

The Renin-Angiotensin System: The Challenge behind Autoimmune Dermatological Diseases
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
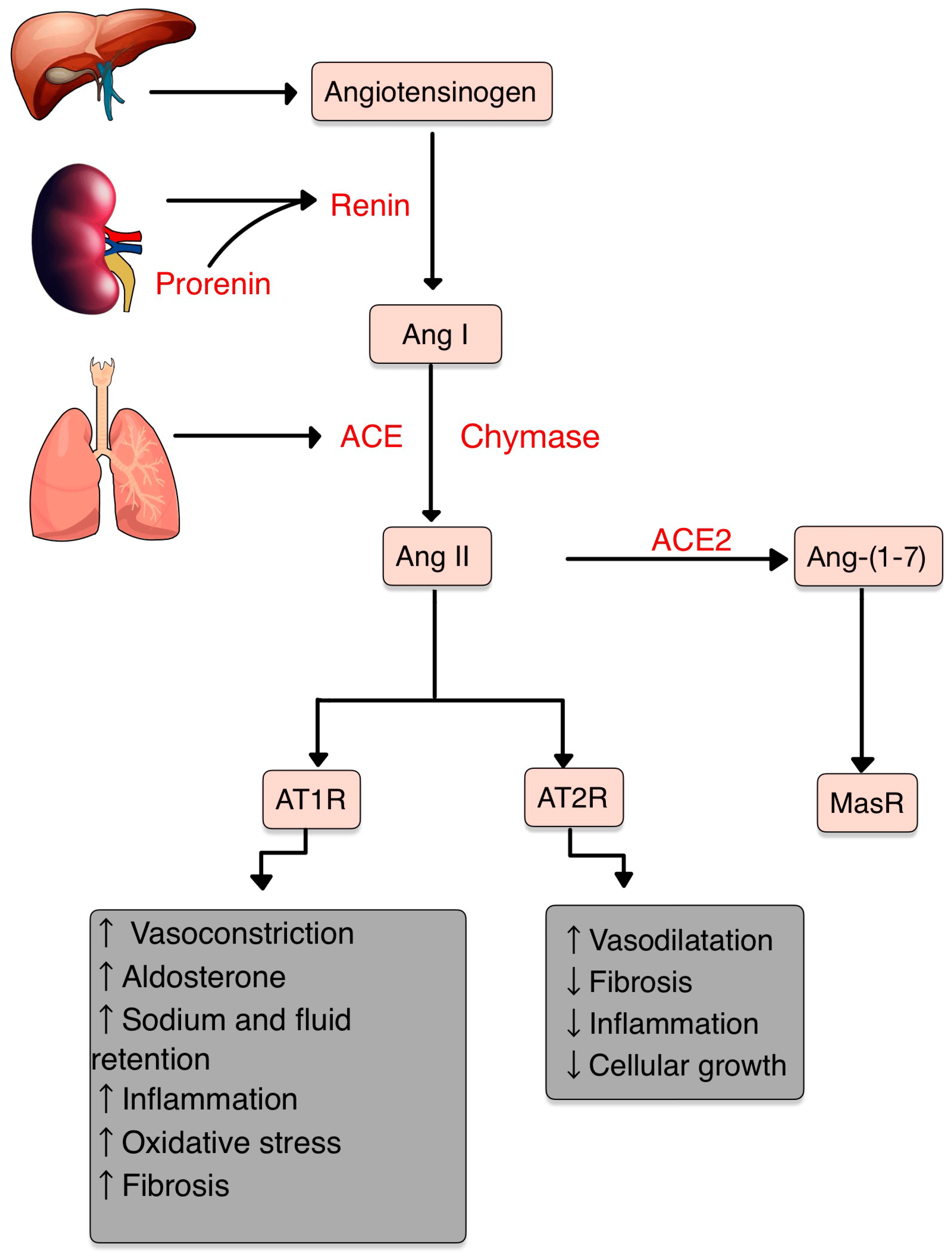
3. The Renin–Angiotensin–Aldosterone System

4. RAS and Inflammation
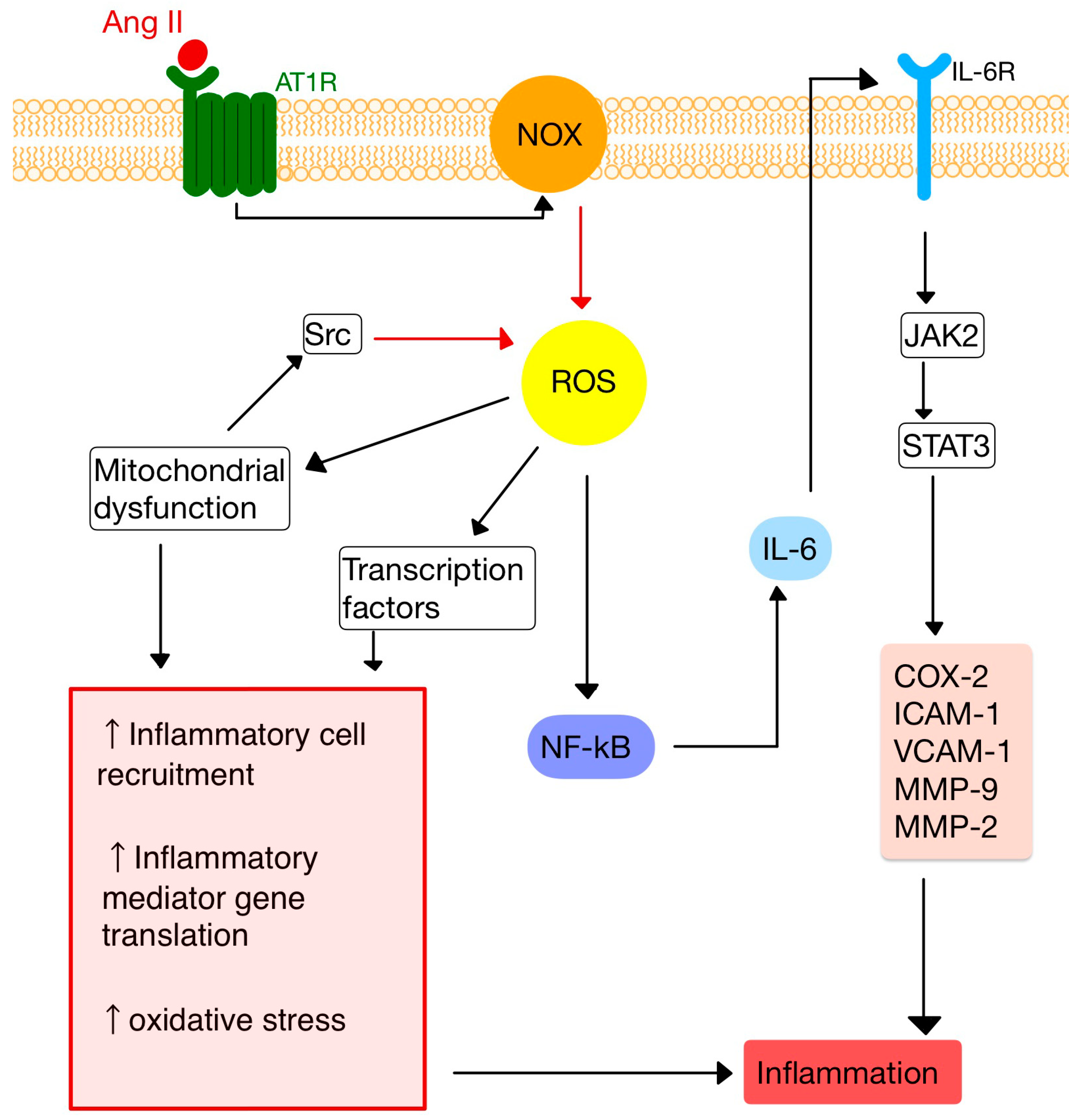
5. RAS and the Immune System
5.1. T Cells
5.2. Dendritic Cells
5.3. Macrophages
5.4. Neutrophils
6. RAS in the Skin
7. The Implication of RAS in AIDD
7.1. Psoriasis
7.1.1. Pathogenesis
7.1.2. ACE/AT1R Gene Polymorphisms
7.1.3. ACEIs and ARBs
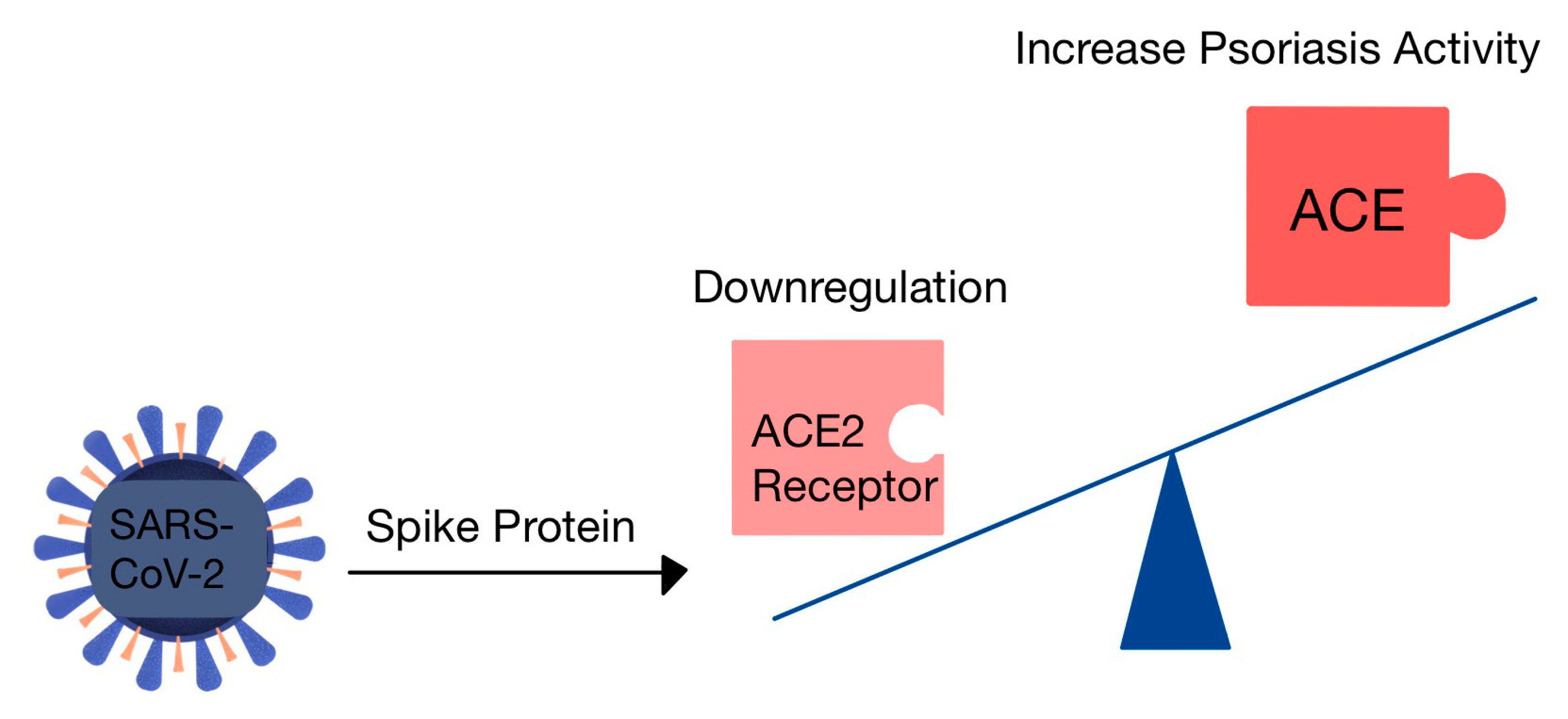
7.1.4. The Risk of Psoriasis in COVID-19
7.2. Systemic Sclerosis (SSc)
7.3. Lupus Erythematosus (LE)
7.4. Vitiligo
7.5. Alopecia Areata (AA)
7.6. Pemphigus
7.7. Bullous Pemphigoid (BP)
7.8. Lichen Planus (LP)
7.9. Other AI Diseases with Dermatological Implication
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrario, C.M.; Groban, L.; Wang, H.; Cheng, C.P.; VonCannon, J.L.; Wright, K.N.; Sun, X.; Ahmad, S. The Angiotensin-(1-12)/Chymase axis as an alternate component of the tissue renin angiotensin system. Mol. Cell. Endocrinol. 2021, 529, 111119. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Boron, W.F.; Boulpaep, E.L. Medical Physiology: A Cellular and Molecular Approach, 3rd ed.; Saunders: Philadelphia, PA, USA, 2016. [Google Scholar]
- Hall, J.E. Guyton and Hall Textbook of Physiology, 13th ed.; Saunders: Philadelphia, PA, USA, 2016. [Google Scholar]
- Soler, M.J.; Batlle, D. Revisiting the renin-angiotensin system. Mol. Cell. Endocrinol. 2021, 529, 111268. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Skorecki, K.; Hamo-Giladi, D.B.; Kruzel-Davila, E.; Heyman, S.N. Kinins and chymase: The forgotten components of the renin-angiotensin system and their implications in COVID-19 disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L422–L429. [Google Scholar] [CrossRef]
- Baranowska, I.; Gawrys, O.; Roszkowska-Chojecka, M.M.; Badzynska, B.; Tymecka, D.; Olszynski, K.H.; Kompanowska-Jezierska, E. Chymase Dependent Pathway of Angiotensin II Generation and Rapeseed Derived Peptides for Antihypertensive Treatment of Spontaneously Hypertensive Rats. Front. Pharmacol. 2021, 12, 658805. [Google Scholar] [CrossRef]
- Andone, S.; Bajko, Z.; Motataianu, A.; Maier, S.; Barcutean, L.; Balasa, R. Neuroprotection in Stroke-Focus on the Renin-Angiotensin System: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 3876. [Google Scholar] [CrossRef]
- Akbarzadeh, R.; Müller, A.; Humrich, J.Y.; Riemekasten, G. When natural antibodies become pathogenic: Autoantibodies targeted against G protein-coupled receptors in the pathogenesis of systemic sclerosis. Front. Immunol. 2023, 14, 1213804. [Google Scholar] [CrossRef]
- Lumbers, E.R.; Head, R.; Smith, G.R.; Delforce, S.J.; Jarrott, B.; Martin, H.J.; Pringle, K.G. The interacting physiology of COVID-19 and the renin-angiotensin-aldosterone system: Key agents for treatment. Pharmacol. Res. Perspect. 2022, 10, e00917. [Google Scholar] [CrossRef]
- Restrepo, Y.M.; Noto, N.M.; Speth, R.C. CGP42112: The full AT2 receptor agonist and its role in the renin-angiotensin-aldosterone system: No longer misunderstood. Clin. Sci. 2022, 136, 1513–1533. [Google Scholar] [CrossRef]
- Chaudhary, M. Anti-Hypertensive Potential and Epigenetics of Angiotensin II type 2 Receptor (AT2R). Curr. Hypertens. Rev. 2021, 17, 176–180. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef]
- Lima, M.L.S.; Martins, A.A.; Medeiros, C.A.C.X.; Guerra, G.C.B.; Santos, R.; Bader, M.; Pirih, F.Q.; Araújo Júnior, R.F.; Brito, G.A.C.; Leitão, R.F.C.; et al. The Receptor AT1 Appears to Be Important for the Maintenance of Bone Mass and AT2 Receptor Function in Periodontal Bone Loss Appears to Be Regulated by AT1 Receptor. Int. J. Mol. Sci. 2021, 22, 12849. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; Santos, A.C.P.M.; Galvão, I.; Souto, G.R.; Mesquita, R.A.; Sá, M.A.; Ferreira, A.J. The angiotensin converting enzyme 2/angiotensin-(1-7)/Mas Receptor axis as a key player in alveolar bone remodeling. Bone 2019, 128, 115041. [Google Scholar] [CrossRef]
- Maranduca, M.A.; Vamesu, C.G.; Tanase, D.M.; Clim, A.; Drochioi, I.C.; Pinzariu, A.C.; Filip, N.; Dima, N.; Tudorancea, I.; Serban, D.N.; et al. The RAAS Axis and SARS-CoV-2: From Oral to Systemic Manifestations. Medicina 2022, 58, 1717. [Google Scholar] [CrossRef]
