Transition from Endothermic to Exothermic Dissolution of Hydroxyapatite Ca5(PO4)3OH–Johnbaumite Ca5(AsO4)3OH Solid Solution Series at Temperatures Ranging from 5 to 65 °C


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Dissolution Experiments
2.3. Analytical Methods
3. Results
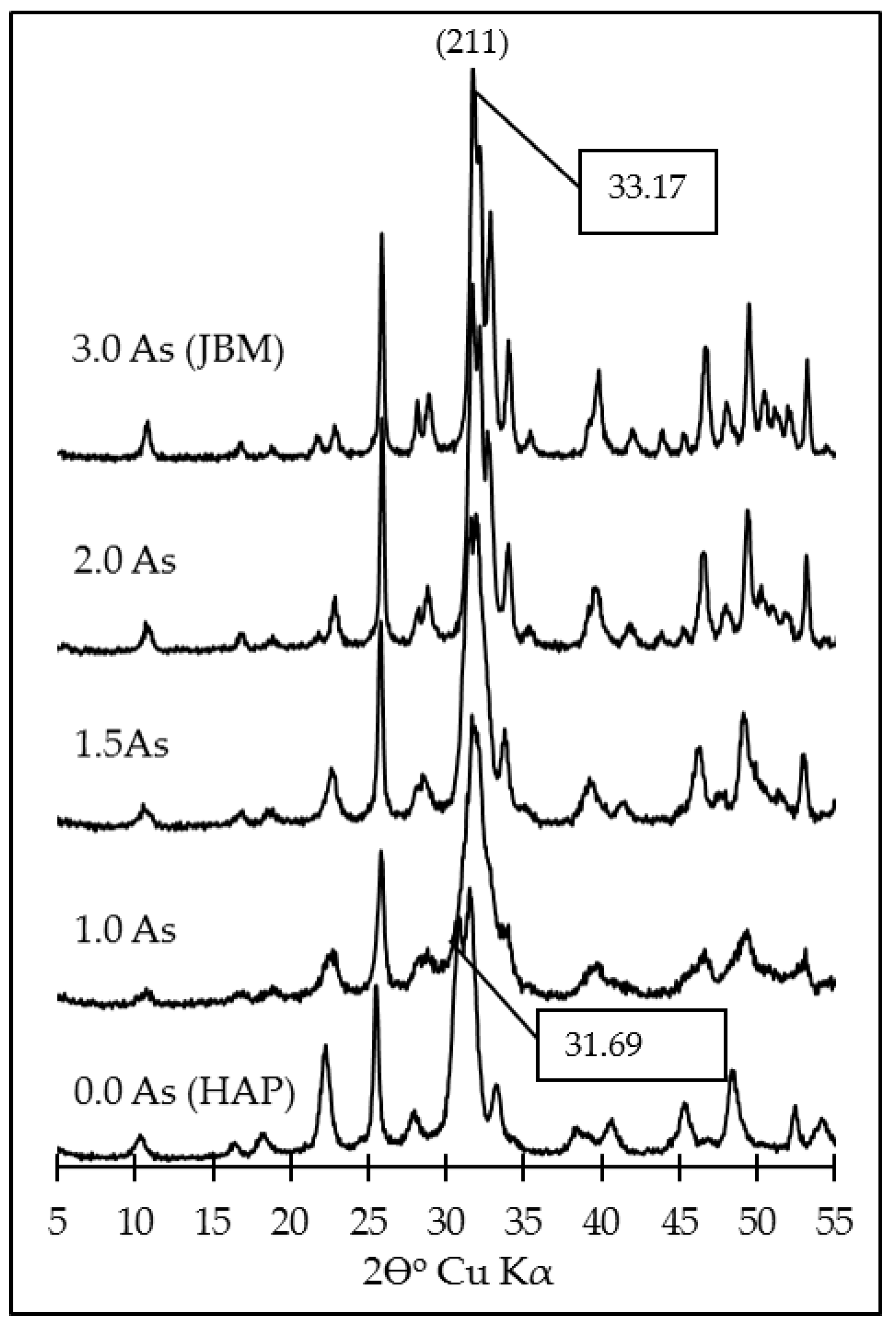
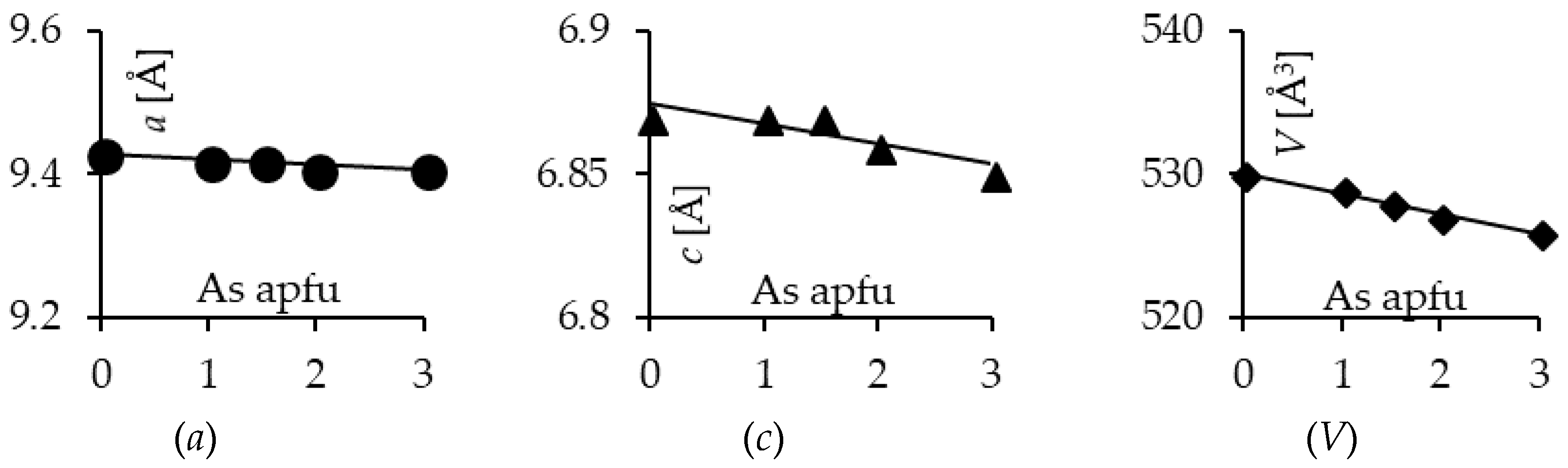
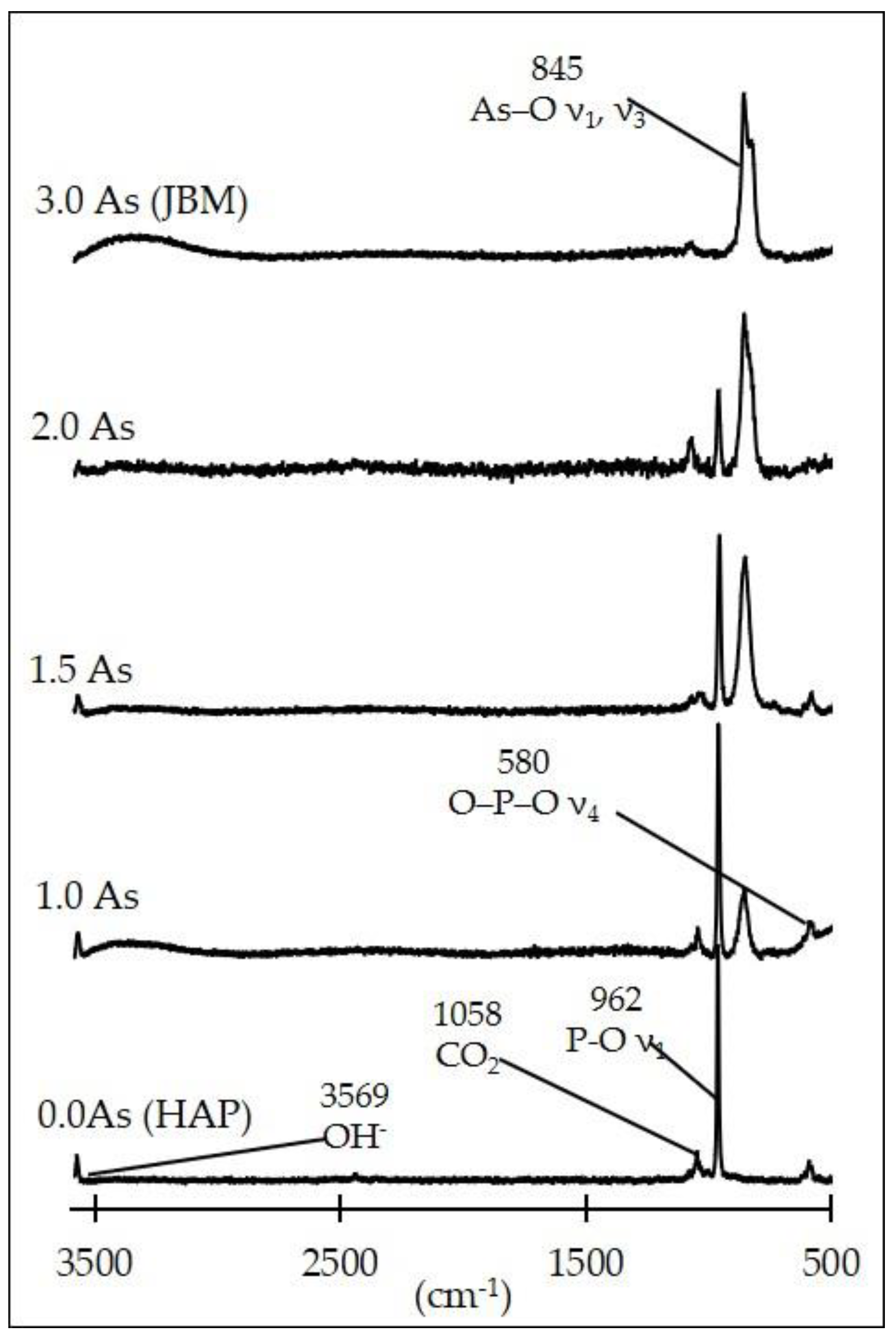
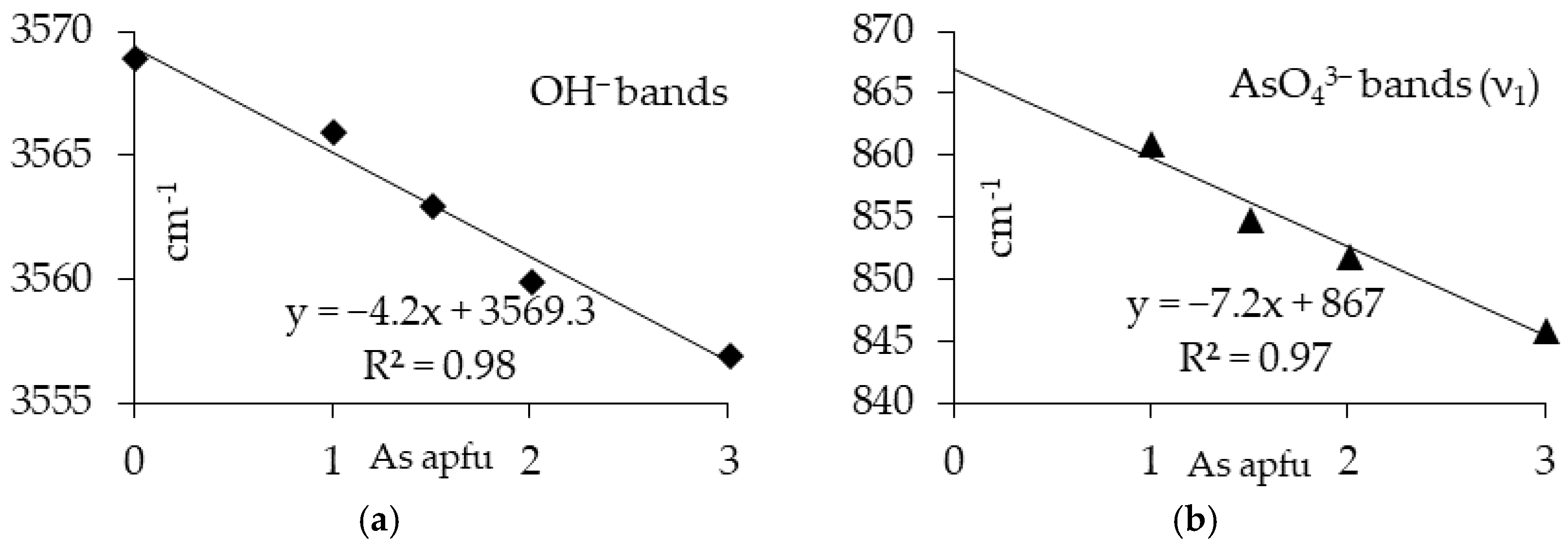
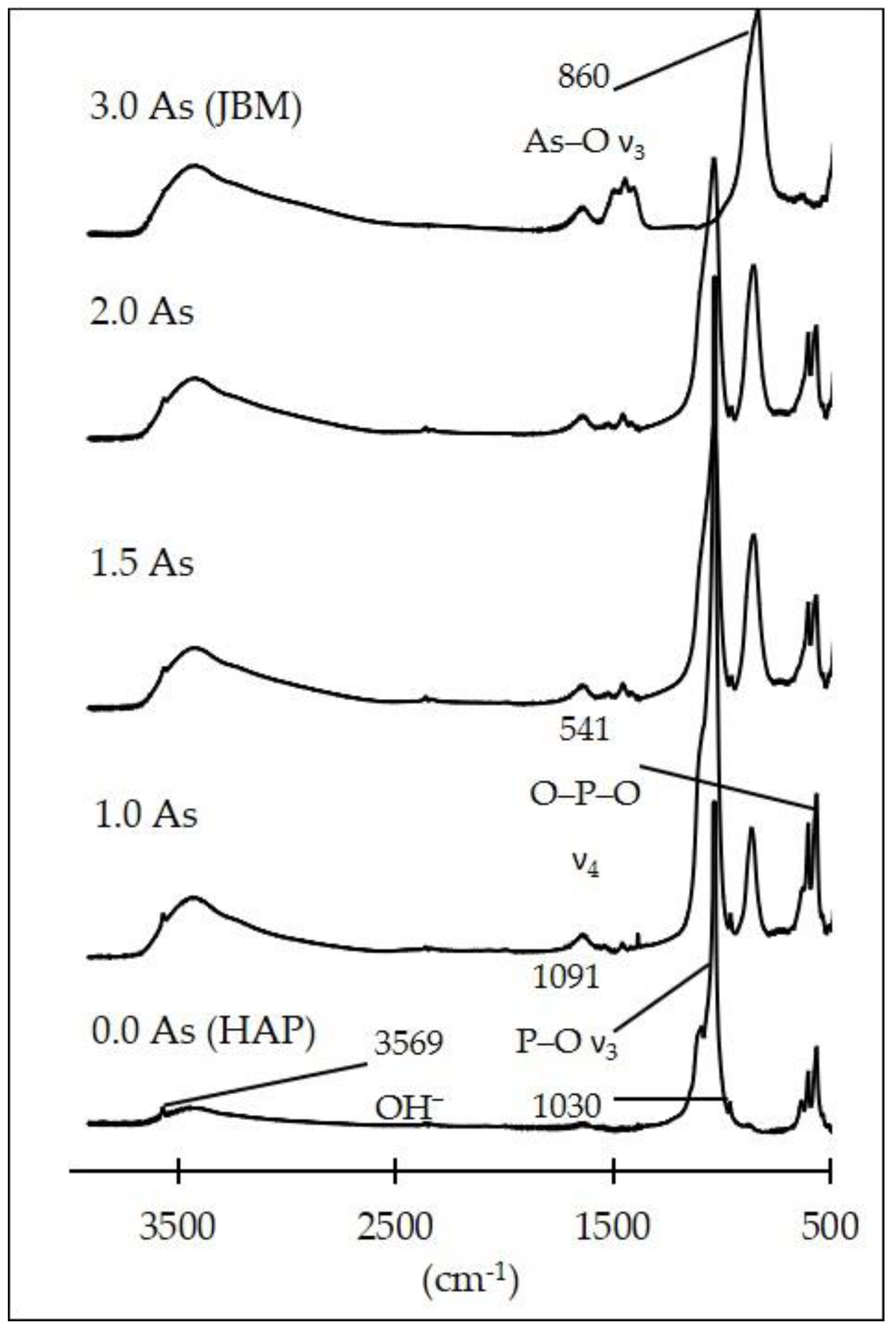
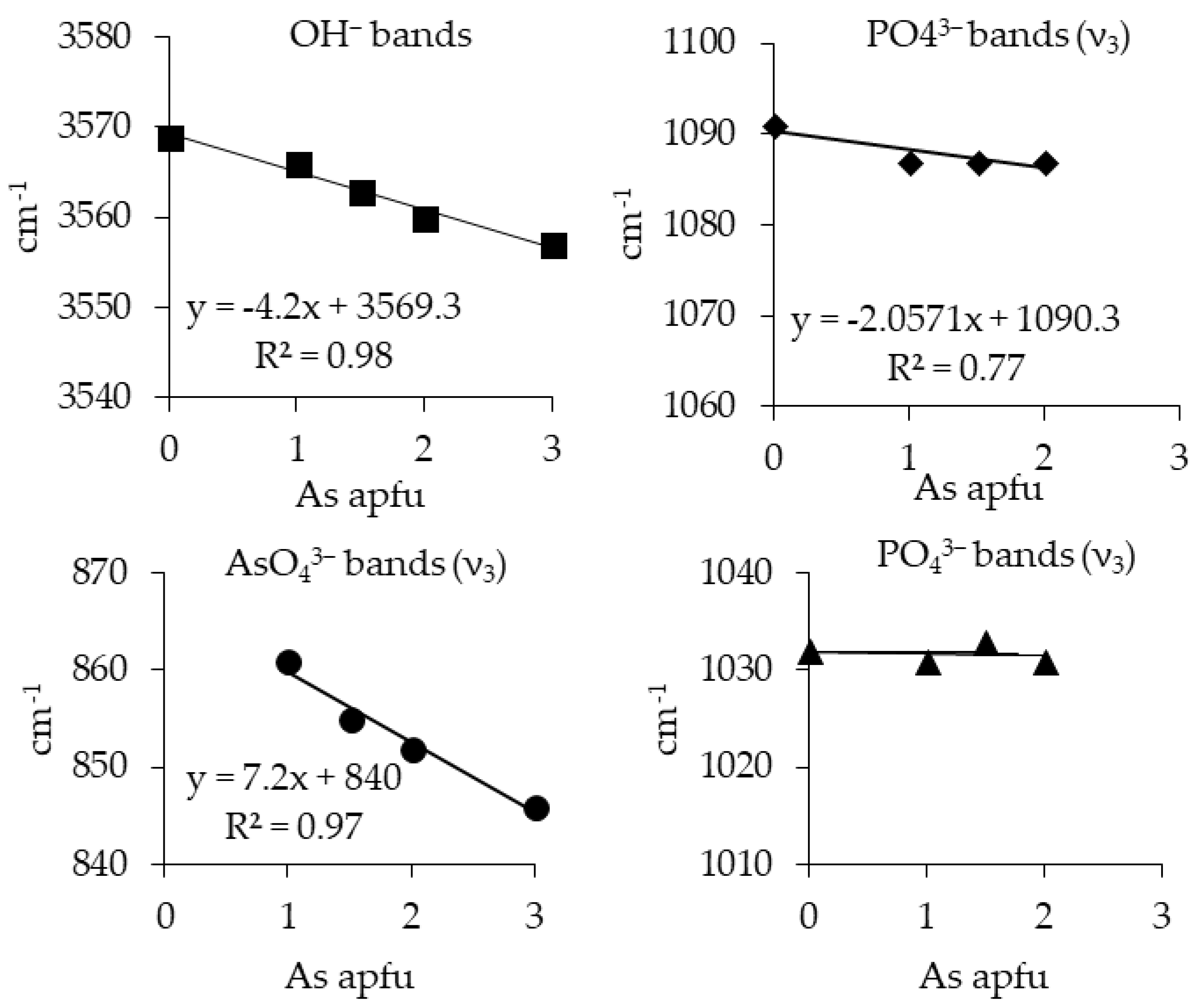
3.1. Characterization of the Synthesis Products
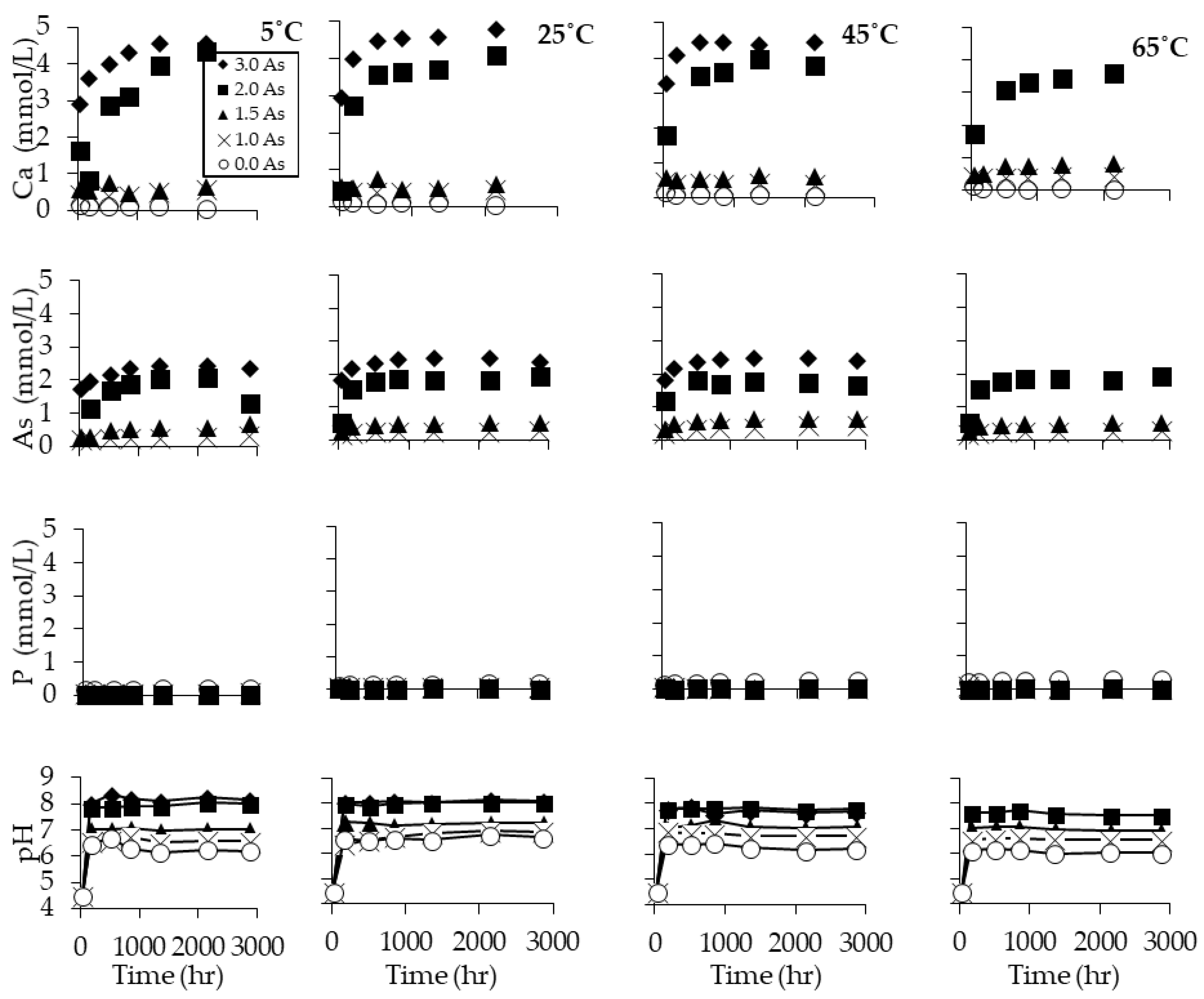
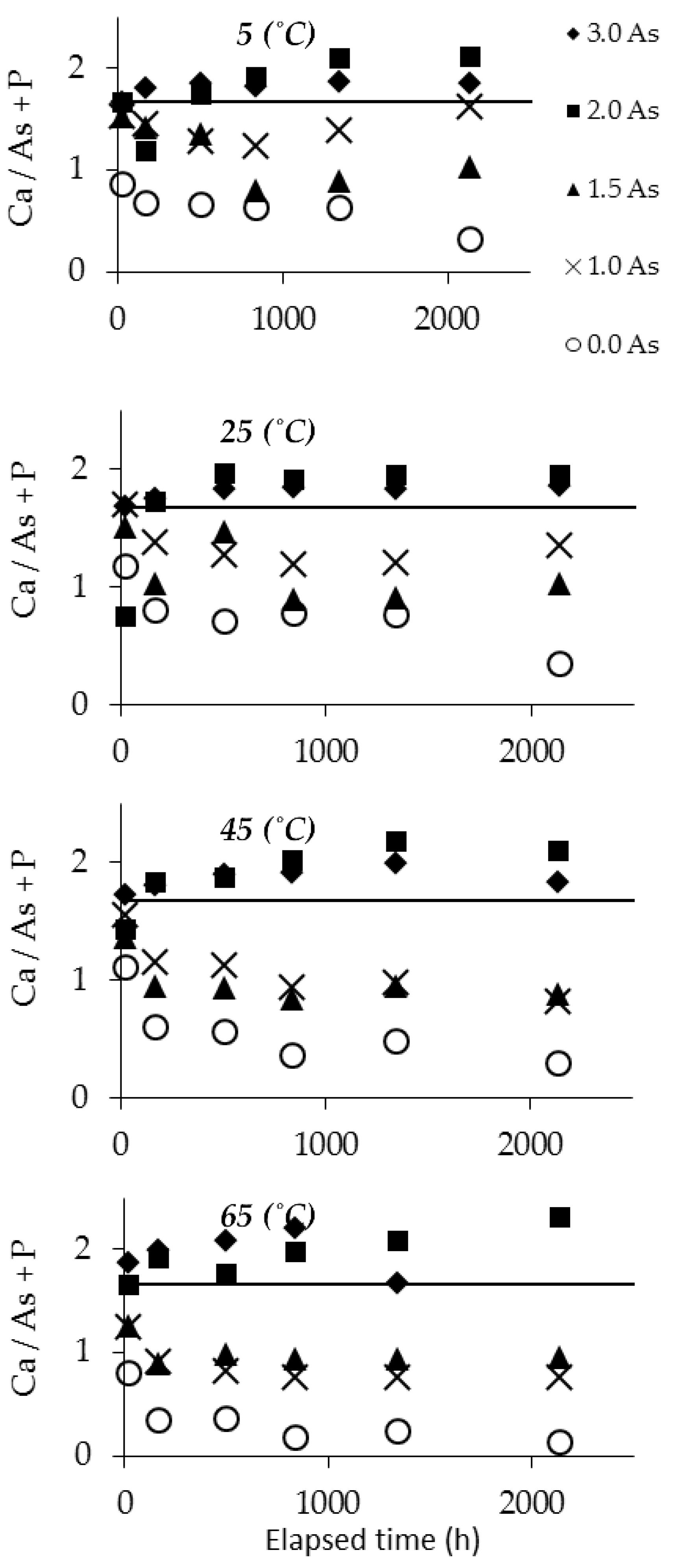
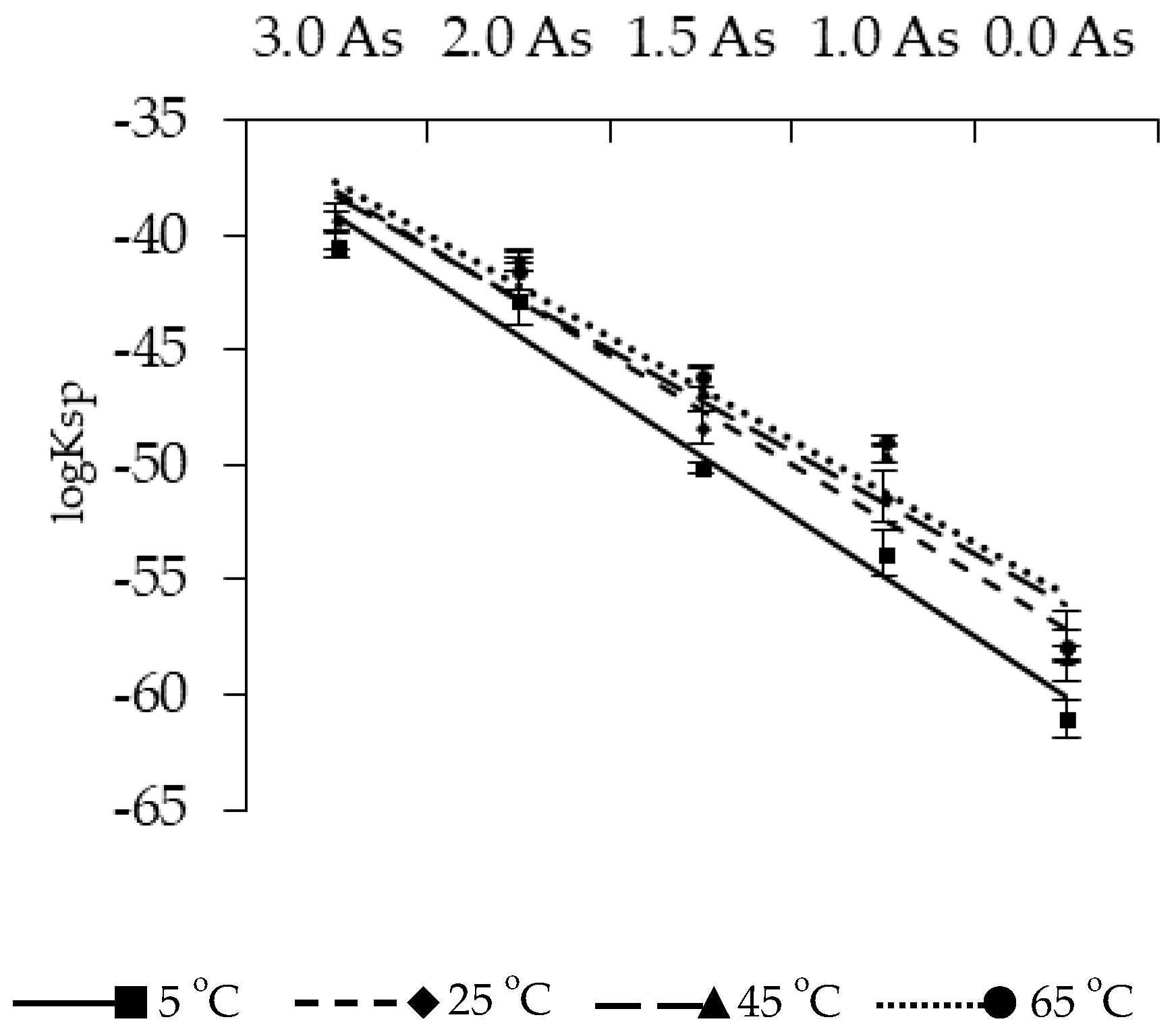
3.2. Dissolution Experiments Results
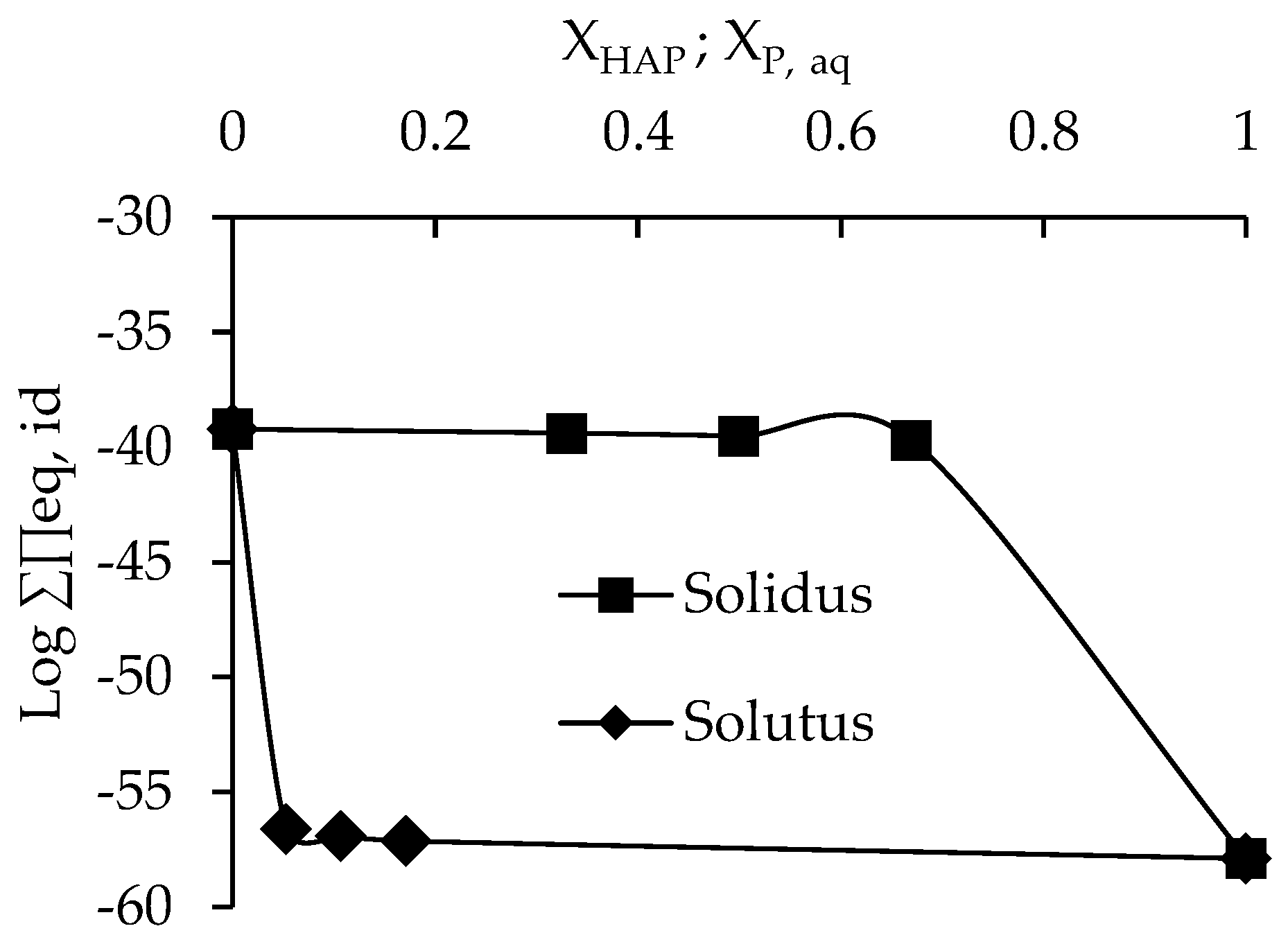
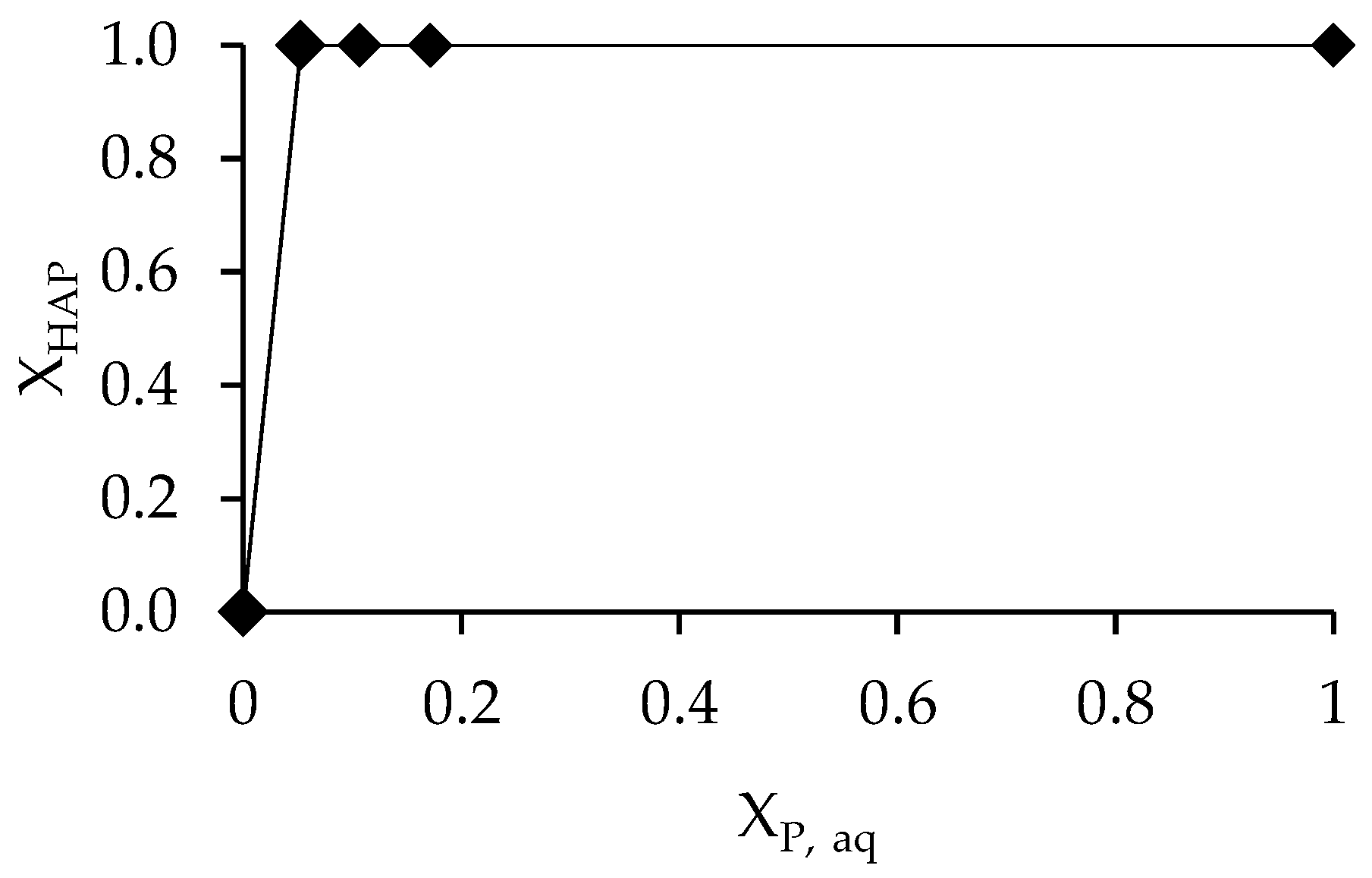
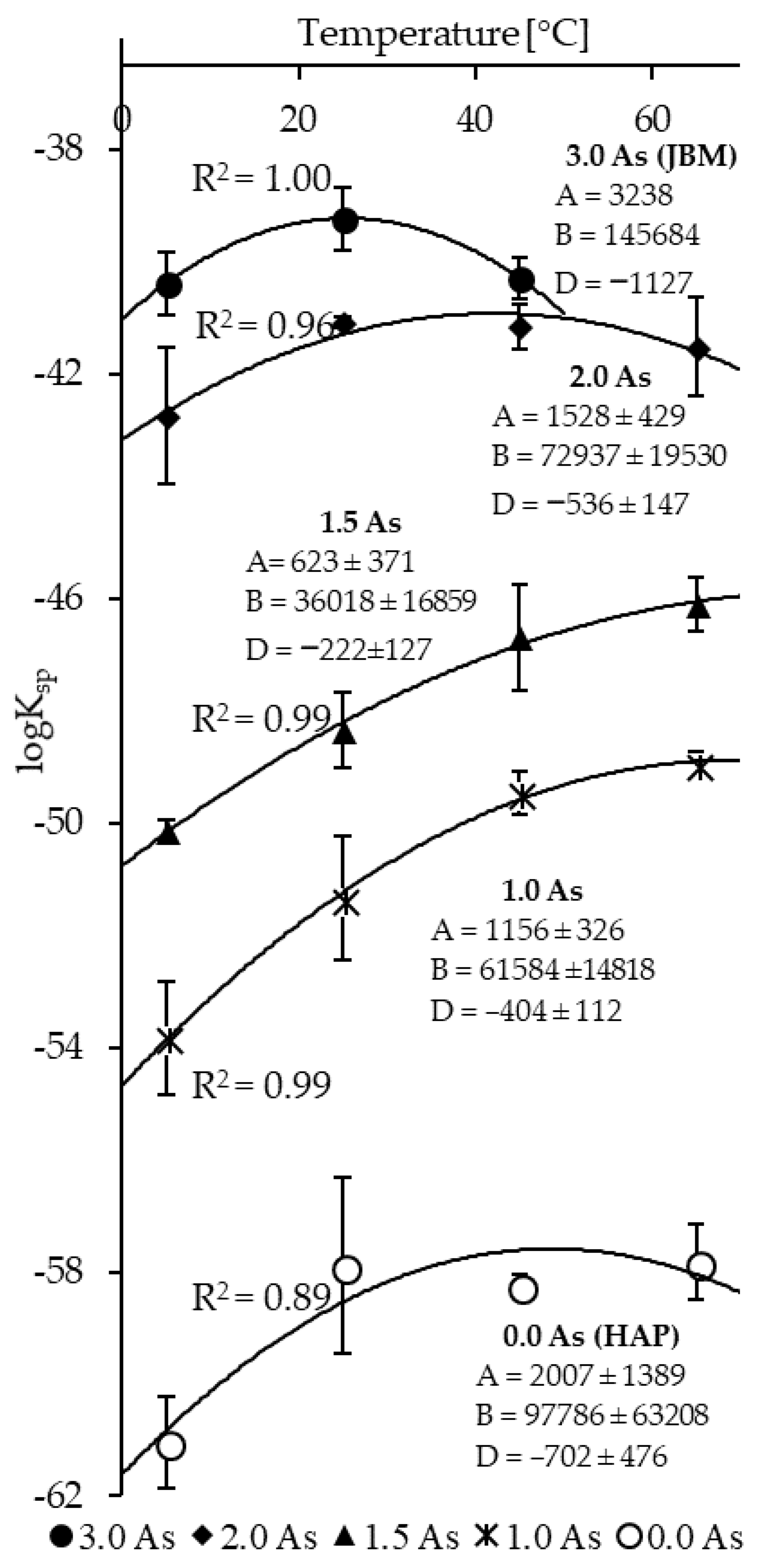
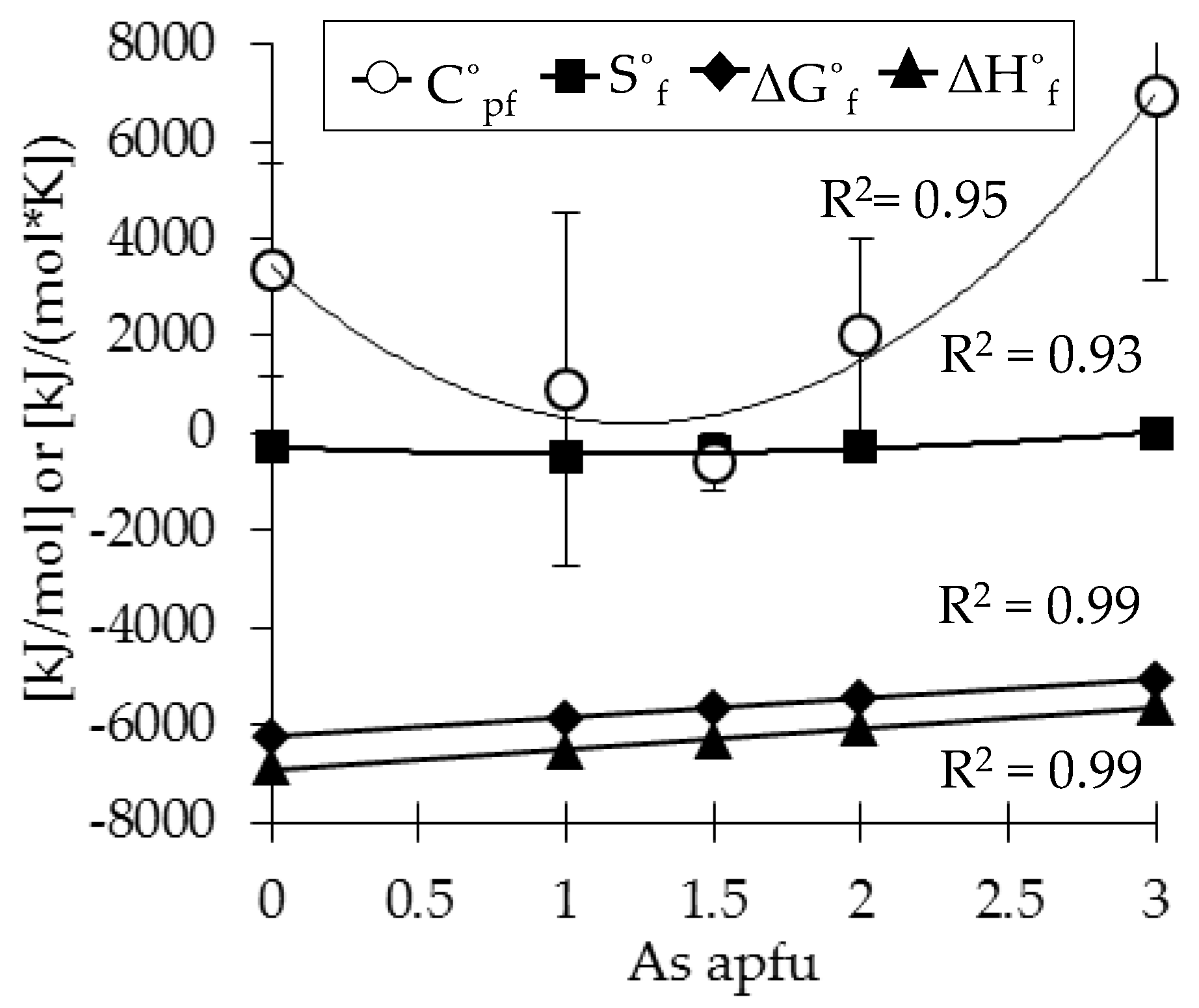
3.3. Determination of Solubility and Thermodynamic Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgment
Conflicts of Interest
