Identification of the Potential Prognostic Markers from the miRNA-lncRNA-mRNA Interactions for Metastatic Renal Cancer via Next-Generation Sequencing and Bioinformatics

 and
and
Abstract
:1. Introduction
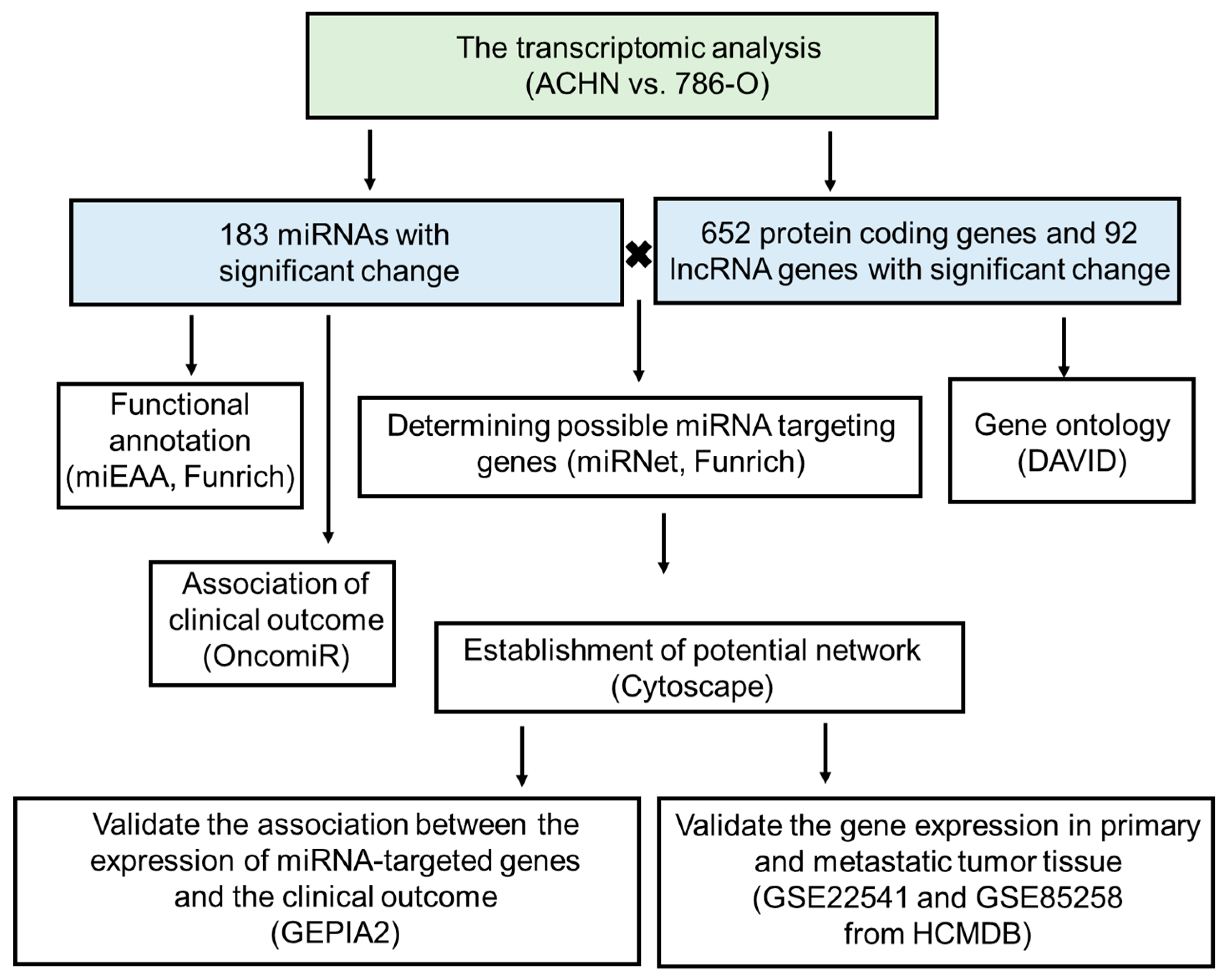
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of RNA Sequencing
2.3. Sequencing, Alignment, and Differential Gene and miRNA Expressions
2.4. miRNA Enrichment Analysis
2.5. Gene Ontology (GO) Analysis of Protein-Coding Genes
2.6. Prediction of miRNA-Targeted Genes
2.7. Selection of Genes with Differential Expression
2.8. Assessment of Gene Expression and Patient’s Prognosis
2.9. Determine the Gene Expression from Online Datasets
2.10. Statistical Analysis
3. Results
3.1. The Results of miRNA Sequencing
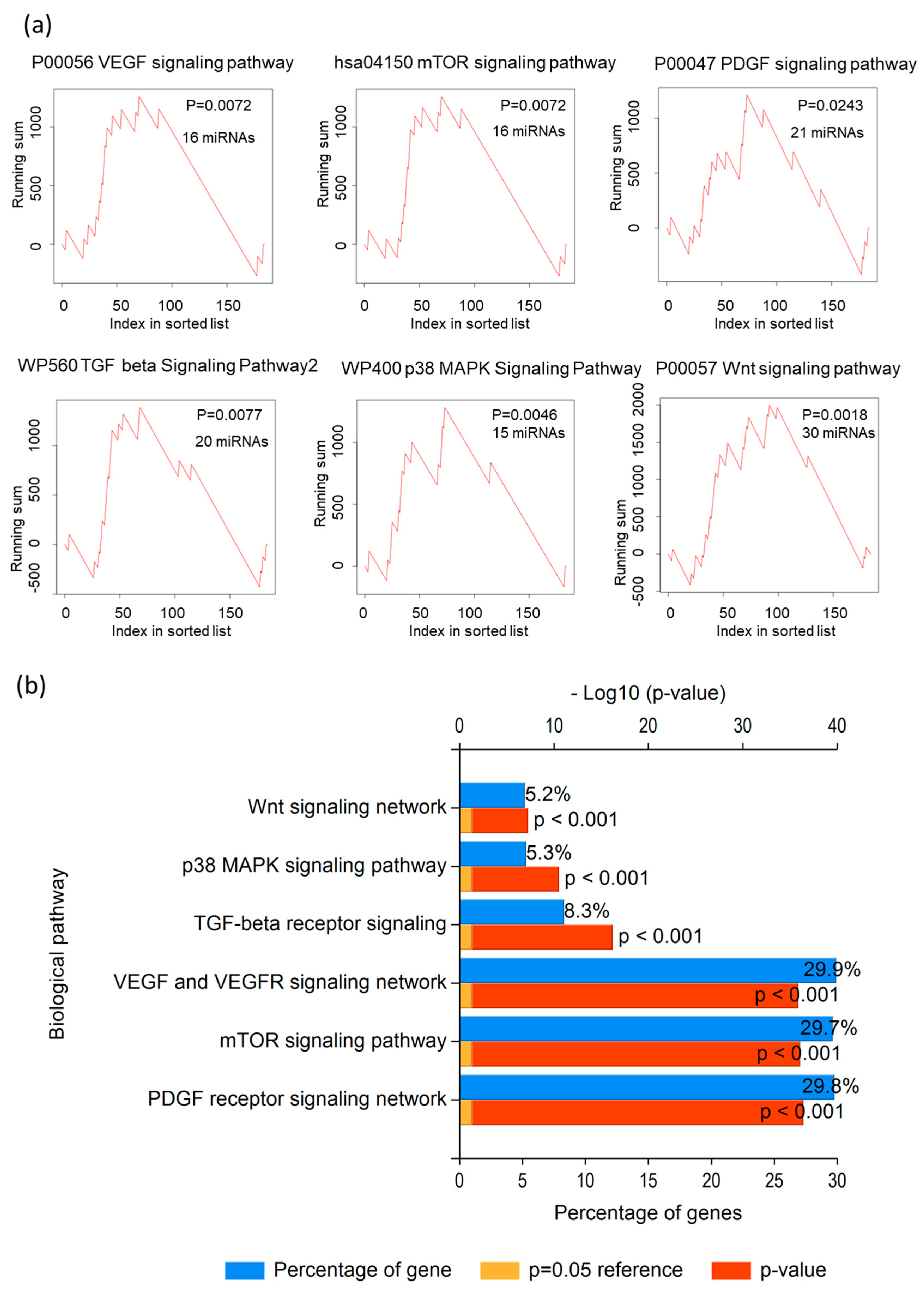
3.2. The miRNA Enrichment Analysis of Differential miRNA Expressions
3.3. The Gene Ontology (GO) Analysis of Differential Protein-Coding Gene Expressions
3.4. Identification of Potential miRNA-Targeting mRNA and lncRNA
3.5. Assessment of whether the miRNA Expression Was Associated with the Survival Outcome in Patients with RCC
3.6. Evaluation of the Potential Interaction of miRNA and Genes
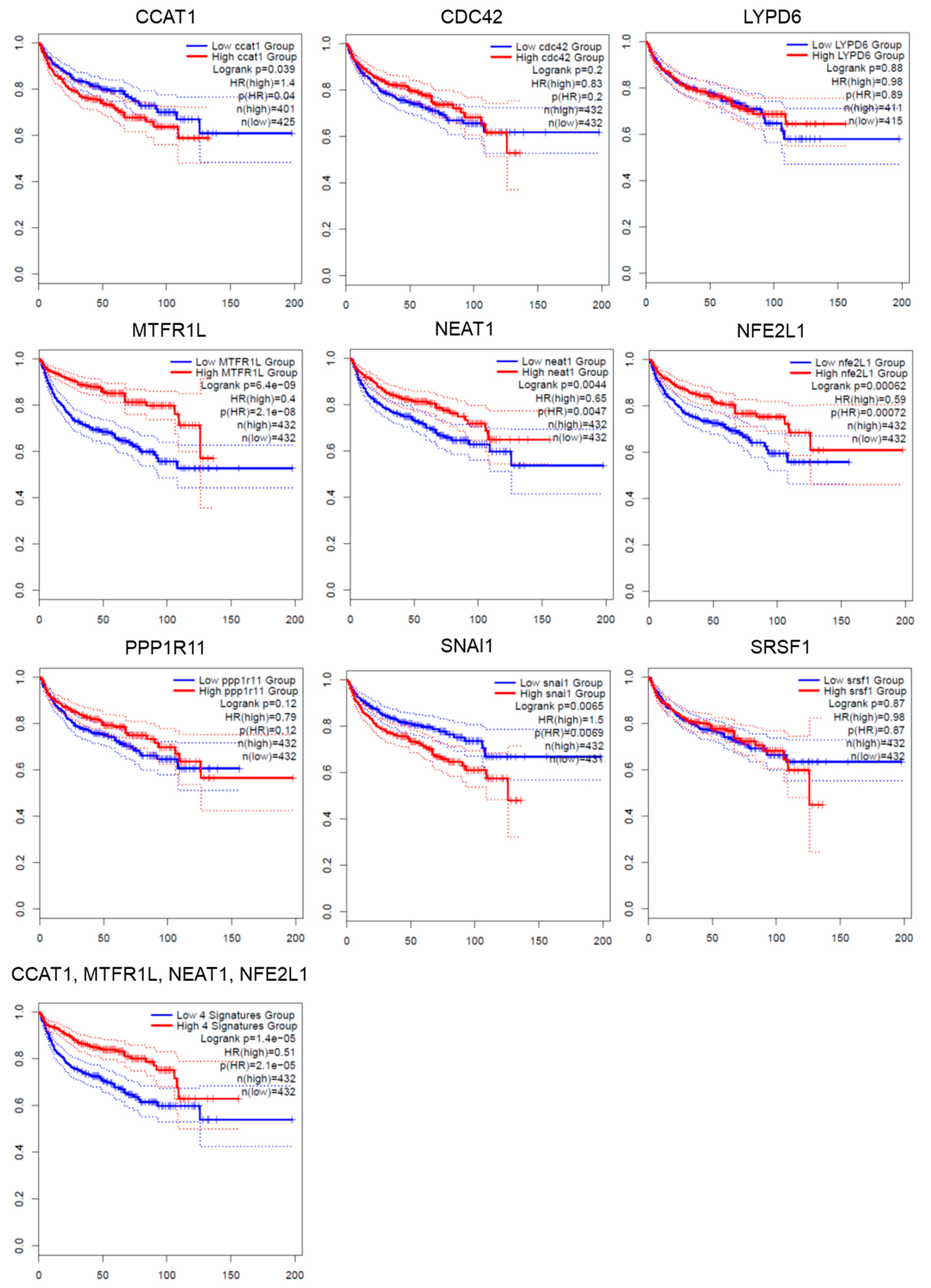
3.7. Validation of the Association of Predicted miRNA-Targeted Gene Expression and Survival Outcome
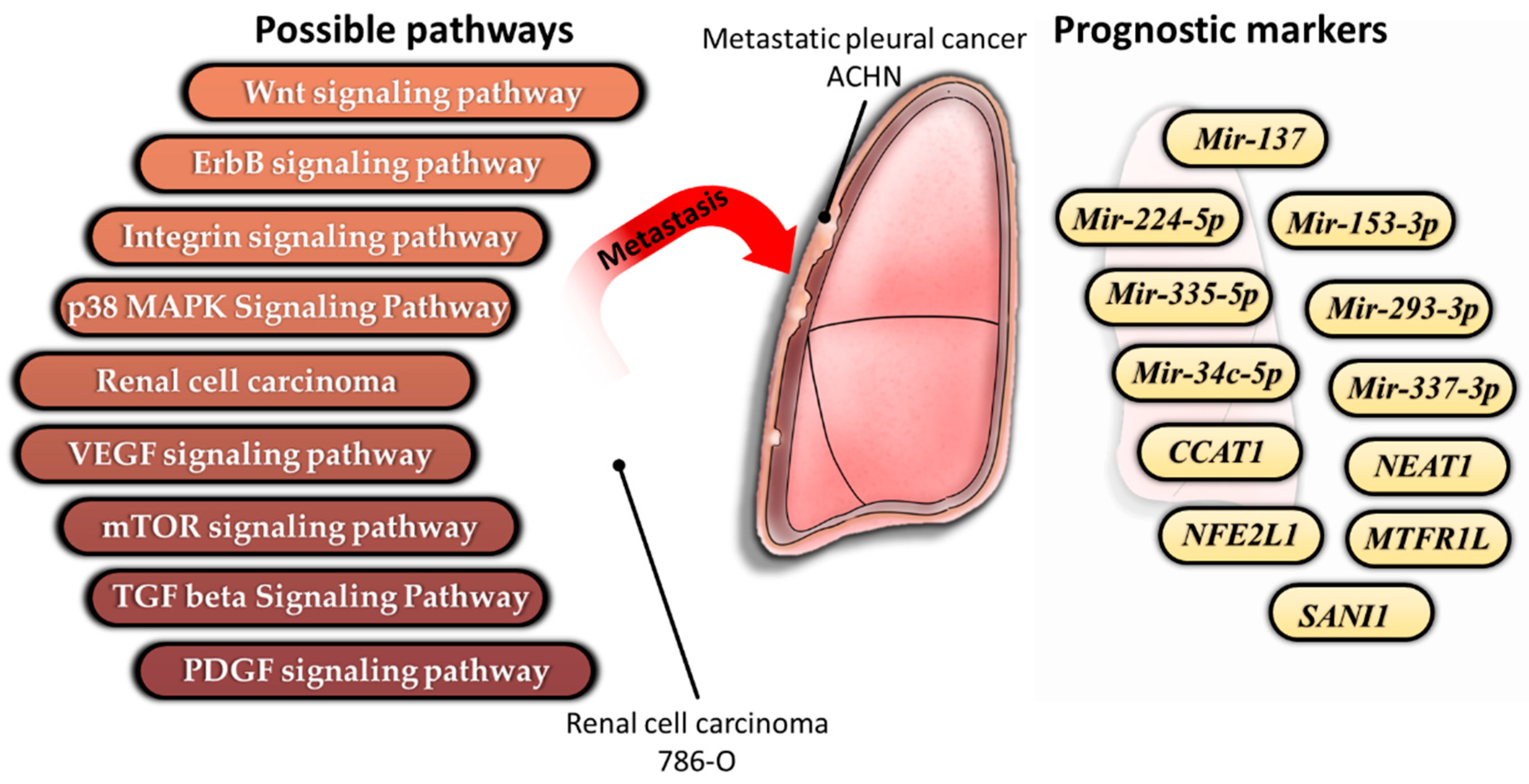
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.R.; Humphrey, P.A.; Catena, J.R.; Narra, V.R.; Srigley, J.R.; Cortez, A.D.; Dalrymple, N.C.; Chintapalli, K.N. Common and uncommon histologic subtypes of renal cell carcinoma: Imaging spectrum with pathologic correlation. Radiographics 2006, 26, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Athar, U.; Gentile, T.C. Treatment options for metastatic renal cell carcinoma: A review. Can. J. Urol. 2008, 15, 3954–3966. [Google Scholar]
- Patard, J.J.; Leray, E.; Rioux-Leclercq, N.; Cindolo, L.; Ficarra, V.; Zisman, A.; De La Taille, A.; Tostain, J.; Artibani, W.; Abbou, C.C.; et al. Prognostic value of histologic subtypes in renal cell carcinoma: A multicenter experience. J. Clin. Oncol. 2005, 23, 2763–2771. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar] [PubMed]
- Schodel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Small, E.J. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J. Clin. Oncol. 2005, 23, 1028–1043. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [Green Version]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaly, Z.; Sztupinszki, Z.; Surowiak, P.; Gyorffy, B. A comprehensive overview of targeted therapy in metastatic renal cell carcinoma. Curr. Cancer Drug. Targets 2012, 12, 857–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerdes, I.; Tolia, M.; Tsoukalas, N.; Mitsis, M.; Kardamakis, D.; Pistevou-Gombaki, K.; Tsekeris, P.; Kyrgias, G. Systemic therapy of metastatic renal cell carcinoma: Review of the current literature. Urologia 2019, 86, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yao, F.; Xiao, Z.; Sun, Y.; Ma, L. MicroRNAs and metastasis: Small RNAs play big roles. Cancer Metastasis Rev. 2018, 37, 5–15. [Google Scholar] [CrossRef]
- Mytsyk, Y.; Dosenko, V.; Skrzypczyk, M.A.; Borys, Y.; Diychuk, Y.; Kucher, A.; Kowalskyy, V.; Pasichnyk, S.; Mytsyk, O.; Manyuk, L. Potential clinical applications of microRNAs as biomarkers for renal cell carcinoma. Cent. Eur. J. Urol. 2018, 71, 295–303. [Google Scholar] [CrossRef]
- Braga, E.A.; Fridman, M.V.; Loginov, V.I.; Dmitriev, A.A.; Morozov, S.G. Molecular Mechanisms in Clear Cell Renal Cell Carcinoma: Role of miRNAs and Hypermethylated miRNA Genes in Crucial Oncogenic Pathways and Processes. Front. Genet. 2019, 10, 320. [Google Scholar] [CrossRef]
- Liu, X.; Hao, Y.; Yu, W.; Yang, X.; Luo, X.; Zhao, J.; Li, J.; Hu, X.; Li, L. Long Non-Coding RNA Emergence During Renal Cell Carcinoma Tumorigenesis. Cell Physiol. Biochem. 2018, 47, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Cheng, L.; Niu, W.; Zhao, G.; Raju, J.K.; Huo, J.; Wu, B.; Yin, B.; Song, Y.; et al. Long non-coding RNAs in renal cell carcinoma: A systematic review and clinical implications. Oncotarget 2017, 8, 48424–48435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic. Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Backes, C.; Khaleeq, Q.T.; Meese, E.; Keller, A. miEAA: microRNA enrichment analysis and annotation. Nucleic. Acids Res. 2016, 44, W110–W116. [Google Scholar] [CrossRef] [Green Version]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Fan, Y.; Xia, J. miRNet-Functional Analysis and Visual Exploration of miRNA-Target Interactions in a Network Context. Methods Mol. Biol. 2018, 1819, 215–233. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic. Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic. Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic. Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Wong, N.W.; Chen, Y.; Chen, S.; Wang, X. OncomiR: An online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 2018, 34, 713–715. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic. Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Ma, Y.; Zou, Y.; Yin, A.; Li, W.; Dong, D. HCMDB: The human cancer metastasis database. Nucleic. Acids Res. 2018, 46, D950–D955. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Krause, M.; Samoylenko, A.; Vainio, S. Wnt Signaling in Renal Cell Carcinoma. Cancers 2016, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, J.; Sun, G. Identification of key genes and pathways in renal cell carcinoma through expression profiling data. Kidney Blood Press Res. 2015, 40, 288–297. [Google Scholar] [CrossRef]
- Feldkoren, B.; Hutchinson, R.; Rapoport, Y.; Mahajan, A.; Margulis, V. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-beta1) dependent down-regulation of E-Cadherin expression—Important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp. Cell Res. 2017, 355, 57–66. [Google Scholar] [CrossRef]
- Liu, M.; Lang, N.; Qiu, M.; Xu, F.; Li, Q.; Tang, Q.; Chen, J.; Chen, X.; Zhang, S.; Liu, Z.; et al. miR-137 targets Cdc42 expression, induces cell cycle G1 arrest and inhibits invasion in colorectal cancer cells. Int. J. Cancer 2011, 128, 1269–1279. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Zhang, M.; Fan, Q.; Luo, S.; Cao, X. miR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42. Dig. Dis. Sci. 2011, 56, 2009–2016. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Y.; Shen, H.; Li, H.; Long, L.; Hui, L.; Xu, W. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 2013, 587, 73–81. [Google Scholar] [CrossRef]
- Tamim, S.; Vo, D.T.; Uren, P.J.; Qiao, M.; Bindewald, E.; Kasprzak, W.K.; Shapiro, B.A.; Nakaya, H.I.; Burns, S.C.; Araujo, P.R.; et al. Genomic analyses reveal broad impact of miR-137 on genes associated with malignant transformation and neuronal differentiation in glioblastoma cells. PLoS ONE 2014, 9, e85591. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Lipchina, I.; Elkabetz, Y.; Hafner, M.; Sheridan, R.; Mihailovic, A.; Tuschl, T.; Sander, C.; Studer, L.; Betel, D. Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Genes Dev. 2011, 25, 2173–2186. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Krell, J.; Stebbing, J.; Carissimi, C.; Dabrowska, A.F.; de Giorgio, A.; Frampton, A.E.; Harding, V.; Fulci, V.; Macino, G.; Colombo, T.; et al. TP53 regulates miRNA association with AGO2 to remodel the miRNA-mRNA interaction network. Genome Res. 2016, 26, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.P.; Rajapakshe, K.I.; Bader, D.A.; Cerne, J.Z.; Smith, E.A.; Coarfa, C.; Hartig, S.M.; McGuire, S.E. The Landscape of microRNA Targeting in Prostate Cancer Defined by AGO-PAR-CLIP. Neoplasia 2016, 18, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Sun, Q.; Zhang, J.; Yu, J.; Chen, W.; Zhang, Z. Downregulation