- Song, G.; Kim, J.Y.; Yoon, H.Y.; Yee, J.; Gwak, H.S. A systematic review and meta-analysis of angiotensin-converting enzyme inhibitor use and psoriasis incidence. Sci. Rep. 2021, 11, 10037. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hamabe, L.; Hirose, M.; Watanabe, M.; Yokoi, A.; Takeuchi, A.; Ozai, Y.; Yoshida, T.; Takai, S.; Jin, D.; et al. Plasma Chymase Activity Reflects the Change in Hemodynamics Observed after the Surgical Treatment of Patent Ductus Arteriosus in Dogs. Vet. Sci. 2022, 9, 682. [Google Scholar] [CrossRef]
- Takai, S.; Jin, D. Chymase as a Possible Therapeutic Target for Amelioration of Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 7543. [Google Scholar] [CrossRef]
- Pereira, E.J.; Smolko, C.M.; Janes, K.A. Computational Models of Reactive Oxygen Species as Metabolic Byproducts and Signal-Transduction Modulators. Front. Pharmacol. 2016, 7, 457. [Google Scholar] [CrossRef]
- Zhang, Z.; Dalan, R.; Hu, Z.; Wang, J.W.; Chew, N.W.; Poh, K.K.; Tan, R.S.; Soong, T.W.; Dai, Y.; Ye, L.; et al. Reactive Oxygen Species Scavenging Nanomedicine for the Treatment of Ischemic Heart Disease. Adv. Mater. 2022, 34, e2202169. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Lee, D.; Song, C.G.; Kang, P.M. Reactive oxygen species-activated nanomaterials as theranostic agents. Nanomedicine 2015, 10, 2709–2723. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Sun, X. Reactive Oxygen Species-Based Nanomaterials for Cancer Therapy. Front. Chem. 2021, 9, 650587. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Rodríguez-Reyna, T.S.; Núñez-Alvarez, C.; Cruz-Lagunas, A.; Posadas-Sánchez, R.; Pérez-Hernández, N.; Jiménez-Alvarez, L.; Ramírez-Martínez, G.; Granados, J.; Vargas-Alarcón, G.; Zúñiga, J. Angiotensin II Type 1 receptor (AGTR1) gene polymorphisms are associated with vascular manifestations in patients with systemic sclerosis (SSc). J. Renin Angiotensin Aldosterone Syst. 2016, 17, 1470320316659954. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Vo, T.T.T.; Lee, C.W.; Chen, Y.L.; Lin, W.N.; Cheng, H.C.; Vo, Q.C.; Lee, I.T. Carbon monoxide releasing molecule-2 attenuates angiotensin II-induced IL-6/Jak2/Stat3-associated inflammation by inhibiting NADPH oxidase- and mitochondria-derived ROS in human aortic smooth muscle cells. Biochem. Pharmacol. 2022, 198, 114978. [Google Scholar] [CrossRef]
- Tanhapour, M.; Falahi, B.; Vaisi-Raygani, A.; Bahrehmand, F.; Kiani, A.; Rahimi, Z.; Vaisi-Raygani, A.A.; Shakiba, E.; Pourmotabbed, T. Angiotensin-converting enzyme insertion/deletion (rs106180) and angiotensin type 1 receptor A1166 C (rs106165) genotypes and psoriasis: Correlation with cellular immunity, lipid profile, and oxidative stress markers. J. Cell. Biochem. 2019, 120, 2627–2633. [Google Scholar] [CrossRef]
- Rashed, L.; Abdel Hay, R.; Mahmoud, R.; Hasan, N.; Zahra, A.; Fayez, S. Association of Angiotensin-Converting Enzyme (ACE) Gene Polymorphism with Inflammation and Cellular Cytotoxicity in Vitiligo Patients. PLoS ONE 2015, 10, e0132915. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Lin, J.; Xu, Y.; Guo, P.; Chen, Y.J.; Zhou, J.; Xia, M.; Tan, B.; Liu, X.; Feng, H.; Chen, Y. CCL5/CCR5-mediated peripheral inflammation exacerbates blood–brain barrier disruption after intracerebral hemorrhage in mice. J. Transl. Med. 2023, 21, 196. [Google Scholar] [CrossRef]
- Leone, G.M.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; Fagone, P. Past, Present and (Foreseeable) Future of Biological Anti-TNF Alpha Therapy. J. Clin. Med. 2023, 12, 1630. [Google Scholar] [CrossRef] [PubMed]
- Galeone, A.; Grano, M.; Brunetti, G. Tumor Necrosis Factor Family Members and Myocardial Ischemia-Reperfusion Injury: State of the Art and Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 4606. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Zhang, Y.L.; Chi, Y.F.; Yan, B.; Zeng, X.J.; Li, H.H.; Liu, Y. Angiotensin II Regulates Th1 T Cell Differentiation Through Angiotensin II Type 1 Receptor-PKA-Mediated Activation of Proteasome. Cell. Physiol. Biochem. 2018, 45, 1366–1376. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, H.; He, L.; Wang, Q.; Li, Y.; Weng, J.; Zhang, R. The Correlation Between IFNG Gene Methylation and Th1|Th2 Cell Balance in ROU and the Interventional Study of Jiaweidaochi Powder. Appl. Biochem. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska-Durczynska, K.; Pacholczyk, M.; Zygmunt, A.; Krawczyk-Rusiecka, K.; Ferenc, T.; Lewinski, A. Angiotensinogen gene T174M polymorphism is related to Hashimoto’s thyroiditis. Neuro Endocrinol. Lett. 2019, 39, 579–585. [Google Scholar] [PubMed]