References
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Phosphates; Elsevier: Amsterdam, The Netherlands, 1994; ISBN 0-444-81582-1. [Google Scholar]
- Li, Z.; Zhou, M.; Lin, W. The Research of Nanoparticle and Microparticle Hydroxyapatite Amendment in Multiple Heavy Metals Contaminated Soil Remediation. J. Nanomater. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Valsami-Jones, E.; Ragnarsdottir, K.V.; Putnis, A.; Bosbach, D.; Kemp, A.J.; Cressey, G. The dissolution of apatite in the presence of aqueous metal cations at pH 2–7. Chem. Geol. 1998, 151, 215–233. [Google Scholar] [CrossRef]
- Matusik, J.; Bajda, T.; Manecki, M. Immobilization of aqueous cadmium by addition of phosphates. J. Hazard. Mater. 2008, 152, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Giammar, D.E.; Xie, L.Y.; Pasteris, J.D. Immobilization of lead with nanocrystalline carbonated apatite present in fish bone. Environ. Eng. Sci. 2008, 25, 725–735. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Chen, Y.; Xie, Q.; Lan, J.; Qian, M.; He, N. A comparative study on the dissolution and solubility of hydroxylapatite and fluorapatite at 25 °C and 45 °C. Chem. Geol. 2009, 268, 89–96. [Google Scholar] [CrossRef]
- Lee, Y.J.; Stephens, P.W.; Tang, Y. Arsenate substitution in hydroxylapatite: Structural characterization of the Ca5(PxAs1−xO4)3(OH) solid solution. Am. Mineral. 2009, 94, 666–675. [Google Scholar] [CrossRef]
- Mushak, P.; Crocetti, A.F. Risk and revisionism in arsenic cancer risk assessment. Environ. Health Perspect. 1995, 103, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.E. An X-ray diffraction study of synthetic members of the pyromorphite series. Am. Mineral. 1966, 51, 1712–1721. [Google Scholar]
- Marciniak, H.; Diduszko, R.; Kozak, M. XRAYAN. Program do Rentgenowskiej Analizy Fazowej, Wersja 4.0.1; Koma: Warszawa, Poland, 2006. (In Polish) [Google Scholar]
- Holland, J.B.; Redfern, S.A.T. UNITCELL: A nonlinear least-squares program for cell parameter refinement implementing regression and deletion diagnostics. J. Appl. Cryst. 1997, 30, 84. [Google Scholar] [CrossRef]
