
Neuroendocrine Carcinomas of the Gastroenteropancreatic System: A Comprehensive Review


 and
and
Abstract
:1. Introduction
2. Method

3. Classification and Histology

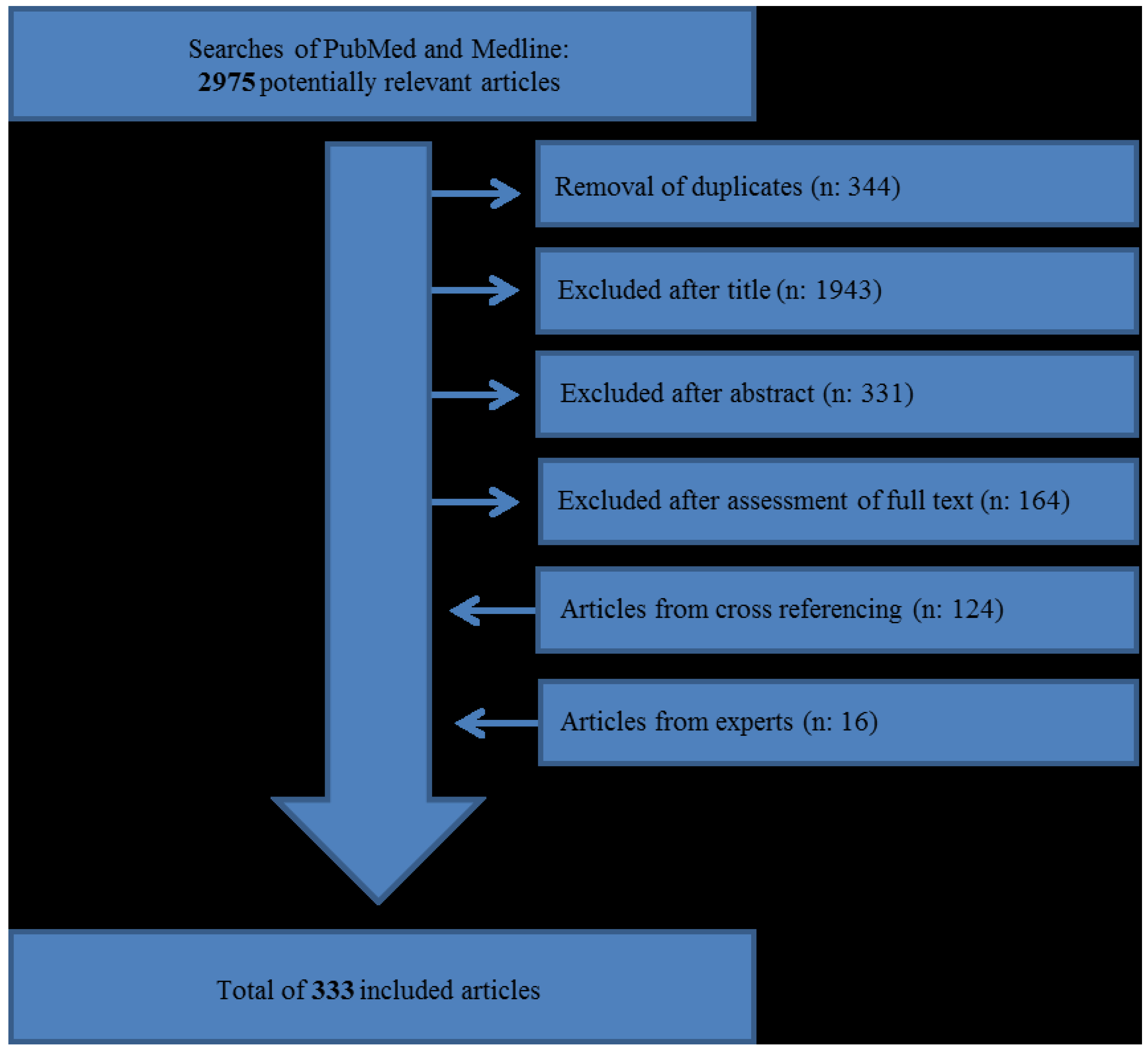{kind=link}
| Neuroendocrine Neoplasm Type | Grade | Ki-67 Index * | Mitotic Count (per 10 HPF **) |
|---|---|---|---|
| Neuroendocrine Tumour (Carcinoid) | G1 | ≤2% | <2 |
| Neuroendocrine Tumour | G2 | 3%–20% | 2–20 |
| Neuroendocrine Carcinoma | G3 | >20% | >20 |
| Mixed adenoneuroendocrine carcinoma (MANEC) | G1–G3 (mostly G3 component) | All ranges | All ranges |
3.1. Small and Large Cell NECs
3.2. Immunohistochemistry (IHC)
3.3. Genetic Profile
4. Origin
5. Epidemiology
6. Biochemistry
7. Ki-67 Index
8. Imaging
9. Treatment
9.1. Surgery
9.2. Chemotherapy
9.3. Other Treatment Modalities
9.4. Metastases
9.5. Follow-Up
10. Survival
11. Oesophagus
12. Stomach
13. Pancreas
14. Gallbladder
15. Small Intestine and Ampulla of Vater
16. Appendix
17. Colon and Rectum
18. Conclusions and Future Directions
18.1. The Challenge of Terminology
18.2. Different Entities
18.3. Incidence
18.4. Treatment and Survival
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lepage, C.; Rachet, B.; Coleman, M.P. Survival From Malignant Digestive Endocrine Tumors in England and Wales: A Population-Based Study. Gastroenterology 2007, 132, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.Y.; Kim, J.M.; Sohn, J.H.; Kim, M.J.; Kim, K.M.; Kim, W.H.; Kim, H.; Kook, M.C.; Park, D.Y.; Lee, J.H.; et al. Current Trends of the Incidence and Pathological Diagnosis of Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs) in Korea 2000–2009: Multicenter Study. Cancer Res. Treat. 2012, 44, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Taal, B.G.; van Velthuysen, M.L.; Visser, O. Incidence and survival of neuroendocrine tumours in the Netherlands according to histological grade: Experience of two decades of cancer registry. Eur. J. Cancer 2013, 49, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann. Oncol. 2013, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; O’Connell, J.; Leonardi, M.; Maggard, M.; McGory, M.; Ko, C. Rare tumors of the colon and rectum: A national review. Int. J. Colorectal Dis. 2007, 22, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.M.; Gregory, D.L.; Stillie, A.; Herschtal, A.; Mac, M.M.; Ball, D.L. Should extrapulmonary small cell cancer be managed like small cell lung cancer? Cancer 2010, 116, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Shah, M.A.; Gonen, M.; Klimstra, D.S.; Shia, J.; Kelsen, D.P. Small-cell carcinoma of the gastrointestinal tract: A retrospective study of 64 cases. Br. J. Cancer 2004, 90, 1720–1726. [Google Scholar] [PubMed]
- Formica, V.; Wotherspoon, A.; Cunningham, D.; Norman, A.R.; Sirohi, B.; Oates, J.; Chong, G. The prognostic role of WHO classification, urinary 5-hydroxyindoleacetic acid and liver function tests in metastatic neuroendocrine carcinomas of the gastroenteropancreatic tract. Br. J. Cancer 2007, 96, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Frytak, S.; Lloyd, R.V. Extrapulmonary small cell carcinoma. Cancer 1997, 79, 1729–1736. [Google Scholar] [PubMed]