- De Angelis, E.; Pecoraro, M.; Rusciano, M.R.; Ciccarelli, M.; Popolo, A. Cross-Talk between Neurohormonal Pathways and the Immune System in Heart Failure: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 1698. [Google Scholar] [CrossRef] [PubMed]
- Pietraforte, I.; Frasca, L. Autoreactive T-Cells in Psoriasis: Are They Spoiled Tregs and Can Therapies Restore Their Functions? Int. J. Mol. Sci. 2023, 24, 4348. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, C.; Liu, Y.; Tian, C.; Li, H.H. Angiotensin II Regulates Dendritic Cells through Activation of NF-κB /p65, ERK1/2 and STAT1 Pathways. Cell. Physiol. Biochem. 2017, 42, 1550–1558. [Google Scholar] [CrossRef]
- Campana, P.; Palaia, M.E.; Conte, M.; Cante, T.; Petraglia, L.; Femminella, G.D.; Parisi, V.; Leosco, D. The elderly at risk: Aldosterone as modulator of the immune response to SARS-CoV-2 infection. Geroscience 2022, 44, 567–572. [Google Scholar] [CrossRef]
- Takayama, S.; Inoue, K.; Ogura, Y.; Hoshino, S.; Sugaya, T.; Ohata, K.; Kotake, H.; Ichikawa, D.; Watanabe, M.; Kimura, K.; et al. Angiotensin II type 1a receptor deficiency alleviates muscle atrophy after denervation. Sci. Rep. 2023, 13, 519. [Google Scholar] [CrossRef]
- Peng, H.; Wang, J.; Li, S. MiR-15a-5p accelerated vascular smooth muscle cells viabilities and migratory abilities via targeting Bcl-2. Physiol. Res. 2022, 71, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ling, S.; Hu, K.; Liu, J.; Xu, J.W. Role of the renin-angiotensin system in NETosis in the coronavirus disease 2019 (COVID-19). Biomed. Pharmacother. 2022, 148, 112718. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Fang, Y.N.; Wu, C.C.; Chen, M.C.; Chang, J.P.; Lin, Y.S.; Pan, K.L.; Ho, W.C.; Chang, T.H.; Huang, Y.K.; et al. Differential Gene Expression Profile of Renin-Angiotensin System in the Left Atrium in Mitral Regurgitation Patients. Dis. Markers 2018, 2018, 6924608. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Liao, Z.; Mu, Y.; Smith, P.G. Inflammatory Renin-Angiotensin System Disruption Attenuates Sensory Hyperinnervation and Mechanical Hypersensitivity in a Rat Model of Provoked Vestibulodynia. J. Pain. 2018, 19, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Fahim, S.; Montazer, F.; Tohidinik, H.R.; Naraghi, Z.S.; Abedini, R.; Nasimi, M.; Ghandi, N. Serum and tissue angiotensin-converting enzyme in patients with alopecia areata. Indian J. Dermatol. Venereol. Leprol. 2019, 85, 295–299. [Google Scholar]
- Hedayatyanfard, K.; Khalili, A.; Karim, H.; Nooraei, S.; Khosravi, E.; Haddadi, N.S.; Dehpour, A.R.; Bayat, G. Potential use of angiotensin receptor blockers in skin pathologies. Iran J. Basic Med. Sci. 2023, 26, 732–737. [Google Scholar]
- Aleksiejczuk, M.; Gromotowicz-Poplawska, A.; Marcinczyk, N.; Przylipiak, A.; Chabielska, E. The expression of the renin-angiotensin-aldosterone system in the skin and its effects on skin physiology and pathophysiology. J. Physiol. Pharmacol. 2019, 70, 325–336. [Google Scholar]
- Akershoek, J.J.; Vlig, M.; Brouwer, K.; Talhout, W.; Beelen, R.H.J.; Middelkoop, E.; Ulrich, M.M.W. The presence of tissue renin-angiotensin system components in human burn wounds and scars. Burns Open 2018, 2, 114–121. [Google Scholar] [CrossRef]
- Silva, I.M.S.; Assersen, K.B.; Willadsen, N.N.; Jepsen, J.; Artuc, M.; Steckelings, U.M. The role of the renin-angiotensin system in skin physiology and pathophysiology. Exp. Dermatol. 2020, 29, 891–901. [Google Scholar] [CrossRef]
- Liao, X.; Xiao, J.; Li, S.H.; Xiao, L.L.; Cheng, B.; Fu, X.B.; Cui, T.; Liu, H.W. Critical role of the endogenous renin-angiotensin system in maintaining self-renewal and regeneration potential of epidermal stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2647–2656. [Google Scholar] [CrossRef]
- Matsuura-Hachiya, Y.; Arai, K.Y.; Ozeki, R.; Kikuta, A.; Nishiyama, T. Angiotensin-converting enzyme inhibitor (enalapril maleate) accelerates recovery of mouse skin from UVB-induced wrinkles. Biochem. Biophys. Res. Commun. 2013, 442, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hedayatyanfard, K.; Haddadi, N.S.; Ziai, S.A.; Karim, H.; Niazi, F.; Steckelings, U.M.; Habibi, B.; Modarressi, A.; Dehpour, A.R. The renin-angiotensin system in cutaneous hypertrophic scar and keloid formation. Exp. Dermatol. 2020, 29, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Faghih, M.; Hosseini, S.M.; Smith, B.; Ansari, A.M.; Lay, F.; Ahmed, A.K.; Inagami, T.; Marti, G.P.; Harmon, J.W.; Walston, J.D.; et al. Knockout of Angiotensin AT2 receptors accelerates healing but impairs quality. Aging 2015, 7, 1185–1197. [Google Scholar] [CrossRef]