- Lenoble, V.; Deluchat, V.; Serpaud, B.; Bollinger, J.C. Arsenite oxidation and arsenate determination by the molybdene blue method. Talanta 2003, 61, 267–276. [Google Scholar] [CrossRef]
- Leroy, N.; Bres, E. Structure and Substitutions in Fluorapatite. Eur. Cell Mater. 2001, 2, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, X.; Long, F.; Liu, H.; Qian, M.; He, N. Synthesis and characterization of arsenate/phosphate hydroxyapatite solid solution. Mater. Lett. 2009, 63, 1185–1188. [Google Scholar] [CrossRef]
- Kim, I.Y.; Koichi, K.; Ohtsuki, C. Hydroxyapatite formation through dissolution-precipitation reaction: Effects of solubility of starting materials. Ceram. Int. 2014, 40, 14385–14390. [Google Scholar] [CrossRef]
- Giera, A.; Manecki, M.; Bajda, T.; Rakovan, J.; Kwaśniak-Kominek, M.; Marchlewski, T. Arsenate substitution in lead hydroxyl apatites: A Raman spectroscopic study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 152, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwaśniak-Kominek, M.; Manecki, M.; Matusik, J.; Lempart, M. Carbonate substitution in lead hydroxyapatite Pb5(PO4)3OH. J. Mol. Struct. 