- Garcia-Carbonero, R.; Capdevila, J.; Crespo-Herrero, G.; Diaz-Perez, J.A.; Martinez del Prado, M.P.; Alonso, O.V.; Sevilla-Garcia, I.; Villabona-Artero, C.; Beguiristain-Gomez, A.; Llanos-Munoz, M.; et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): Results from the National Cancer Registry of Spain (RGETNE). Ann. Oncol. 2010, 21, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.A.; Pedroso, F.E.; Byrne, M.M.; Koniaris, L.G.; Misra, S. Does surgery or radiation therapy impact survival for patients with extrapulmonary small cell cancers? J. Surg. Oncol. 2011, 104, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.; Shahid, R.K.; Finch, D.; Sami, A.; Ahmad, I.; Yadav, S.; Alvi, R.; Popkin, D.; Ahmed, S. Extrapulmonary small cell cancer. Cancer 2006, 107, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.O.; Lee, H.Y.; Chun, S.H.; Shin, S.J.; Kim, M.K.; Lee, K.H.; Hyun, M.S.; Bae, S.H.; Ryoo, H.M. Clinical overview of extrapulmonary small cell carcinoma. J. Korean Med. Sci. 2006, 21, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Konishi, K.; Maeda, K.; Yabushita, K.; Tsuji, M.; Miwa, A. Highly aggressive behavior and poor prognosis of small-cell carcinoma in the alimentary tract: Flow-cytometric analysis and immunohistochemical staining for the p53 protein and proliferating cell nuclear antigen. Dig. Surg. 1999, 16, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, J.L.; Ryu, M.H.; Chang, H.M.; Kim, T.W.; Kim, W.K.; Lee, J.S.; Jang, S.J.; Khang, S.K.; Kang, Y.K.; et al. Extrapulmonary small cell carcinoma: Single center experience with 61 patients. Acta Oncol. 2007, 46, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Li, A.F.-Y.; Hsu, H.S.; Hsu, C.Y.; Li, A.C.-H.; Li, W.Y.; Liang, W.Y.; Chen, J.Y. A 20-year retrospective study of small-cell carcinomas in Taiwan. J. Surg. Oncol. 2010, 102, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Chung, C.Y.; Chang, C.S.; Wu, J.S.; Kuo, K.T.; Kuo, S.H.; Cheng, A.L. Prognostic Factors in Extrapulmonary Small Cell Carcinomas. Oncology 2007, 72, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Pape, U.F.; Bohmig, M.; Berndt, U.; Tiling, N.; Wiedenmann, B.; Plockinger, U. Survival and clinical outcome of patients with neuroendocrine tumors of the gastroenteropancreatic tract in a german referral center. Ann. N. Y. Acad. Sci. 2004, 1014, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One Hundred Years After “Carcinoid”: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2010. [Google Scholar]
- Capelli, P.; Fassan, M.; Scarpa, A. Pathology—Grading and staging of GEP-NETs. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Solcia, E.; Kloeppel, G.; Sobin, L.H. Histological Typing of Endocrine Tumours; Springer: New York, NY, USA, 2000. [Google Scholar]
- Hamilton, S.R.; Aaltonen, L.A. World Health Oranization Classification of Tumours, Pathology and Genetics, Tumours of the Digestive System; IARC: Lyon, France, 2000. [Google Scholar]
- DeLellis, R.A.; Lloyd, R.V.; Heitz, P.U.; Eng, C. Pathology and Genetics. Tumours of Endocrine Organs; IARC Press: Lyon, France, 2004. [Google Scholar]
- Lee, J.L.; Yu, C.S.; Kim, M.; Hong, S.M.; Lim, S.B.; Kim, J.C. Prognostic impact of diagnosing colorectal neuroendocrine carcinoma using the World Health Organization 2010 classification. Surgery 2014, 155, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Ozkara, S.; Aker, F.; Yesil, A.; Senates, E.; Canbey, C.; Yitik, A.; Gonen, C. Re-evaluation of cases with gastroenteropancreatic neuroendocrine tumors between 2004 and 2012 according to the 2010 criteria. Hepatogastroenterology 2013, 60, 1665–1672. [Google Scholar] [PubMed]