- Tan, W.Q.; Fang, Q.Q.; Shen, X.Z.; Giani, J.F.; Zhao, T.V.; Shi, P.; Zhang, L.Y.; Khan, Z.; Li, Y.; Li, L.; et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-β-activated kinase 1 (TAK1) pathways in mice. Br. J. Pharmacol. 2018, 175, 4239–4252. [Google Scholar] [CrossRef]
- Fang, Q.Q.; Wang, X.F.; Zhao, W.Y.; Ding, S.L.; Shi, B.H.; Xia, Y.; Yang, H.; Wu, L.H.; Li, C.Y.; Tan, W.Q. Angiotensin-converting enzyme inhibitor reduces scar formation by inhibiting both canonical and noncanonical TGF-β1 pathways. Sci. Rep. 2018, 8, 3332. [Google Scholar] [CrossRef]
- Petit, R.G.; Cano, A.; Ortiz, A.; Espina, M.; Prat, J.; Muñoz, M.; Severino, P.; Souto, E.B.; García, M.L.; Pujol, M.; et al. Psoriasis: From Pathogenesis to Pharmacological and Nano-Technological-Based Therapeutics. Int. J. Mol. Sci. 2021, 22, 4983. [Google Scholar] [CrossRef]
- Korman, N.J. Management of psoriasis as a systemic disease: What is the evidence? Br. J. Dermatol. 2020, 182, 840–848. [Google Scholar] [CrossRef]
- Gao, N.; Kong, M.; Li, X.; Zhu, X.; Wei, D.; Ni, M.; Wang, Y.; Hong, Z.; Dong, A. The Association Between Psoriasis and Risk of Cardiovascular Disease: A Mendelian Randomization Analysis. Front. Immunol. 2022, 13, 918224. [Google Scholar] [CrossRef]
- Weber, B.; Merola, J.F.; Husni, M.E.; Di Carli, M.; Berger, J.S.; Garshick, M.S. Psoriasis and Cardiovascular Disease: Novel Mechanisms and Evolving Therapeutics. Curr. Atheroscler. Rep. 2021, 23, 67. [Google Scholar] [CrossRef]
- Piros, É.A.; Szilveszter, B.; Vattay, B.; Maurovich-Horvat, P.; Szalai, K.; Dósa, E.; Merkely, B.; Holló, P. Novel anti-inflammatory therapies to reduce cardiovascular burden of psoriasis. Dermatol. Ther. 2021, 34, e14721. [Google Scholar] [CrossRef]
- Shokrian Zeini, M.; Haddadi, N.S.; Shayan, M.; Shokrian Zeini, M.; Kazemi, K.; Solaimanian, S.; Abdollahifar, M.A.; Hedayatyanfard, K.; Dehpour, A.R. Losartan ointment attenuates imiquimod-induced psoriasis-like inflammation. Int. Immunopharmacol. 2021, 100, 108160. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Zhao, Y.; Wei, Y.; Zhang, X.; Ji, J.; Yang, S. Exploring the Pathogenesis of Psoriasis Complicated With Atherosclerosis via Microarray Data Analysis. Front. Immunol. 2021, 12, 667690. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.R.; Cho, D.H.; Park, H.J. Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis. Int. J. Mol. Sci. 2017, 18, 2684. [Google Scholar] [CrossRef]
- Zhu, H.; Lou, F.; Yin, Q.; Gao, Y.; Sun, Y.; Bai, J.; Xu, Z.; Liu, Z.; Cai, W.; Ke, F.; et al. RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease. EMBO Mol. Med. 2017, 9, 589–604. [Google Scholar] [CrossRef]
- ElGhareeb, M.I.; Khater, M.H.; Fakhr, A.; Khedr, H.A. Risk and severity of psoriasis vulgaris in relation to angiotensin II type 1 receptor gene polymorphism and metabolic syndrome. Clin. Cosmet. Investig. Dermatol. 2019, 12, 683–690. [Google Scholar] [CrossRef]
- Mohammadi, Y.; Vaisi-Raygani, A.; Shakiba, E.; Bahrehmand, F.; Khodarahmi, R.; Nemati, H.; Rahimi, Z.; Kiani, A.; Rahimi, Z.; Vaisi-Raygani, H.; et al. Angiotensin II type 1 receptor A1166 C (rs5186) gene polymorphism increased risk and severity of psoriasis, contribution to oxidative stress, antioxidant statues, lipid peroxidation and correlation with vascular adhesion protein 1, preliminary report. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1395–1397. [Google Scholar] [CrossRef]
- Song, G.; Yoon, H.Y.; Yee, J.; Kim, M.G.; Gwak, H.S. Antihypertensive drug use and psoriasis: A systematic review, meta- and network meta-analysis. Br. J. Clin. Pharmacol. 2022, 88, 933–941. [Google Scholar] [CrossRef]
- Ohyama, K.; Arai, H.; Sugiura, M.; Hori, Y. Psoriasis associated with ACE inhibitors: An analysis of the FAERS database. Pharmazie 2020, 75, 524–526. [Google Scholar]
- Xu, Q.; Zhang, L.; Chen, L.; Zhao, X.; Wang, X.; Hu, M.; Le, Y.; Xue, F.; Li, X.; Zheng, J. SARS-CoV-2 might transmit through the skin while the skin barrier function could be the mediator. Med. Hypotheses 2022, 159, 110752. [Google Scholar] [CrossRef]