of miR-153 contributes to epithelial-mesenchymal transition and tumor metastasis in human epithelial cancer. Carcinogenesis 2013, 34, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, J.; Bai, Z.; Li, H.; He, S.; Chen, R.; Che, X. MicroRNA-153 acts as a prognostic marker in gastric cancer and its role in cell migration and invasion. Onco Targets Ther. 2015, 8, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, C. MiR-153 regulates metastases of gastric cancer through Snail. Tumour. Biol. 2015. [Google Scholar] [CrossRef]
- Zeng, H.F.; Yan, S.; Wu, S.F. MicroRNA-153-3p suppress cell proliferation and invasion by targeting SNAI1 in melanoma. Biochem. Biophys. Res. Commun. 2017, 487, 140–145. [Google Scholar] [CrossRef]
- Zhu, S.; Sachdeva, M.; Wu, F.; Lu, Z.; Mo, Y.Y. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene 2010, 29, 1763–1772. [Google Scholar] [CrossRef] [Green Version]
- Ke, T.W.; Hsu, H.L.; Wu, Y.H.; Chen, W.T.; Cheng, Y.W.; Cheng, C.W. MicroRNA-224 suppresses colorectal cancer cell migration by targeting Cdc42. Dis. Markers 2014, 2014, 617150. [Google Scholar] [CrossRef]
- Zhang, Y.; Takahashi, S.; Tasaka, A.; Yoshima, T.; Ochi, H.; Chayama, K. Involvement of microRNA-224 in cell proliferation, migration, invasion, and anti-apoptosis in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2013, 28, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic. Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whisnant, A.W.; Bogerd, H.P.; Flores, O.; Ho, P.; Powers, J.G.; Sharova, N.; Stevenson, M.; Chen, C.H.; Cullen, B.R. In-depth analysis of the interaction of HIV-1 with cellular microRNA biogenesis and effector mechanisms. mBio 2013, 4, e000193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Gao, H.; Qu, H.; Li, J.; Liu, K.; Han, Z. MiR-137 suppresses tumor growth and metastasis in clear cell renal cell carcinoma. Pharmacol. Rep. 2018, 70, 963–971. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Luo, M.; Cui, W.; Zhou, X.; Miao, L. Long Noncoding RNA Small Nucleolar RNA Host Gene 1 (SNHG1) Promotes Renal Cell Carcinoma Progression and Metastasis by Negatively Regulating miR-137. Med. Sci. Monit. 2018, 24, 3824–3831. [Google Scholar] [CrossRef]
- Wang, K.; Jin, W.; Jin, P.; Fei, X.; Wang, X.; Chen, X. miR-211-5p Suppresses Metastatic Behavior by Targeting SNAI1 in Renal Cancer. Mol. Cancer Res. 2017, 15, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhang, H.; Li, W.; Yan, Y.; Yao, X.; Gu, W. FOXM1-Activated LINC01094 Promotes Clear Cell Renal Cell Carcinoma Development via MicroRNA 224-5p/CHSY1. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef]