- Klimstra, D.S.; Modlin, I.R.; Adsay, N.V.; Chetty, R.; Deshpande, V.; Gonen, M.; Jensen, R.T.; Kidd, M.; Kulke, M.H.; Lloyd, R.V.; et al. Pathology reporting of neuroendocrine tumors: Application of the Delphic consensus process to the development of a minimum pathology data set. Am. J. Surg. Pathol. 2010, 34, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Dousei, T.; Yoshikawa, Y.; Hatanaka, N.; Taniyama, K.; Yamauchi, A.; Kamiike, W.; Nishijima, J. Gastric Neuroendocrine Tumors in Our Institutions according to the WHO 2010 Classification. Int. Surg. 2012, 97, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Nakamura, K.I.; Sawabe, M.; Arai, T.; Esaki, Y.; Miyashita, M.; Mafune, K.I.; Tanaka, Y.; Sasajima, K. Primary undifferentiated small cell carcinoma of the esophagus. Hum. Pathol. 1999, 30, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Janson, E.T.; Sorbye, H.; Welin, S.; Federspiel, B.; Grønbæk, H.; Hellman, P.; Ladekarl, M.; Langer, S.W.; Mortensen, J.; Schalin-Jäntti, C.; et al. Nordic Guidelines 2014 for Diagnosis and Treatment of Gastroeneropancreatic Neuroendocrine Neoplasias. Acta Oncol. 2014, 53, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Shia, J.; Tang, L.H.; Weiser, M.R.; Brenner, B.; Adsay, N.V.; Stelow, E.B.; Saltz, L.B.; Qin, J.; Landmann, R.; Leonard, G.D.; et al. Is nonsmall cell type high-grade neuroendocrine carcinoma of the tubular gastrointestinal tract a distinct disease entity? Am. J. Surg. Pathol. 2008, 32, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Wiedenmann, B. Neuroendocrine neoplasms of the gut and pancreas: New insights. Nat. Rev. Endocrinol. 2012, 8, 54–64. [Google Scholar] [CrossRef]
- Rindi, G.; Bordi, C.; La, R.S.; Solcia, E.; Delle, F.G. Gastroenteropancreatic (neuro)endocrine neoplasms: The histology report. Dig. Liver Dis. 2011, 43 (Suppl. 4), S356–S360. [Google Scholar] [CrossRef] [PubMed]
- Li, A.F.; Li, A.C.; Hsu, C.Y.; Li, W.Y.; Hsu, H.S.; Chen, J.Y. Small cell carcinomas in gastrointestinal tract: Immunohistochemical and clinicopathological features. J. Clin. Pathol. 2010, 63, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, W.; Kwan, M.Y.; Suster, S.; Chan, J.K.C. Immunostaining for Thyroid Transcription Factor 1 and Cytokeratin 20 Aids the Distinction of Small Cell Carcinoma from Merkel Cell Carcinoma, But Not Pulmonary from Extrapulmonary Small Cell Carcinomas. Arch. Pathol. Lab. Med. 2001, 125, 228–231. [Google Scholar] [PubMed]
- Ordonez, N.G. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am. J. Surg. Pathol. 2000, 24, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Uccella, S.; Cerutti, R.; Vigetti, D.; Furlan, D.; Oldrini, R.; Carnevali, I.; Pelosi, G.; Rosa, S.L.; Passi, A.; Capella, C.; et al. Histidine Decarboxylase, DOPA Decarboxylase, and Vesicular Monoamine Transporter 2 Expression in Neuroendocrine Tumors: Immunohistochemical Study and Gene Expression Analysis. J. Histochem. Cytochem. 2006, 54, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, S.; Azzoni, C.; Bassi, D.; Bottarelli, L.; Milione, M.; Bordi, C. Genetic alterations in poorly differentiated endocrine carcinomas of the gastrointestinal tract. Cancer 2003, 98, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Furlan, D.; Cerutti, R.; Uccella, S.; La, R.S.; Rigoli, E.; Genasetti, A.; Capella, C. Different molecular profiles characterize well-differentiated endocrine tumors and poorly differentiated endocrine carcinomas of the gastroenteropancreatic tract. Clin. Cancer Res. 2004, 10, 947–957. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, D.; Couvelard, A.; Rebours, V.; Zappa, M.; Hentic, O.; Hammel, P.; Levy, P.; Bedossa, P.; Raymond, E.; Ruszniewski, P.; et al. Molecular markers associated with response to chemotherapy in gastro-entero-pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Dacic, S.; Finkelstein, S.D.; Baksh, F.K.; Swalsky, P.A.; Barnes, L.E.; Yousem, S.A. Small-cell neuroendocrine carcinoma displays unique profiles of tumor-suppressor gene loss in relationship to the primary site of formation. Hum. Pathol. 2002, 33, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Li, A.F.-Y.; Tsay, S.H.; Liang, W.Y.; Li, W.Y.; Chen, J.Y. Clinical Significance of p16INK4A and p53 Overexpression in Endocrine Tumors of the Gastrointestinal Tract. Am. J. Clin. Pathol. 2006, 126, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Furlan, D.; Bernasconi, B.; Uccella, S.; Cerutti, R.; Carnevali, I.; Capella, C. Allelotypes and Fluorescence in Situ Hybridization Profiles of Poorly Differentiated Endocrine Carcinomas of Different Sites. Clin. Cancer Res. 2005, 11, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Moyana, T.N.; Xiang, J.; Senthilselvan, A.; Kulaga, A. The spectrum of neuroendocrine differentiation among gastrointestinal carcinoids: Importance of histologic grading, MIB-1, p53, and bcl-2 immunoreactivity. Arch. Pathol. Lab. Med. 2000, 124, 570–576. [Google Scholar] [PubMed]
- Grabowski, P.; Griss, S.; Arnold, C.N.; Horsch, D.; Goke, R.; Arnold, R.; Heine, B.; Stein, H.; Zeitz, M.; Scherubl, H.; et al. Nuclear survivin is a powerful novel prognostic marker in gastroenteropancreatic neuroendocrine tumor disease. Neuroendocrinology 2005, 81, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Kishimoto, T.; Furuya, M.; Nikaido, T.; Koda, K.; Takano, S.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother. Pharmacol. 2010, 65, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Catena, L.; Bajetta, E.; Milione, M.; Ducceschi, M.; Valente, M.; Dominoni, F.; Colonna, V. Mammalian target of rapamycin expression in poorly differentiated endocrine carcinoma: Clinical and therapeutic future challenges. Target. Oncol. 2011, 6, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, P.; Schrader, J.; Wagner, J.; Hörsch, D.; Arnold, R.; Arnold, C.N.; Georgieva, I.; Stein, H.; Zeitz, M.; Daniel, P.T.; et al. Loss of Nuclear p27 Expression and Its Prognostic Role in Relation to Cyclin E and p53 Mutation in Gastroenteropancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2008, 14, 7378–7384. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Volante, M.; Ferrero, A.; Righi, L.; Rapa, I.; Rosas, R.; Berruti, A.; Dogliotti, L.; Scagliotti, G.V.; Papotti, M.; et al. Thymidylate Synthase Expression in Gastroenteropancreatic and Pulmonary Neuroendocrine Tumors. Clin. Cancer Res. 2008, 14, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Chen, M.; Kim, J.H.; Kim, W.H.; Ahn, S.; Maeng, K.; Allegra, C.J.; Kaye, F.J.; Hochwald, S.N.; Zajac-Kaye, M.; et al. Analysis of 320 gastroenteropancreatic neuroendocrine tumors identifies TS expression as independent biomarker for survival. Int. J. Cancer 2014, 135, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; Astudillo, A.; Vallina, A.; Fonseca, P.J.; Gómez-Izquierdo, L.; García-Carbonero, R.; González, M.V. Epithelial-Mesenchymal Transition Markers in the Differential Diagnosis of Gastroenteropancreatic Neuroendocrine Tumors. Am. J. Clin. Pathol. 2013, 140, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Furuya, M.; Nikaido, T.; Kishimoto, T.; Koda, K.; Oda, K.; Nakatani, Y.; Miyazaki, M.; Ishikura, H. Aberrant Expression of Human Achaete-Scute Homologue Gene 1 in the Gastrointestinal Neuroendocrine Carcinomas. Clin. Cancer Res. 2005, 11, 450–458. [Google Scholar] [PubMed]
