, L.G.; Kellermeier, M. New Perspectives on Mineral Nucleation and Growth; Van Driessche, A.E.S., Kellermeier, M., Benning, L.G., Gebauer, D., Eds.; Springer: Berlin, Germany, 2017; pp. 227–256. [Google Scholar]
- Singh, N.B.; Middendorf, B. Calcium sulphate hemihydrate hydration leading to gypsum crystallization Prog. Cryst. Growth Charact. Mater. 2007, 53, 57–77. [Google Scholar] [CrossRef]
- Wang, Y.W.; Kim, Y.Y.; Christenson, H.K.; Meldrum, F.C. A new precipitation pathway for calcium sulfate dihydrate (gypsum) via amorphous and hemihydrate intermediates. Chem. Commun. 2012, 48, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.E.S.; Benning, L.G.; Rodríguez-Blanco, J.D.; Ossorio, M.; Bots, P.; García-Ruiz, J.M. The Role and Implications of Bassanite as a Stable Precursor Phase to Gypsum Precipitation. Science 2012, 336, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Jones, F. Infrared investigation of barite and gypsum crystallization: Evidence for an amorphous to crystalline transition. Cryst. Eng. Commun. 2012, 14, 8374–8381. [Google Scholar] [CrossRef]
- Saha, A.; Lee, J.; Pancera, S.M.; Bräeu, M.F.; Kempter, A.; Tripathi, A.; Bose, A. New Insights into the transformation of calcium sulfate hemihydrate to gypsum using time-resolved cryogenic transmission electron microscopy. Langmuir 2012, 28, 11182–11187. [Google Scholar] [CrossRef] [PubMed]
- Stawski, T.M.; Benning, L.G. SAXS in Inorganic and Bioispired Research. Methods Enzymol. 2013, 532, 95–127. [Google Scholar] [PubMed]
- Stawski, T.M.; Van Driessche, A.E.S.; Ossorio, M.; Rodriguez-Blanco, J.D.; Besselink, R.; Benning, L.G. Formation of calcium sulfate through the aggregation of sub-3 nanometre primary species. Nat. Commun. 2016, 7, 11177. [Google Scholar] [CrossRef] [PubMed]
- Tadros, M.E.; Mayes, I.J. Linear growth rates of calcium sulfate dihydrate crystals in the presence of additives. J. Colloid Interf. Sci. 1979, 72, 245–254. [Google Scholar] [CrossRef]
- Prisciandaro, M.; Lancia, A.; Musmarra, D. The Retarding Effect of Citric Acid on Calcium Sulfate Nucleation Kinetics. Ind. Eng. Chem. Res. 2003, 42, 6647–6652. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC: A Computer Program for Speciation, Reaction-Path, 1D-Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; U.S. Geological Survey: Denver, CO, USA, 1999.
- Huang, T.C.; Toraya, H.; Blanton, T.N.; Wu, Y. X-ray Powder Diffraction Analysis of Silver Behenate, a Possible Low-Angle Diffraction Standard. J. Appl. Crystallogr. 1993, 26, 180–184. [Google Scholar] [CrossRef]
- Fratzl, P.; Misof, K.; Zizak, I.; Rapp, G.; Amenitsch, H.; Bernstorff, S. Fibrillar structure and mechanical properties of collagen. J. Struct. Biol. 1998, 122, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Smith, A. Software Manuals for SAXS. Available online: http://confluence.diamond.ac.uk/display/SCATTERWEB/Software+Manuals+for+SAXS (accessed on 3 November 2013).
- Besselink, R.; Stawski, T.M.; Van Driessche, A.E.S.; Benning, L.G. Not just fractal surfaces, but surface fractal aggregates: Derivation of the expression for the structure factor and its applications. J. Chem. Phys. 2016, 145, 211908. [Google Scholar] [CrossRef]
- Cheary, R.W.; Coelho, A.A. Programs XFIT and FOURYA, Deposited in CCP14 Powder Diffraction Library; Engineering and Physical Sciences Research Council: Swindon, UK; Daresbury Laboratory: Warrington, UK, 1996. [Google Scholar]
- Yee, N.; Shaw, S.; Benning, L.G.; Nguyen, T.H. The rate of ferrihydrite transformation to goethite via the Fe (II) pathway. Am. Mineral. 2006, 91, 92–96. [Google Scholar] [CrossRef]
- Wang, F.; Richards, V.N.; Shields, S.P.; Buhro, W.E. Kinetics and Mechanisms of Aggregative Nanocrystal Growth. Chem. Mat. 2014, 26, 5–21. [Google Scholar] [CrossRef]
- Liebrecht, L.J.H. Sol-Gel Derived Barium Titanate Thin Films. Maste’s Thesis, University of Twente, Enschede, The Netherlands, 2006. [Google Scholar]
- Chernov, A.A. Modern Crystallography III, Crystal Growth; Springer: Berlin, Germany, 1984. [Google Scholar]
- Van Driessche, A.E.S.; García-Ruiz, J.M.; Delgado-López, J.M.; Sazaki, G. In Situ Observation of Step Dynamics on Gypsum Crystals. Cryst. Growth Des. 2010, 10, 3909–3916. [Google Scholar] [CrossRef]