- Tembhre, M.K.; Parihar, A.S.; Sharma, V.K.; Imran, S.; Bhari, N.; Lakshmy, R.; Bhalla, A. Enhanced expression of angiotensin-converting enzyme 2 in psoriatic skin and its upregulation in keratinocytes by interferon-γ: Implication of inflammatory milieu in skin tropism of SARS-CoV-2. Br. J. Dermatol. 2021, 184, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Murrell, D.F.; Garcet, S.; Navrazhina, K.; Lee, P.C.; Muscianisi, E.; Blauvelt, A. Secukinumab lowers expression of ACE2 in affected skin of patients with psoriasis. J. Allergy Clin. Immunol. 2021, 147, 1107–1109.e2. [Google Scholar] [CrossRef] [PubMed]
- Shahidi-Dadras, M.; Tabary, M.; Robati, R.M.; Araghi, F.; Dadkhahfar, S. Psoriasis and risk of the COVID-19: Is there a role for angiotensin converting enzyme (ACE)? J. Dermatolog. Treat. 2022, 33, 1175–1176. [Google Scholar] [CrossRef]
- Sauza-Sosa, J.C.; Zenteno-Langle, R.; Zamora-Medina, M.D.C. Hypertensive Emergency in a Woman with Systemic Sclerosis. High Blood Press. Cardiovasc. Prev. 2020, 27, 597–599. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Bruni, C.; Tomelleri, A.; Dagna, L.; Matucci-Cerinic, M. Interleukin-1 and Systemic Sclerosis: Getting to the Heart of Cardiac Involvement. Front. Immunol. 2021, 12, 653950. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, C.; Pène, F.; Charpentier, J.; Mouthon, L.; Chaigne, B. Scleroderma cardiac crisis: A-life-threatening but reversible complication of systemic sclerosis. Autoimmun. Rev. 2022, 21, 103162. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.Y.; Wang, X.D.; Zhang, T.; Xue, J. Cardiac complications in systemic sclerosis: Early diagnosis and treatment. Chin. Med. J. 2019, 132, 2865–2871. [Google Scholar] [CrossRef]
- Bankamp, L.; Preuß, B.; Pecher, A.C.; Vogel, W.; Henes, J.; Klein, R. Functional autoantibodies in systemic sclerosis: Influence of autologous stem cell transplantation and correlation with clinical outcome. Rheumatology 2023, 62, 2168–2177. [Google Scholar] [CrossRef]
- Höppner, J.; Tabeling, C.; Casteleyn, V.; Kedor, C.; Windisch, W.; Burmester, G.R.; Huscher, D.; Siegert, E. Comprehensive autoantibody profiles in systemic sclerosis: Clinical cluster analysis. Front. Immunol. 2023, 13, 1045523. [Google Scholar] [CrossRef]
- Miziołek, B.; Sieńczyk, M.; Grzywa, R.; Łupicka-Słowik, A.; Kucharz, E.; Kotyla, P.; Bergler-Czop, B. The prevalence and role of functional autoantibodies to angiotensin-converting-enzyme-2 in patients with systemic sclerosis. Autoimmunity 2021, 54, 181–186. [Google Scholar] [CrossRef]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kühl, A.A.; Günther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res. Ther. 2014, 16, R29. [Google Scholar] [CrossRef]
- Khan, M.A.; Khan, F.H.; Khan, H.B.; Saadeh, C.; Davey, N. Role of Anifrolumab in Refractory Cutaneous Manifestations of Lupus Erythematosus: A Case Series and Literature Review. Cureus 2023, 15, e39553. [Google Scholar] [CrossRef]
- Ene, C.D.; Nicolae, I. The Inflammatory Profile Orchestrated by Inducible Nitric Oxide Synthase in Systemic Lupus Erythematosus. J. Pers. Med. 2023, 13, 934. [Google Scholar] [CrossRef]
- Hinduja, N.; Mv, P.; Padhee, S.; Maikap, D.; Padhan, P.; Kar, H.K.; Misra, R.; Srinivas, C.R.; Ahmed, S. Assessment of cutaneous disease activity in early lupus and its correlation with quality of life: A cross-sectional study. Rheumatol. Int. 2023, 43, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Zagelbaum Ward, N.K.; Linares-Koloffon, C.; Posligua, A.; Gandrabur, L.; Kim, W.Y.; Sperber, K.; Wasserman, A.; Ash, J. Cardiac Manifestations of Systemic Lupus Erythematous: An Overview of the Incidence, Risk Factors, Diagnostic Criteria, Pathophysiology and Treatment Options. Cardiol. Rev. 2022, 30, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Xiang, W.; He, X. IFN-I Mediates Dysfunction of Endothelial Progenitor Cells in Atherosclerosis of Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 581385. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, Y.; Zhu, M.; Xu, A. Heart involvement in systemic lupus erythematosus: A systemic review and meta-analysis. Clin. Rheumatol. 2016, 35, 2437–2448. [Google Scholar] [CrossRef]
- Soto, M.; Delatorre, N.; Hurst, C.; Rodgers, K.E. Targeting the Protective Arm of the Renin-Angiotensin System to Reduce Systemic Lupus Erythematosus Related Pathologies in MRL-lpr Mice. Front. Immunol. 2020, 11, 1572. [Google Scholar] [CrossRef]