- Shida, T.; Furuya, M.; Kishimoto, T.; Nikaido, T.; Tanizawa, T.; Koda, K.; Oda, K.; Takano, S.; Kimura, F.; Shimizu, H.; et al. The expression of NeuroD and mASH1 in the gastroenteropancreatic neuroendocrine tumors. Mod. Pathol. 2008, 21, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Mia-Jan, K.; Munkhdelger, J.; Lee, M.R.; Ji, S.Y.; Kang, T.Y.; Choi, E.; Cho, M.Y. Expression of CD133 in Neuroendocrine Neoplasms of the Digestive Tract: A Detailed Immunohistochemical Analysis. Tohoku J. Exp. Med. 2013, 229, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Knosel, T.; Chen, Y.; Altendorf-Hofmann, A.; Danielczok, C.; Freesmeyer, M.; Settmacher, U.; Wurst, C.; Schulz, S.; Yang, L.L.; Petersen, I.; et al. High KIT and PDGFRA are associated with shorter patients survival in gastroenteropancreatic neuroendocrine tumors, but mutations are a rare event. J. Cancer Res. Clin. Oncol. 2012, 138, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Ishikubo, T.; Akagi, K.; Kurosumi, M.; Yamaguchi, K.; Fujimoto, T.; Sakamoto, H.; Tanaka, Y.; Ochiai, A. Immunohistochemical and Mutational Analysis of c-kit in Gastrointestinal Neuroendocrine Cell Carcinoma. Jpn. J. Clin. Oncol. 2006, 36, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, S.; Azzoni, C.; Bottarelli, L.; Campanini, N.; D’Adda, T.; Pasquali, C.; Rossi, G.; Rindi, G.; Bordi, C. RASSF1A promoter methylation and 3p21.3 loss of heterozygosity are features of foregut, but not midgut and hindgut, malignant endocrine tumours. J. Pathol. 2005, 206, 409–416. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Rigoli, E.; Uccella, S.; Chiaravalli, A.; Capella, C. CDX2 as a marker of intestinal EC-cells and related well-differentiated endocrine tumors. Virchows Arch. 2004, 445, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Sciandivasci, A.; Intrivici, C.; Pascucci, A.; del Vecchio, M.T.; Marsili, S.; Savelli, V.; Voltolini, L.; di Bisceglie, M.; Guarnieri, A.; et al. Chemo-hormone therapy of non-well-differentiated endocrine tumours from different anatomic sites with cisplatinum, etoposide and slow release lanreotide formulation. Br. J. Cancer 2007, 96, 1343–1347. [Google Scholar] [PubMed]
- Helpap, B.; Köllermann, J. Immunohistochemical analysis of the proliferative activity of neuroendocrine tumors from various organs. Virchows Arch. 2001, 438, 86–91. [Google Scholar] [CrossRef] [PubMed]
- La, R.S.; Inzani, F.; Vanoli, A.; Klersy, C.; Dainese, L.; Rindi, G.; Capella, C.; Bordi, C.; Solcia, E. Histologic characterization and improved prognostic evaluation of 209 gastric neuroendocrine neoplasms. Hum. Pathol. 2011, 42, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Kim, M.J.; Cho, B.K.; Choi, S.W.; Rhyu, M.G. Genetic evidence for the multi-step progression of mixed glandular-neuroendocrine gastric carcinomas. Virchows Arch. 2002, 440, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Azzoni, C.; La, R.S.; Klersy, C.; Paolotti, D.; Rappel, S.; Stolte, M.; Capella, C.; Bordi, C.; Solcia, E.; et al. ECL cell tumor and poorly differentiated endocrine carcinoma of the stomach: Prognostic evaluation by pathological analysis. Gastroenterology 1999, 116, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Annenkov, A.; Nishikura, K.; Domori, K.; Ajioka, Y. Alpha-methylacyl-coenzyme A racemase expression in neuroendocrine neoplasms of the stomach. Virchows Arch. 2012, 461, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.N.; Jack, R.H.; Mak, V.; Henrik, M.; Davies, E.A. The epidemiology and survival of extrapulmonary small cell carcinoma in South East England, 1970–2004. BMC Cancer 2009, 9, 209. [Google Scholar] [CrossRef] [PubMed]