- Wang, K.; Chen, X.; Zhan, Y.; Jiang, W.; Liu, X.; Wang, X.; Wu, B. miR-335 inhibits the proliferation and invasion of clear cell renal cell carcinoma cells through direct suppression of BCL-W. Tumour. Biol. 2015, 36, 6875–6882. [Google Scholar] [CrossRef]
- Xue, D.; Wang, H.; Chen, Y.; Shen, D.; Lu, J.; Wang, M.; Zebibula, A.; Xu, L.; Wu, H.; Li, G.; et al. Circ-AKT3 inhibits clear cell renal cell carcinoma metastasis via altering miR-296-3p/E-cadherin signals. Mol. Cancer 2019, 18, 151. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Boateng, L.A.; Friedman, J.R.; Baldwin, D.A.; Tobias, J.W.; Judkins, A.R.; Mourelatos, Z.; Lal, P. MiRNA-9 and MiRNA-200a distinguish hemangioblastomas from metastatic clear cell renal cell carcinomas in the CNS. Brain Pathol. 2012, 22, 522–529. [Google Scholar] [CrossRef]
- Ning, L.; Li, Z.; Wei, D.; Chen, H.; Yang, C. LncRNA, NEAT1 is a prognosis biomarker and regulates cancer progression via epithelial-mesenchymal transition in clear cell renal cell carcinoma. Cancer Biomark. 2017, 19, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, Q.; Hann, S.S. The functions and oncogenic roles of CCAT1 in human cancer. Biomed. Pharmacother. 2019, 115, 108943. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ma, P.; Li, B.; Zhu, D.; Chen, X.; Xiang, Y.; Wang, T.; Ren, X.; Liu, C.; Jin, X. LncRNA CCAT1 inhibits cell apoptosis of renal cell carcinoma through up-regulation of Livin protein. Mol. Cell Biochem. 2017, 434, 135–142. [Google Scholar] [CrossRef]
- Jiang, B.; Chen, W.; Qin, H.; Diao, W.; Li, B.; Cao, W.; Zhang, Z.; Qi, W.; Gao, J.; Chen, M.; et al. TOX3 inhibits cancer cell migration and invasion via transcriptional regulation of SNAI1 and SNAI2 in clear cell renal cell carcinoma. Cancer Lett. 2019, 449, 76–86. [Google Scholar] [CrossRef]
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear Factor Erythroid-2 Like 1 (NFE2L1): Structure, function and regulation. Gene 2016, 584, 17–25. [Google Scholar] [CrossRef]
- Chen, S.C.; Kuo, P.L. Bone Metastasis from Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 987. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; Sun, M.; Jeldres, C.; Shariat, S.F.; Trinh, Q.D.; Briganti, A.; Tian, Z.; Schmitges, J.; Graefen, M.; Perrotte, P.; et al. Distribution of metastatic sites in renal cell carcinoma: A population-based analysis. Ann. Oncol. 2012, 23, 973–980. [Google Scholar] [CrossRef]
- Gudas, L.J.; Fu, L.; Minton, D.R.; Mongan, N.P.; Nanus, D.M. The role of HIF1alpha in renal cell carcinoma tumorigenesis. J. Mol. Med. 2014, 92, 825–836. [Google Scholar] [CrossRef]