- Khan, S.; Dar, S.A.; Mandal, R.K.; Jawed, A.; Wahid, M.; Panda, A.K.; Lohani, M.; Mishra, B.N.; Akhter, N.; Haque, S. Angiotensin-Converting Enzyme Gene I/D Polymorphism Is Associated With Systemic Lupus Erythematosus Susceptibility: An Updated Meta-Analysis and Trial Sequential Analysis. Front. Physiol. 2018, 9, 1793. [Google Scholar] [CrossRef]
- Shoaib, R.M.S.; Hammad, A.; Yahia, S.; Elsaid, A.; Abdel-Malak, C.A. Angiotensin II type 1 receptor gene polymorphism and serum angiotensin-converting enzyme level in Egyptian children with systemic lupus erythematosus. Clin. Rheumatol. 2018, 37, 3309–3317. [Google Scholar] [CrossRef]
- Shoaib, R.M.S.; Yahia, S.; Elsaid, A.; Abdel-Malak, C.; Hammad, A. Angiotensin II type 2 receptor gene polymorphisms and serum angiotensin-converting enzyme level in Egyptian children with systemic lupus erythematosus. Lupus 2019, 28, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, D.; Sturrock, E.D. Exploring the Impact of ACE Inhibition in Immunity and Disease. J. Renin Angiotensin Aldosterone Syst. 2022, 2022, 9028969. [Google Scholar] [CrossRef] [PubMed]
- Nocito, C.; Lubinsky, C.; Hand, M.; Khan, S.; Patel, T.; Seliga, A.; Winfield, M.; Zuluaga-Ramirez, V.; Fernandes, N.; Shi, X.; et al. Centrally Acting Angiotensin-Converting Enzyme Inhibitor Suppresses Type I Interferon Responses and Decreases Inflammation in the Periphery and the CNS in Lupus-Prone Mice. Front. Immunol. 2020, 11, 573677. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, G.; Su, M.; Leung, G.; Lui, H.; Zhou, P.; Wu, Y.; Zhou, J.; Xu, J.; Zhang, X.; et al. Vitiligo Skin Biomarkers Associated With Favorable Therapeutic Response. Front. Immunol. 2021, 12, 613031. [Google Scholar] [CrossRef]
- Azzazi, Y.; Mostafa, W.Z.; Sayed, K.S.; Alhelf, M.; Safwat, M.; Mahrous, A.; El Lawindi, M.; Ragab, N. Support for increased cardiovascular risk in non-segmental vitiligo among Egyptians: A hospital-based, case-control study. Pigment Cell Melanoma Res. 2021, 34, 598–604. [Google Scholar] [CrossRef]
- Xia, J.; Melian, C.; Guo, W.; Usmani, H.; Clark, R.; Lozeau, D. Vitiligo and Metabolic Syndrome: Systematic Review and Meta-Analysis. JMIR Dermatol. 2022, 5, e34772. [Google Scholar] [CrossRef]
- Almohideb, M. Associations of Angiotensin-Converting Enzyme Gene Insertion/Deletion (ACE Gene I/D) Polymorphism With Vitiligo: An Updated Systematic Review and Meta-Analysis. Cureus 2020, 12, e8046. [Google Scholar] [CrossRef]
- Basher, N.S.; Malik, A.; Aldakheel, F.; Chaudhary, A.A.; Rudayni, H.A.; Alkholief, M.; Alshamsan, A. Deleterious effect of angiotensin-converting enzyme gene polymorphism in vitiligo patients. Saudi J. Biol. Sci. 2021, 28, 4478–4483. [Google Scholar] [CrossRef]
- Custurone, P.; Di Bartolomeo, L.; Irrera, N.; Borgia, F.; Altavilla, D.; Bitto, A.; Pallio, G.; Squadrito, F.; Vaccaro, M. Role of Cytokines in Vitiligo: Pathogenesis and Possible Targets for Old and New Treatments. Int. J. Mol. Sci. 2021, 22, 11429. [Google Scholar] [CrossRef]
- Zhou, C.; Li, X.; Wang, C.; Zhang, J. Alopecia Areata: An Update on Etiopathogenesis, Diagnosis, and Management. Clin. Rev. Allergy Immunol. 2021, 61, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Sterkens, A.; Lambert, J.; Bervoets, A. Alopecia areata: A review on diagnosis, immunological etiopathogenesis and treatment options. Clin. Exp. Med. 2021, 21, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, A.; Albino-Teixeira, A.; Magina, S. Systematic review-alopecia areata and tofacitinib in paediatric patients. Cutan. Ocul. Toxicol. 2022, 41, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Pagan, A.D.; Jung, S.; Caldas, S.; Ungar, J.; Gulati, N.; Ungar, B. Cross-Sectional Study of Psoriasis, Atopic Dermatitis, Rosacea, and Alopecia Areata Suggests Association With Cardiovascular Diseases. J. Drugs Dermatol. 2023, 22, 576–581. [Google Scholar] [CrossRef]
- Glickman, J.W.; Dubin, C.; Renert-Yuval, Y.; Dahabreh, D.; Kimmel, G.W.; Auyeung, K.; Estrada, Y.D.; Singer, G.; Krueger, J.G.; Pavel, A.B.; et al. Cross-sectional study of blood biomarkers of patients with moderate to severe alopecia areata reveals systemic immune and cardiovascular biomarker dysregulation. J. Am. Acad. Dermatol. 2021, 84, 370–380. [Google Scholar] [CrossRef]
- Wang, E.H.; Santos, L.; Li, X.Y.; Tran, A.; Kim, S.S.Y.; Woo, K.; Shapiro, J.; McElwee, K.J. Alopecia Areata is Associated with Increased Expression of Heart Disease Biomarker Cardiac Troponin I. Acta Derm. Venereol. 2018, 98, 776–782. [Google Scholar] [CrossRef]