- Niederle, M.B.; Hackl, M.; Kaserer, K.; Niederle, B. Gastroenteropancreatic neuroendocrine tumours: The current incidence and staging based on the WHO and European Neuroendocrine Tumour Society classification: An analysis based on prospectively collected parameters. Endocr. Relat. Cancer 2010, 17, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Delektorskaya, V.V.; Kozlov, N.A.; Chemeris, G.Y. Clinico-morphological analysis of the neuroendocrine neoplasms of the gastroenteropancreatic system. Klin. Lab. Diagn. 2013, 48–50, 10–13. [Google Scholar]
- Faggiano, A.; Ferolla, P.; Grimaldi, F.; Campana, D.; Manzoni, M.; Davi, M.V.; Bianchi, A.; Valcavi, R.; Papini, E.; Giuffrida, D.; et al. Natural history of gastro-entero-pancreatic and thoracic neuroendocrine tumors. Data from a large prospective and retrospective Italian epidemiological study: The NET management study. J. Endocrinol. Investig. 2012, 35, 817–823. [Google Scholar]
- Fjällskog, M.L.; Ludvigsen, E.; Stridsberg, M.; Öberg, K.; Eriksson, B.; Janson, E. Expression of somatostatin receptor subtypes 1 to 5 in tumor tissue and intratumoral vessels in malignant endocrine pancreatic tumors. Med. Oncol. 2003, 20, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Choi, J.; An, J.S.; Kim, H.; Shin, B.K.; Kim, A.; Kim, H.; Kim, I. The Clinicopathological Characteristics of Gastrointestinal Neuroendocrine Tumors: An Analysis of 65 cases. Korean J. Pathol. 2007, 41, 3:149–3:157. [Google Scholar]
- Lombard-Bohas, C.; Mitry, E.; O’Toole, D.; Louvet, C.; Pillon, D.; Cadiot, G.; Borson-Chazot, F.; Aparicio, T.; Ducreux, M.; Lecomte, T.; et al. Thirteen-Month Registration of Patients with Gastroenteropancreatic Endocrine Tumours in France. Neuroendocrinology 2009, 89, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Papotti, M.; Bongiovanni, M.; Volante, M.; Allìa, E.; Landolfi, S.; Helboe, L.; Schindler, M.; Cole, S.; Bussolati, G. Expression of somatostatin receptor types 1–5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. Virchows Arch. 2002, 440, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Ploeckinger, U.; Kloeppel, G.; Wiedenmann, B.; Lohmann, R. The German NET-Registry: An Audit on the Diagnosis and Therapy of Neuroendocrine Tumors. Neuroendocrinology 2009, 90, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, F.; Carnaghi, C.; Di, T.L.; Rodari, M.; Destro, A.; Rimassa, L.; Giordano, L.; Chiti, A.; Roncalli, M.; Santoro, A.; et al. Detection of somatostatin receptor subtypes 2 and 5 by somatostatin receptor scintigraphy and immunohistochemistry: Clinical implications in the diagnostic and therapeutic management of gastroenteropancreatic neuroendocrine tumors. Tumori 2011, 97, 620–628. [Google Scholar] [PubMed]