2017, 1147, 594–602. [Google Scholar] [CrossRef]
- Flis, J.; Manecki, M.; Bajda, T.; Klasa, J. Synchrotron-based X-ray diffraction of the lead apatite series Pb10(PO4)6Cl2–Pb10(AsO4)6Cl2. J. Synchrotron Radiat. 2010, 17, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ding, Y.; Leng, Y. Infrared spectroscopic characterization of carbonated apatite: A combined experimental and computational study. J. Biomed. Mater. Res. 2014, 102, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Farzadi, A.; Bakhshi, F.; Solati-Hashjin, M.; Asadi-Eydivand, M.; Azuan abu Osman, N. Magnesium incorporated hydroxyapatite: Synthesis and structural properties characterization. Ceram. Int. 2014, 40, 6021–6029. [Google Scholar] [CrossRef]
- Masaoka, M.; Kyono, A.; Hatta, T.; Kimata, M. Single crystal growth of Pb5(PxAs1−xO4)3Cl solid solution with apatite type structure. J. Cryst. Growth 2004, 292, 129–135. [Google Scholar] [CrossRef]
- Zhang, Y.; Reddy Venugopal, J.; El-Turki, A.; Ramakrishna, S.; Su, B.; Lim, C.T. Electrospun biomimetic nanocomposite nanofibers of hydroxyapatite/chitosan for bone tissue engineering. Biomaterials 2008, 29, 4314–4322. [Google Scholar] [CrossRef] [PubMed]
- Bajda, T.; Mozgawa, W.; Manecki, M.; Flis, J. Vibrational spectroscopic study of mimetite-pyromorphite solid solutions. Polyhedron 2011, 30, 2479–2485. [Google Scholar] [CrossRef]
- Flis, J.; Manecki, M.; Bajda, T. Solubility of pyromorphite Pb5(PO4)3Cl-mimetite Pb5(AsO4)3Cl solid solution series. Geochim. Cosmochim. Acta 2011, 75, 1858–1868. [Google Scholar] [CrossRef]
- Bakan, F.; Laçin, O.; Sarac, H. A novel low temperature sol-gel synthesis process for thermally stable nanocrystalline hydroxyapatite. Powder Technol. 2013, 233, 295–302. [Google Scholar] [CrossRef]
- Johnson, J.; Anderson, G.; Parkhurst, D. Database ‘Thermo. com. V8. R6. 230,’Rev. 1.11; Lawrence Livermore National Laboratory: Livermore, CA, USA, 2000.