- Namazi, M.R.; Ashraf, A.; Handjani, F.; Eftekhar, E.; Kalafi, A. Angiotensin converting enzyme activity in alopecia areata. Enzyme Res. 2014, 2014, 694148. [Google Scholar] [CrossRef]
- Pietkiewicz, P.; Gornowicz-Porowska, J.; Bowszyc-Dmochowska, M.; Dmochowski, M. A retrospective study of antihypertensives in pemphigus: A still unchartered odyssey particularly between thiols, amides and phenols. Arch. Med. Sci. 2015, 11, 1021–1027. [Google Scholar]
- Malik, A.M.; Tupchong, S.; Huang, S.; Are, A.; Hsu, S.; Motaparthi, K. An Updated Review of Pemphigus Diseases. Medicina 2021, 57, 1080. [Google Scholar] [CrossRef]
- Amber, K.T.; Valdebran, M.; Grando, S.A. Non-Desmoglein Antibodies in Patients With Pemphigus Vulgaris. Front. Immunol. 2018, 9, 1190. [Google Scholar] [CrossRef]
- Wada, N.; Nishifuji, K.; Yamada, T.; Kudoh, J.; Shimizu, N.; Matsumoto, M.; Peltonen, L.; Nagafuchi, S.; Amagai, M. Aire-dependent thymic expression of desmoglein 3, the autoantigen in pemphigus vulgaris, and its role in T-cell tolerance. J. Investig. Dermatol. 2011, 131, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Rokni, A.M.; Ayasse, M.; Ahmed, A.; Guggina, L.; Kantor, R.W.; Silverberg, J.I. Association of autoimmune blistering disease, and specifically, pemphigus vulgaris, with cardiovascular disease and its risk factors: A systematic review and meta-analysis. Arch. Dermatol. Res. 2023, 315, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Francone, M.; Verardo, R.; Scialla, R.; Bagnato, G.; Alfarano, M.; Chimenti, C.; Frustaci, E.; Sansone, L.; Russo, M. Pemphigus-associated cardiomyopathy: Report of autoimmune myocarditis and review of literature. ESC Heart Fail. 2021, 8, 3690–3695. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Melduni, R.M. Autoimmunity and cardiac arrhythmias in endemic pemphigus foliaceus-Association, correlation, or causation? Heart Rhythm. 2018, 15, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Robati, R.M.; Ayatollahi, A.; Toossi, P.; Younespour, S. Serum Angiotensin converting enzyme in pemphigus vulgaris. Indian J. Dermatol. 2014, 59, 348–351. [Google Scholar]
- Cozzani, E.; Rosa, G.M.; Drosera, M.; Intra, C.; Barsotti, A.; Parodi, A. ACE inhibitors can induce circulating antibodies directed to antigens of the superficial epidermal cells. Arch. Dermatol. Res. 2011, 303, 327–332. [Google Scholar] [CrossRef]
- Miyamoto, D.; Santi, C.G.; Aoki, V.; Maruta, C.W. Bullous pemphigoid. An. Bras. Dermatol. 2019, 94, 133–146. [Google Scholar] [CrossRef]
- Bulger, D.A.; Minhas, S.; Asbeutah, A.A.; Kayali, S.; Shirwany, H.A.K.; Patel, J.R.; Seitz, M.P.; Clark, K.; Patel, T.; Khouzam, R.N. Chronic Systemic Inflammatory Skin Disease as a Risk Factor for Cardiovascular Disease. Curr. Probl. Cardiol. 2021, 46, 100799. [Google Scholar] [CrossRef]
- Shen, W.C.; Chiang, H.Y.; Chen, P.S.; Lin, Y.T.; Kuo, C.C.; Wu, P.Y. Risk of All-Cause Mortality, Cardiovascular Disease Mortality, and Cancer Mortality in Patients With Bullous Pemphigoid. JAMA Dermatol. 2022, 158, 167–175. [Google Scholar] [CrossRef]
- Kalińska-Bienias, A.; Kowalczyk, E.; Jagielski, P.; Bienias, P.; Kowalewski, C.; Woźniak, K. The association between neurological diseases, malignancies and cardiovascular comorbidities among patients with bullous pemphigoid: Case-control study in a specialized Polish center. Adv. Clin. Exp. Med. 2019, 28, 637–642. [Google Scholar] [CrossRef]
- Kalińska-Bienias, A.; Rogoziński, T.T.; Woźniak, K.; Kowalewski, C. Can pemphigoid be provoked by lisinopril? Br. J. Dermatol. 2006, 155, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Ballout, R.A.; Musharrafieh, U.; Khattar, J. Lisinopril-associated bullous pemphigoid in an elderly woman: A case report of a rare adverse drug reaction. Br. J. Clin. Pharmacol. 2018, 84, 2678–2682. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Suzuki, T.; Kayanuma, G.; Yamamoto, H.; Nagayasu, K.; Shirakawa, H.; Kaneko, S. Lisinopril prevents bullous pemphigoid induced by dipeptidyl peptidase 4 inhibitors via the Mas receptor pathway. Front. Immunol. 2023, 13, 1084960. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Mansoori, S.; Sircar, K.; Popli, D.B. Isolated Lichen Planus of the Lower Lip: Report of a Rare Case with an Updated Literature Review. Curr. Health Sci. J. 2022, 48, 345–352. [Google Scholar]