- Cicin, I.; Karagol, H.; Uzunoglu, S.; Uygun, K.; Usta, U.; Kocak, Z.; Caloglu, M.; Saynak, M.; Tokatli, F.; Uzal, C.; et al. Extrapulmonary small-cell carcinoma compared with small-cell lung carcinoma. Cancer 2007, 110, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A.; Sabourin, J.C.; Ducreux, M.; Lumbroso, J.; Duvillard, P.; Leboulleux, S.; Dromain, C.; Colao, A.; Schlumberger, M.; Baudin, E.; et al. Pulmonary and extrapulmonary poorly differentiated large cell neuroendocrine carcinomas. Cancer 2007, 110, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Gregory, D.L.; Brennan, S.M.; Stillie, A.; Herschtal, A.; Hicks, R.J.; MacManus, M.P.; Ball, D.L. Impact of 18F-fluorodeoxyglucose positron emission tomography in the staging and treatment response assessment of extra-pulmonary small-cell cancer. J. Med. Imaging Radiat. Oncol. 2010, 54, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Quinn, A.M.; Blackhall, F.; Wilson, G.; Danson, S.; Clamp, A.; Ashcroft, L.; Brierley, J.; Hasleton, P. Extrapulmonary small cell carcinoma: A clinicopathological study with identification of potential diagnostic mimics. Histopathology 2012, 61, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Ciccolallo, L.; De, A.R.; Bouvier, A.M.; Faivre, J.; Gatta, G. European disparities in malignant digestive endocrine tumours survival. Int. J. Cancer 2010, 126, 2928–2934. [Google Scholar] [PubMed]
- Cimitan, M.; Buonadonna, A.; Cannizzaro, R.; Canzonieri, V.; Borsatti, E.; Ruffo, R.; de Apollonia, L. Somatostatin receptor scintigraphy versus chromogranin A assay in the management of patients with neuroendocrine tumors of different types: Clinical role. Ann. Oncol. 2003, 14, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Wang, Y.; Zhou, Y.; Wen, F.; Li, Q. First-line irinotecan combined with 5-fluorouracil and leucovorin for high-grade metastatic gastrointestinal neuroendocrine carcinoma. Tumori 2013, 99, 57–60. [Google Scholar] [PubMed]
- Feng, S.T.; Luo, Y.; Chan, T.; Peng, Z.; Chen, J.; Chen, M.; Li, Z.P. CT Evaluation of Gastroenteric Neuroendocrine Tumors: Relationship Between CT Features and the Pathologic Classification. Am. J. Roentgenol. 2014, 203, W260–W266. [Google Scholar] [CrossRef]
- Fjällskog, M.L.; Granberg, D.P.; Welin, S.L.; Eriksson, C.; Oberg, K.E.; Janson, E.T.; Eriksson, B.K. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001, 92, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Spigel, D.R.; Litchy, S.; Greco, F.A. Phase II Trial of Paclitaxel, Carboplatin, and Etoposide in advanced Poorly Differentiated Neuroendocrine Carcinoma: A Minnie Pearl Cancer Research Network Study. J. Clin. Oncol. 2006, 24, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Hentic, O.; Hammel, P.; Couvelard, A.; Rebours, V.; Zappa, M.; Palazzo, M.; Maire, F.; Goujon, G.; Gillet, A.; Levy, P.; et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr. Relat. Cancer 2012, 19, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wang, T.; Chen, X.; Xu, B.; Sun, J.; Guo, R.; Shu, Y. Phase II Study of Weekly Irinotecan plus Cisplatin in Patients with Previously Untreated Extensive-Stage Extrapulmonary Small Cell Carcinoma. Oncol. Res. Treat. 2011, 34, 378–381. [Google Scholar]
- Lu, Z.H.; Li, J.; Lu, M.; Zhang, X.T.; Li, J.; Zhou, J.; Wang, X.C.; Gong, J.F.; Gao, J.; Li, Y.; et al. Feasibility and efficacy of combined cisplatin plus irinotecan chemotherapy for gastroenteropancreatic neuroendocrine carcinomas. Med. Oncol. 2013, 30, 1–5. [Google Scholar]