- Chen, S.C.; Chen, F.W.; Hsu, Y.L.; Kuo, P.L. Systematic Analysis of Transcriptomic Profile of Renal Cell Carcinoma under Long-Term Hypoxia Using Next-Generation Sequencing and Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2657. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Eberhardt, M.; Schmitz, U.; Vera, J. Systems biology-based investigation of cooperating microRNAs as monotherapy or adjuvant therapy in cancer. Nucleic. Acids Res. 2019, 47, 7753–7766. [Google Scholar] [CrossRef]
- Lai, X.; Gupta, S.K.; Schmitz, U.; Marquardt, S.; Knoll, S.; Spitschak, A.; Wolkenhauer, O.; Putzer, B.M.; Vera, J. MiR-205-5p and miR-342-3p cooperate in the repression of the E2F1 transcription factor in the context of anticancer chemotherapy resistance. Theranostics 2018, 8, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet 2016, 17, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wolkenhauer, O.; Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic. Acids Res. 2016, 44, 6019–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | p Value | miRNA |
|---|---|---|
| P00057 Wnt signaling pathway | 0.001861 | miR-100-5p; miR-135a-5p; miR-137; miR-148a-3p; miR-192-5p; miR-194-5p; miR-195-5p; miR-203a-3p; miR-205-5p; miR-211-5p; miR-215-5p; miR-218-5p; miR-224-5p; miR-296-3p; miR-296-5p; miR-31-3p; miR-31-5p; miR-335-5p; miR-337-3p; miR-34b-3p; miR-34b-5p; miR-34c-5p; miR-376a-3p; miR-376a-5p; miR-376b-3p; miR-376c-3p; miR-380-5p; miR-485-5p; miR-9-5p; miR-935 |
| WP673 ErbB signaling pathway | 0.003884 | miR-100-5p; miR-129-1-3p; miR-135a-5p; miR-192-5p; miR-194-5p; miR-199b-5p; miR-203a-3p; miR-205-5p; miR-20b-5p; miR-215-5p; miR-218-5p; miR-296-5p; miR-299-5p; miR-326; miR-335-5p; miR-34b-3p; miR-34b-5p; miR-34c-5p; miR-363-3p; miR-433-3p; miR-497-5p; miR-548d-3p; miR-654-3p; miR-99a-5p |
| P00034 Integrin signaling pathway | 0.004683 | miR-100-5p; miR-129-1-3p; miR-137; miR-148a-3p; miR-192-5p; miR-194-5p; miR-195-5p; miR-199b-5p; miR-205-5p; miR-215-5p; miR-218-5p; miR-224-5p; miR-296-3p; miR-31-3p; miR-31-5p; miR-326; miR-335-5p; miR-337-3p; miR-34b-5p; miR-34c-5p; miR-376a-3p; miR-433-3p; miR-497-5p; miR-9-3p; miR-9-5p; miR-935; miR-99a-5p |
| WP400 p38 MAPK Signaling Pathway | 0.004683 | miR-100-5p; miR-135a-5p; miR-137; miR-148a-3p; miR-192-5p; miR-194-5p; miR-195-5p; miR-203a-3p; miR-224-5p; miR-335-5p; miR-34b-3p; miR-34b-5p; miR-34c-5p; miR-433-3p; miR-99a-5p |