- Rashed, L.; Abdel Hay, R.; AlKaffas, M.; Ali, S.; Kadry, D.; Abdallah, S. Studying the association between methylenetetrahydrofolate reductase (MTHFR) 677 gene polymorphism, cardiovascular risk and lichen planus. J. Oral Pathol. Med. 2017, 46, 1023–1029. [Google Scholar] [CrossRef]
- Mushtaq, S.; Dogra, D.; Dogra, N.; Shapiro, J.; Fatema, K.; Faizi, N.; Gupta, G. Cardiovascular and Metabolic Risk Assessment in Patients with Lichen Planus: A Tertiary Care Hospital-based Study from Northern India. Indian Dermatol. Online J. 2020, 11, 158–166. [Google Scholar] [CrossRef]
- Nasiri, S.; Sadeghzadeh-Bazargan, A.; Robati, R.M.; Haghighatkhah, H.R.; Younespour, S. Subclinical atherosclerosis and cardiovascular markers in patients with lichen planus: A case-control study. Indian J. Dermatol. Venereol. Leprol. 2019, 85, 138–144. [Google Scholar]
- Huskić, J.; Mulabegović, N.; Alendar, F.; Ostojić, L.; Ostojić, Z.; Simić, D.; Milicević, R.; Naletilić, M. Serum and tissue angiotensin converting enzyme in patients with psoriasis. Coll. Antropol. 2008, 32, 1215–1219. [Google Scholar]
- Alendar, F.; Huskić, J.; Babić, N.; Mulabegović, N. Serum and tissue angiotensin converting enzyme in patients with lichen planus. Bosn. J. Basic Med. Sci. 2005, 5, 59–62. [Google Scholar] [CrossRef]
- Ben Salem, C.; Chenguel, L.; Ghariani, N.; Denguezli, M.; Hmouda, H.; Bouraoui, K. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol. Drug Saf. 2008, 17, 722–724. [Google Scholar] [CrossRef]
- Ogg, G.S.; Bhogal, B.S.; Hashimoto, T.; Coleman, R.; Barker, J.N. Ramipril-associated lichen planus pemphigoides. Br. J. Dermatol. 1997, 136, 412–414. [Google Scholar] [CrossRef]
- Shinjo, S.K.; Uno, M.; Oba-Shinjo, S.M.; Marie, S.K. Angiotensin-converting enzyme insertion/deletion gene polymorphism is associated with dermatomyositis. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, H. Comment on: Cardiovascular events in adult polymyositis and dermatomyositis: A meta-analysis of observational studies. Rheumatology 2022, 61, e178–e179. [Google Scholar] [CrossRef] [PubMed]
- Mecoli, C.A.; Yoshida, A.; Paik, J.J.; Lin, C.T.; Danoff, S.; Hanaoka, H.; Rosen, A.; Christopher-Stine, L.; Kuwana, M.; Casciola-Rosen, L. Presence and Implications of Anti-Angiotensin Converting Enzyme-2 Immunoglobulin M Antibodies in Anti-Melanoma-Differentiation-Associated 5 Dermatomyositis. ACR Open Rheumatol. 2022, 4, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Yigit, S.; Tural, S.; Rüstemoglu, A.; Inanir, A.; Gul, U.; Kalkan, G.; Akkanet, S.; Karakuş, N.; Ateş, O. DD genotype of ACE gene I/D polymorphism is associated with Behcet disease in a Turkish population. Mol. Biol. Rep. 2013, 40, 365–368. [Google Scholar] [CrossRef]
- Rouhi, N.; Nazm, S.A.; Bonyadi, M.; Jabbarpoor Bonyadi, M.H.; Soheilian, M. Angiotensin-converting enzyme gene polymorphism in Behçet’s disease in Iranian population. Ophthalmic Genet. 2019, 40, 388–389. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Tang, S.; Cai, X.; Bai, J.; Sun, Q.; Qiao, J.; Fang, H. Risk factors of cardiovascular involvement in patients with Behcet’s disease. J. Transl. Autoimmun. 2023, 6, 100195. [Google Scholar] [CrossRef]
- Mandal, R.K.; Yaday, S.S.; Panda, A.K.; Khattri, S. Insertion/deletion polymorphism of the ACE gene increased risk of Behcet disease: Evidence from a meta-analysis. Ann. Saudi Med. 2013, 33, 437–442. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maranduca, M.A.; Cosovanu, M.A.; Clim, A.; Pinzariu, A.C.; Filip, N.; Drochioi, I.C.; Vlasceanu, V.I.; Timofte, D.V.; Nemteanu, R.; Plesa, A.; et al. The Renin-Angiotensin System: The Challenge behind Autoimmune Dermatological Diseases. Diagnostics 2023, 13, 3398. https://doi.org/10.3390/diagnostics13223398
Maranduca MA, Cosovanu MA, Clim A, Pinzariu AC, Filip N, Drochioi IC, Vlasceanu VI, Timofte DV, Nemteanu R, Plesa A, et al. The Renin-Angiotensin System: The Challenge behind Autoimmune Dermatological Diseases. Diagnostics. 2023; 13(22):3398. https://doi.org/10.3390/diagnostics13223398
Chicago/Turabian StyleMaranduca, Minela Aida, Mihai Andrei Cosovanu, Andreea Clim, Alin Constantin Pinzariu, Nina Filip, Ilie Cristian Drochioi, Vlad Ionut Vlasceanu, Daniel Vasile Timofte, Roxana Nemteanu, Alina Plesa, and et al. 2023. "The Renin-Angiotensin System: The Challenge behind Autoimmune Dermatological Diseases" Diagnostics 13, no. 22: 3398. https://doi.org/10.3390/diagnostics13223398