- Glynn, P.D.; Reardon, E.J. Solid solution aqueous solution equilibria: Thermodynamic theory and representation. Am. J. Sci. 1990, 278, 164–201. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Zeng, H.; Wang, D.; Liu, J.; Liu, H.; Qian, M.; Xu, L. Dissolution and solubility of the arsenate-phosphate hydroxylapatite solid solution [Ca5(PxAs1−xO4)3(OH)] at 25 °C. Environ. Chem. 2011, 8, 133–145. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s guide to PHREEQC-A computer program for speciation, reaction-path, 1D-transport, and inverse geochemical calculations. US Geol. Surv. Water Resour. Investig. Rep. 1999, 95–4227. [Google Scholar]
- Bothe, J.V., Jr.; Brown, P.W. The stabilities of calcium arsenates at 23 ± 1 °C. J. Hazard. Mater. 1999, 69, 197–207. [Google Scholar] [CrossRef]
- Wei, C.; Zhu, Y.; Yang, F.; Li, J.; Zhu, Z.; Zhu, H. Dissolution of hydroxylapatite and fluoroapatite at 25 °C at different pH. Res. J. Chem. Environ. 2013, 11, 57–61. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry, Chemical Equilibria and Rates in Natural Waters; Wiley: New York, NY, USA, 1996; ISBN 0-471-511184-6. [Google Scholar]
- Moreno, E.C.; Gregory, T.M.; Brown, W.E. Preparation and Solubility of Hydroxyapatite. J. Res. Natl. Bur. Stand. 1968, 72A, 773–782. [Google Scholar] [CrossRef]
- McDowell, H.; Gregory, T.M.; Brown, W.E. Solubility of Ca5(PO4)3OH in the system Ca(OH)2–H3PO4–H2O at 5, 15, 25, and 37.8 °C. J. Res. Natl. Bur. Stand. 1977, 81, 273. [Google Scholar] [CrossRef]
- Prieto, M. Thermodynamics of solid solution-aqueous solution systems. Rev. Mineral. Geochem. 2009, 70, 47–85. [Google Scholar] [CrossRef]
- Prieto, M.; Putnis, A.; Fernandez-Diaz, L. Crystallization of solid solutions from aqueous solutions in a porous medium: Zoning in (Ba, Sr)SO4. Geol. Mag. 1993, 130, 289–299. [Google Scholar] [CrossRef]
- Chaϊrat, C.; Oelkers, E.H.; Schott, J.; Lartigue, J.E. Fluorapatite surface composition in aqueous solution deduced from potentiometric, electrokinetic, and solubility measurements, and spectroscopic observations. Geochim. Cosmochim. Acta 2007, 71, 5888–5900. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. A review on the dissolution models of calcium apatites. Prog. Cryst. Gr. Char. Mater. 2002, 44, 45–61. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Munoz, J.L. Geochemical Thermodynamics; Blackwell: Boston, MA, USA, 1994. [Google Scholar]
- Topolska, J.; Manecki, M.; Bajda, T.; Borkiewicz, O.; Budzewski, P. Solubility of pyromorphite Pb5(PO4)3Cl at 5–65 °C and its experimentally determined thermodynamic parameters. J. Chem. Thermodyn. 2016, 98, 282–287. [Google Scholar] [CrossRef]
- Marcus, Y. The thermodynamics of solvation of ions. Part 2—The enthalpy of hydration at 298.15 K. J. Chem. Soc. Faraday Trans. 1987, 2, 231–569. [Google Scholar] [CrossRef]
- Jemal, M. Thermochemistry and relative stability of apatite phosphates. Phosphor. Res. Bull. 2004, 15, 119–124. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, B.; Zhu, Z.; Liu, H.; Huang, Y.; Zhao, X.; Liang, M. Characterization, dissolution and solubility of the hydroxypyromorphite-hydroxyapatite solid solution [(PbxCa1−x)5(PO4)3OH] at 25 °C and pH 2–9. Geochem. Trans. 2016, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Drouet, C. A comprehensive guide to experimental and predicted thermodynamic properties of phosphate apatite minerals in view of applicative purposes. J. Chem. Thermodyn. 2015, 81, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Vieillard, P.; Tardy, Y. Thermochemical properties of phosphates. In Phosphate Minerals; Nriagu, J., Moore, P., Eds.; Springer: Berlin, Germany, 1984; pp. 171–198. [Google Scholar]
- Cruz, F.; Da Piedade, M.E.; Calado, J.C.G. Standard molar enthalpies of formation of hydroxy-, chlor-, and bromapatite. J. Chem. Thermodyn. 2005, 37, 1061–1070. [Google Scholar] [CrossRef]
- Shock, E.L.; Sassani, D.C.; Willis, M.; Sverjensky, D.A. Inorganic species in geology fluids: Correlations among standard molal thermodynamics properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 1997, 61, 950–970. [Google Scholar] [CrossRef]
- Novikov, A.N.; Rassokhina, L.Y. Heat Capacity and Density of Solutions of Calcium and Cadmium Nitrates in N-Methylpyrrolidone at 298.15 K. Russ. J. Phys. Chem. 2013, 87, 1318–1321. [Google Scholar] [CrossRef]
- Bianchi, H.; Tremaine, P.R. Thermodynamics of aqueous phosphate solutions: Apparent molar heat capacities and volumes of sodium and tetramethyloammonium salt at 25 °C. J. Solut. Chem. 1995, 24, 439–463. [Google Scholar] [CrossRef]
- Wagman, D.D.; Evans, W.H.; Parker, V.B.; Schumm, R.H.; Halow, I.; Bailey, S.M.; Churney, K.L.; Nuttall, R.L. The NBS Tables of Chemical Thermodynamic Properties. J. Phys. Chem. Ref. Data 1989, 18, 1807–1812. [Google Scholar] [CrossRef]
- Sienko, M.J.; Plane, R.A. Chemia. Podstawy i Własności; Wydawnictwo Naukowo-Techniczne: Warsaw, Poland, 1980; p. 788. (In Polish) [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic Phases | Mass of the Reagents (g/400 mL of Water) | ||
|---|---|---|---|
| Ca(NO3)2∙4H2O | (NH4)H2PO4 | Na2AsHO4∙7H2O | |