- Linnikov, O.D. Investigation of the initial period of sulphate scale formation Part 2. Kinetics of calcium sulphate crystal growth at its crystallization on a heat-exchange surface. Desalination 2000, 128, 35–46. [Google Scholar] [CrossRef]
- Liu, S.; Nancollas, G.H. The kinetics of crystal growth of calcium sulfate dihydrate. J. Cryst. Growth 1970, 6, 281–289. [Google Scholar] [CrossRef]
- Smith, B.R.; Sweett, F. The crystallization of calcium sulfate dehydrate. J. Colloid Interf. Sci. 1971, 37, 612–618. [Google Scholar] [CrossRef]
- Nancollas, G.H.; Reddy, M.M.; Tsai, F. Calcium sulfate dihydrate crystal growth in aqueous solution at elevated temperatures. J. Cryst. Growth 1973, 20, 125–134. [Google Scholar] [CrossRef]
- Witkamp, G.J.; Van der Eerden, J.P.; Van Rosmalen, G.M. Growth of gypsum: I. Kinetics. J. Cryst. Growth 1990, 120, 281–289. [Google Scholar] [CrossRef]
- Klepetsanis, P.G.; Koutsoukos, P.G. Spontaneous precipitation of calcium sulfate at conditions of sustained supersaturation. J. Colloid Interf. Sci. 1991, 143, 299–308. [Google Scholar] [CrossRef]
- Klepetsanis, P.G.; Dalas, E.; Koutsoukos, P.G. Role of Temperature in the Spontaneous Precipitation of Calcium Sulfate Dihydrate. Langmuir 1999, 15, 1534–1540. [Google Scholar] [CrossRef]
- De Yoreo, J.J.; Vekilov, P.G. Principles of Crystal Nucleation and Growth. Mineral. Soc. Am. 2003, 54, 57–93. [Google Scholar] [CrossRef]
- Bosbach, D.; Hochella, M.F. Gypsum growth in the presence of growth inhibitors: A scanning force microscopy study. Chem. Geol. 1996, 132, 227–236. [Google Scholar] [CrossRef]
- Rabizadeh, T.; Stawski, T.M.; Morgan, D.J.; Peacock, C.L.; Benning, L.G. The effects of inorganic additives on the nucleation and growth kinetics of calcium sulfate dihydrate crystals. Cryst. Growth Des. 2017, 17, 582–589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [CaSO4] (mmol·L−1) | [Mg2+] (mmol·L−1) | [C6H8O7] (mmol·L−1) | T (°C) | SIgyp |
|---|---|---|---|---|
| 50 | 4 | 0.59 | ||
| 50 | 12 | 0.56 | ||
| 50 | 21 | 0.53 | ||
| 50 | 30 | 0.49 | ||
| 50 | 40 | 0.46 | ||
| 75 | 21 | 0.75 | ||
| 75 | 75 | 21 | 0.67 | |
| 75 | 2.6 | 21 | 0.74 | |
| 100 | 21 | 0.90 | ||
| 100 | 100 | 21 | 0.83 | |
| 100 | 2.6 | 21 | 0.9 | |
| 150 | 21 | 1.13 | ||
| 150 | 150 | 21 | 1.05 | |
| 150 | 2.6 | 21 | 1.13 |
| Reference | EA/kJ·mol−1 | Observations |
|---|---|---|
| [27] | 62.8 | Crystallization of gypsum on the addition of seed crystals to stable supersaturated solutions was studied from 15–45 °C. |
| [28] | 62.8 | Conductometric study of seeded crystallization at four temperatures and several ionic compositions |
| [29] | 58.6 | Growth of gypsum seed crystals from supersaturated solutions from 60–105 °C. |
| [30] | 60.0 | A suspension of gypsum crystals was grown from electrolyte solution using the constant composition technique |
| [31] | 184.2 | Potentiometric measurements in supersaturated solutions of calcium sulphate. |
| [32] | 46.0–67.0 | The spontaneous precipitation of calcium sulphate in supersaturated solutions over the temperature range between 25.0 and 80.0 °C was investigated by monitoring the solution specific conductivity during desupersaturation. |
| [26] | 55.0–63.0 | Experimental data of the growth rate of the (120) face from electrolytic solutions on a heated metal surface. |
| [25] | 70.7 | Step kinetics on the {010} face were measured as a function of supersaturation at different temperatures |
| This work | 25.0–35.0 | (020) EA = 35.2 ± 0.6 kJ·mol−1; (041) EA = 33.1 ± 0.6 kJ·mol−1; (−221) EA = 24.9 ± 0.7 kJ·mol−1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ossorio, M.; Stawski, T.M.; Rodríguez-Blanco, J.D.; Sleutel, M.; García-Ruiz, J.M.; Benning, L.G.; Van Driessche, A.E.S. Physicochemical and Additive Controls on the Multistep Precipitation Pathway of Gypsum. Minerals 2017, 7, 140. https://doi.org/10.3390/min7080140
Ossorio M, Stawski TM, Rodríguez-Blanco JD, Sleutel M, García-Ruiz JM, Benning LG, Van Driessche AES. Physicochemical and Additive Controls on the Multistep Precipitation Pathway of Gypsum. Minerals. 2017; 7(8):140. https://doi.org/10.3390/min7080140
Chicago/Turabian StyleOssorio, Mercedes, Tomasz M. Stawski, Juan Diego Rodríguez-Blanco, Mike Sleutel, Juan Manuel García-Ruiz, Liane G. Benning, and Alexander E. S. Van Driessche. 2017. "Physicochemical and Additive Controls on the Multistep Precipitation Pathway of Gypsum" Minerals 7, no. 8: 140. https://doi.org/10.3390/min7080140