| hsa05211 Renal cell carcinoma | 0.00488 | miR-100-5p; miR-1262; miR-129-1-3p; miR-134-5p; miR-137; miR-192-5p; miR-194-5p; miR-195-5p; miR-199b-5p; miR-205-5p; miR-20b-5p; miR-215-5p; miR-218-5p; miR-224-5p; miR-296-3p; miR-31-5p; miR-335-5p; miR-337-3p; miR-34b-3p; miR-34b-5p; miR-34c-5p; miR-376a-3p; miR-377-3p; miR-433-3p; miR-489-3p; miR-497-5p; miR-9-5p; miR-935; miR-99a-5p |
| P00056 VEGF signaling pathway | 0.007226 | miR-100-5p; miR-134-5p; miR-137; miR-192-5p; miR-195-5p; miR-199b-5p; miR-205-5p; miR-20b-5p; miR-215-5p; miR-296-3p; miR-31-5p; miR-335-5p; miR-34b-3p; miR-376a-3p; miR-9-5p; miR-99a-5p |
| hsa04150 mTOR signaling pathway | 0.007226 | miR-100-5p; miR-134-5p; miR-192-5p; miR-195-5p; miR-199b-5p; miR-205-5p; miR-20b-5p; miR-215-5p; miR-218-5p; miR-296-3p; miR-31-3p; miR-335-5p; miR-34b-3p; miR-376a-3p; miR-9-5p; miR-99a-5p |
| hsa04350 TGF beta Signaling Pathway | 0.007709 | miR-100-5p; miR-153-3p; miR-192-5p; miR-194-5p; miR-195-5p; miR-203a-3p; miR-205-5p; miR-20b-5p; miR-215-5p; miR-218-5p; miR-224-5p; miR-299-5p; miR-31-3p; miR-335-5p; miR-337-3p; miR-409-3p; miR-433-3p; miR-9-5p; miR-935; miR-99a-5p |
| P00047 PDGF signaling pathway | 0.024344 | miR-100-5p; miR-135a-5p; miR-148a-3p; miR-192-5p; miR-194-5p; miR-195-5p; miR-20b-5p; miR-215-5p; miR-296-3p; miR-31-5p; miR-335-5p; miR-337-3p; miR-34b-3p; miR-34b-5p; miR-34c-5p; miR-376a-3p; miR-433-3p; miR-497-5p; miR-9-5p; miR-935; miR-99a-5p |
| Gene Ontology Term | Count | p Value |
|---|---|---|
| GO:0007155~cell adhesion | 110 | 1.74 × 10−10 |
| GO:0022610~biological adhesion | 110 | 2.18 × 10−10 |
| GO:0030198~extracellular matrix organization | 36 | 6.87 × 10−9 |
| GO:0043062~extracellular structure organization | 36 | 7.45 × 10−9 |
| GO:0040007~growth | 67 | 3.09 × 10−8 |
| GO:0016477~cell migration | 78 | 7.54 × 10−8 |
| GO:0048589~developmental growth | 48 | 8.20 × 10−8 |
| GO:0000904~cell morphogenesis involved in differentiation | 56 | 1.76 × 10−7 |
| GO:2000145~regulation of cell motility | 55 | 2.02 × 10−7 |
| GO:0048870~cell motility | 83 | 2.61 × 10−7 |
| GO:0051674~localization of cell | 83 | 2.61 × 10−7 |
| miRNA | LogRank p Value | Type 1 | Upregulated |
|---|---|---|---|
| hsa-miR-137 | 1.53 × 10−2 | KIRC | Deceased |
| hsa-miR-153-3p | 9.19 × 10−4 | KIRC | Deceased |
| hsa-miR-153-3p | 1.98 × 10−3 | KIRP | Deceased |
| hsa-miR-224-5p | 7.59 × 10−5 | KIRP | Deceased |
| hsa-miR-224-5p | 1.05 × 10−2 | KIRC | Deceased |
| hsa-miR-296-3p | 8.48 × 10−5 | KIRC | Deceased |
| hsa-miR-296-5p | 1.84 × 10−4 | KIRC | Deceased |
| hsa-miR-335-5p | 1.71 × 10−3 | KIRC | Deceased |
| hsa-miR-34c-5p | 3.75 × 10−2 | KICH | Deceased |
| hsa-miR-34c-5p | 1.98 × 10−8 | KIRC | Deceased |
| hsa-miR-377-3p | 4.07 × 10−3 | KICH | Deceased |