| 0.0 As (HAP): Ca5(PO4)3OH | 41.01 | 13.74 | - |
| 1.0 As: Ca5(PO4)2(AsO4)OH | 21.62 | 4.21 | 5.71 |
| 1.5 As: Ca5(PO4)1.5(AsO4)1.5OH | 20.78 | 3.07 | 8.24 |
| 2.0 As: Ca5(PO4)(AsO4)2OH | 20.01 | 2.24 | 10.55 |
| 3.0 As (JBM): Ca5(AsO4)3OH | 37.24 | - | 29.52 |
| Sample | Theoretical Formula | CaO wt % | P2O5 wt % | As2O3 wt % | Ca/(P + As) Molar Ratio |
|---|---|---|---|---|---|
| 0.0 As HAP | Ca5(PO4)3OH | 52.19 | 47.81 | - | 1.67 |
| 1.0 As | Ca5(PO4)2(AsO4)OH | 49.11 | 34.49 | 16.39 | 1.67 |
| 1.5 As | Ca5(PO4)1.5(AsO4)1.5OH | 48.73 | 23.91 | 27.36 | 1.66 |
| 2.0 As | Ca5(PO4)(AsO4)2OH | 52.0 | 13.93 | 34.08 | 1.66 |
| 3.0 As JBM | Ca5(AsO4)3OH | 46.76 | - | 53.25 | 1.67 |
| Equilibrium Reaction | logK298.15 |
|---|---|
| Ca5(PO4)3OH + 4H+ = 5Ca2+(aq) + 3HPO43−(aq) + H2O | −56.00 |
| Ca5(AsO4)3OH + 4H+ = 5Ca2+(aq) + 3HAsO43−(aq) + H2O | 10.75 |
| PO43−(aq) + H+ = HPO42−(aq) | 12.34 |
| PO43−(aq) + 2H+ = H2PO4−(aq) | 19.53 |
| PO43−(aq) + 3H+ = H3PO40(aq) | 21.70 |
| H2AsO4−(aq) = AsO43−(aq) + 2H+ | −18.36 |
| H2AsO4−(aq) = HAsO42−(aq) + H+ | −6.76 |
| H3AsO40(aq) = H2AsO4−(aq) + H+ | 2.25 |
| Sample ID | Temperature | log Activity | log Activity | log Activity | log Activity | Average logKsp |
|---|---|---|---|---|---|---|
| (K) | Ca2+ | PO43− | AsO43− | OH− | ||
| 3.0 As (JBM) | 278.15 | −2.766 | - | −6.667 | −6.542 | −40.37 ± 0.56 |
| 298.15 | −2.761 | - | −6.495 | −5.930 | −39.22 ± 0.56 | |
| 318.15 | −2.800 | - | −6.833 | −5.783 | −40.28 ± 0.37 | |
| 338.15 | - | - | - | - | - | |
| 2.0 As | 278.15 | −2.747 | −8.197 | −7.021 | −6.762 | −42.74 ± 1.20 |
| 298.15 | −2.743 | −7.951 | −6.698 | −6.004 | −41.07 ± 0.11 | |
| 318.15 | −2.770 | −8.065 | −6.798 | −5.637 | −41.15 ± 0.40 | |
| 338.15 | −2.816 | −8.214 | −6.947 | −5.324 | −41.51 ± 0.88 | |
| 1.5 As | 278.15 | −3.088 | −9.436 | −8.550 | −7.702 | −50.12 ± 1.02 |
| 298.15 | −3.134 | −9.066 | −8.144 | −6.850 | −48.33 ± 1.10 | |
| 318.15 | −3.046 | −8.872 | −7.913 | −6.277 | −46.69 ± 0.39 | |
| 338.15 | −2.975 | −8.929 | −7.931 | −5.934 | −46.10 ± 0.47 | |
| 1.0 As | 278.15 | −3.227 | −10.083 | −9.419 | −8.106 | −53.83 ± 1.02 |
| 298.15 | −3.225 | −9.560 | −8.876 | −7.217 | −51.34 ± 1.10 | |
| 318.15 | −3.082 | −9.369 | −8.642 | −6.687 | −49.48 ± 0.39 | |
| 338.15 | −2.998 | −9.455 | −8.685 | −6.698 | −49.28 ± 0.20 | |
| 0.0 As (HAP) | 278.15 | −3.804 | −11.174 | - | −8.496 | −61.04 ± 0.83 |
| 298.15 | −3.897 | −10.339 | - | −7.397 | −57.90 ± 1.57 | |
| 318.15 | −3.758 | −10.442 | - | −7.023 | −57.14 ± 0.32 | |
| 338.15 | −3.681 | −10.857 | - | −6.854 | −57.83 ± 0.68 |
| Sample ID | Log IAP a | Log IAP b | Log IAP c |
|---|---|---|---|
| 3.0 As (JBM) | −39.22 ± 0.56 | −39.22 | −38.04 [30] |
| 2.0 As | −41.07 ± 0.11 | −41.24 | - |
| 1.5 As | −48.33 ± 1.10 | −48.18 | - |
| 1.0 As | −51.34 ± 1.10 | −51.21 | - |
| 0.0 As (HAP) | −57.90 ± 1.57 | −58.46 | −57.00 [32] |
| Sample ID | (kJ·mol−1) | (kJ·mol−1) | (J·mol−1·K−1) | (J·mol−1·K−1) |
|---|---|---|---|---|
| 3.0 As (JBM) | 224 ± 4 | −5 ± 41 | −766 ± 127 | −9370 ± 3790 |
| 2.0 As | 235 ± 2 | 69 ± 81 | −558 ± 268 | −4452 ± 1985 |
| 1.5 As | 275 ± 2 | 139 ± 40 | −457 ± 129 | −1848 ± 591 |
| 1.0 As | 292 ± 5 | 177 ± 18 | −387 ± 64 | −3361 ± 3615 |
| 0.0 As (HAP) | 334 ± 8 | 132 ± 58 | −678 ± 211 | −5838 ± 2206 |
| Species | (kJ·mol−1) | (kJ·mol−1) | (J·mol−1·K−1) | (J·mol−1·K−1) |
|---|---|---|---|---|
| Johnbaumite (JBM) | −5087 [30] | - | - | - |
| Hydroxyapatite (HAP) | −6310.45 [42] −6287 [43] −6255 [44] | −6654.5 [44] −6762.5 [45] | 390.35 [46] | 347 [46] |
| Ca2+ | −553.5 [47] | −542.8 [47] | −53.1 [47] | −169 [48] |
| PO43− | −1001.6 [47] | −1259.6 [47] | −222 [47] | −497 [49] |
| AsO43− | −648.4 [47] | −888.1 [47] | −162.8 [47] | −486.1 [47] |
| OH− | −157.3 [50] | −230 [50] | −10.71 [50] | −138.6 [49] |
| Sample ID | (kJ·mol−1) | (kJ·mol−1) | (J·mol−1·K−1) | (J·mol−1·K−1) |
|---|---|---|---|---|
| 3.0 As (JBM) | −5094 ± 4 | −5604 ± 41 | 2 ± 127 | 6928 ± 3790 |
| 2.0 As | −5459 ± 2 | −6049 ± 81 | −266 ± 268 | 1999 ± 1985 |
| 1.5 As | −5675 ± 2 | −6304 ± 40 | −396 ± 129 | −610 ± 591 |
| 1.0 As | −5869 ± 5 | −6528 ± 18 | −496 ± 64 | 898 ± 3615 |
| 0.0 As (HAP) | −6263 ± 8 | −6855 ± 58 | −264 ± 211 | 3363 ± 2206 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puzio, B.; Manecki, M.; Kwaśniak-Kominek, M. Transition from Endothermic to Exothermic Dissolution of Hydroxyapatite Ca5(PO4)3OH–Johnbaumite Ca5(AsO4)3OH Solid Solution Series at Temperatures Ranging from 5 to 65 °C. Minerals 2018, 8, 281. https://doi.org/10.3390/min8070281
Puzio B, Manecki M, Kwaśniak-Kominek M. Transition from Endothermic to Exothermic Dissolution of Hydroxyapatite Ca5(PO4)3OH–Johnbaumite Ca5(AsO4)3OH Solid Solution Series at Temperatures Ranging from 5 to 65 °C. Minerals. 2018; 8(7):281. https://doi.org/10.3390/min8070281
Chicago/Turabian StylePuzio, Bartosz, Maciej Manecki, and Monika Kwaśniak-Kominek. 2018. "Transition from Endothermic to Exothermic Dissolution of Hydroxyapatite Ca5(PO4)3OH–Johnbaumite Ca5(AsO4)3OH Solid Solution Series at Temperatures Ranging from 5 to 65 °C" Minerals 8, no. 7: 281. https://doi.org/10.3390/min8070281