| hsa-miR-377-3p | 8.33 × 10−6 | KIRC | Deceased |
| microRNA (log2(Fold Change #)) | Target Genes (log2(Fold Change #)) | Experimental Validation | Reference |
|---|---|---|---|
| hsa-miR-137 (3.76) | CDC42 (−15.14) | Luciferase reporter assay | [36,37,38,39] |
| qRT-PCR | |||
| Reporter assay | |||
| Western blot | |||
| hsa-miR-137 (3.76) | LYPD6 (−3.90) | PAR-CLIP | [40,41,42,43,44] |
| hsa-miR-153-3p (2.65) | SNAI1 (−3.73) | Immunohistochemistry | [45,46,47,48] |
| Luciferase reporter assay | |||
| qRT-PCR | |||
| Western blot | |||
| hsa-miR-224-5p (6.88) | CDC42 (−15.14) | Luciferase reporter assay | [49,50,51] |
| Microarray | |||
| qRT-PCR | |||
| Western blot | |||
| hsa-mir-296-3p (−13.01) | CCAT1 (5.99) * | CLIP-Seq | [52] |
| hsa-mir-335-5p (6.16) | NEAT1 (−16.46) * | CLIP-Seq | [52] |
| hsa-miR-34c-5p (9.71) | NFE2L1 (−16.58) | PAR-CLIP | [43] |
| hsa-miR-34c-5p (9.71) | PPP1R11 (−4.25) | PAR-CLIP | [44] |
| hsa-miR-34c-5p (9.71) | NEAT1 (−16.46) * | CLIP-Seq | [52] |
| hsa-mir-377-3p (8.13) | SRSF1 (−17.83) | PAR-CLIP | [53] |
| hsa-mir-377-3p (8.13) | MTFR1L (−15.04) | PAR-CLIP | [41] |
| hsa-mir-377-3p (8.13) | NEAT1 (−16.46) * | CLIP-Seq | [52] |
| GEO Accession Number | Gene Symbol | Fold Change | p Value |
|---|---|---|---|
| GSE22541 | NEAT1 | 1.105 | 0.235 |
| GSE22541 | NFE2L1 | 0.684 | <0.001 |
| GSE22541 | MTFR1L | 0.850 | 0.115 |
| GSE22541 | SNAI1 | 0.886 | 0.194 |
| GSE85258 | NEAT1 | 0.941 | 0.224 |
| GSE85258 | NFE2L1 | 0.941 | 0.057 |
| GSE85258 | MTFR1L | 0.806 | 0.030 |
| GSE85258 | SNAI1 | 1.032 | 0.758 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, I.-J.; Liu, K.-T.; Shen, J.-H.; Wu, Y.-H.; Liu, Y.-H.; Yen, M.-C.; Kuo, P.-L. Identification of the Potential Prognostic Markers from the miRNA-lncRNA-mRNA Interactions for Metastatic Renal Cancer via Next-Generation Sequencing and Bioinformatics. Diagnostics 2020, 10, 228. https://doi.org/10.3390/diagnostics10040228
Yeh I-J, Liu K-T, Shen J-H, Wu Y-H, Liu Y-H, Yen M-C, Kuo P-L. Identification of the Potential Prognostic Markers from the miRNA-lncRNA-mRNA Interactions for Metastatic Renal Cancer via Next-Generation Sequencing and Bioinformatics. Diagnostics. 2020; 10(4):228. https://doi.org/10.3390/diagnostics10040228
Chicago/Turabian StyleYeh, I-Jeng, Kuan-Ting Liu, Jheng-Heng Shen, Yen-Hung Wu, Yao-Hua Liu, Meng-Chi Yen, and Po-Lin Kuo. 2020. "Identification of the Potential Prognostic Markers from the miRNA-lncRNA-mRNA Interactions for Metastatic Renal Cancer via Next-Generation Sequencing and Bioinformatics" Diagnostics 10, no. 4: 228. https://doi.org/10.3390/diagnostics10040228