- Mitry, E.; Baudin, E.; Ducreux, M.; Sabourin, J.C.; Rufie, P.; Aparicio, T.; Aparicio, T.; Lasser, P.; Elias, D.; Duvillard, P.; et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br. J. Cancer 1999, 81, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Strosberg, J.; Baudin, E.; Klimstra, D.S.; Yao, J.C. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer 2014, 120, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Vélayoudom-Céphise, F.L.; Duvillard, P.; Foucan, L.; Hadoux, J.; Chougnet, C.N.; Leboulleux, S.; Malka, D.; Guigay, J.; Goere, D.; Debaere, T.; et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr. Relat. Cancer 2013, 20, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Van der Gaast, A.; Verwey, J.; Prins, E.; Splinter, T.A. Chemotherapy as treatment of choice in extrapulmonary undifferentiated small cell carcinomas. Cancer 1990, 65, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Machida, N.; Morizane, C.; Kasuga, A.; Takahashi, H.; Sudo, K.; Nishina, T.; Tobimatsu, K.; Ishido, K.; Furuse, J.; et al. Multicenter retrospective analysis of systemic chemotherapy for advanced neuroendocrine carcinoma of the digestive system. Cancer Sci. 2014, 105, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Taal, B.G.; Vincent, A.; van Velthuysen, M.L.; Baas, P.; Buning-Kager, J.C.G.M.; Linders, T.C.; Bonfrer, J.M.G. Choice of tumour markers in patients with neuroendocrine tumours is dependent on the histological grade. A marker study of Chromogranin A, Neuron specific enolase, Progastrin-releasing peptide and cytokeratin fragments. Eur. J. Cancer 2012, 48, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Srirajaskanthan, R.; Bhogal, M.; Toubanakis, C.; Meyer, T.; Noonan, A.; Witney-Smith, C.; Amin, T.; Bhogal, P.; Sivathasan, N.; et al. α-Fetoprotein and human chorionic gonadotrophin-β as prognostic markers in neuroendocrine tumour patients. Br. J. Cancer 2008, 99, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, R.; Jiang, S.; Zhu, K.; Feng, R.; Xu, X.; Meng, X. NSE can predict the sensitivity to definitive chemoradiotherapy of small cell carcinoma of esophagus. Med. Oncol. 2013, 31, 1–7. [Google Scholar]
- Young, H.T.; Carr, N.J.; Green, B.; Tilley, C.; Bhargava, V.; Pearce, N. Accuracy of visual assessments of proliferation indices in gastroenteropancreatic neuroendocrine tumours. J. Clin. Pathol. 2013, 66, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Gonen, M.; Hedvat, C.; Modlin, I.M.; Klimstra, D.S. Objective quantification of the Ki67 proliferative index in neuroendocrine tumors of the gastroenteropancreatic system: A comparison of digital image analysis with manual methods. Am. J. Surg. Pathol. 2012, 36, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Hentic, O.; Couvelard, A.; Rebours, V.; Zappa, M.; Dokmak, S.; Hammel, P.; Maire, F.; O’Toole, D.; Levy, P.; Sauvanet, A.; et al. Ki-67 index, tumor differentiation, and extent of liver involvement are independent prognostic factors in patients with liver metastases of digestive endocrine carcinomas. Endocr. Relat. Cancer 2011, 18, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Baudin, E.; Couvelard, A.; Krenning, E.; Oberg, K.; Steinmuller, T.; Anlauf, M.; Wiedenmann, B.; Salazar, R. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.M.; Coppola, D.M.; Klimstra, D.S.; Phan, A.T.M.; Kulke, M.H.M.; Wiseman, G.A.M.; Kvols, L.K.M. The NANETS Consensus Guidelines for the Diagnosis and Management of Poorly Differentiated (High-Grade) Extrapulmonary Neuroendocrine Carcinomas. Pancreas 2010, 39, 799–800. [Google Scholar] [CrossRef] [PubMed]
- McDermott, S.; O’Neill, A.C.; Skehan, S.J. Staging of gastroenteropancreatic neuroendocrine tumors: How we do it based on an evidence-based approach. Clin. Imaging 2013, 37, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Rodallec, M.; Vilgrain, V.; Couvelard, A.; Rufat, P.; O’Toole, D.; Barrau, V.; Sauvanet, A.; Ruszniewski, P.; Menu, Y. Endocrine Pancreatic Tumours and Helical CT: Contrast Enhancement Is Correlated with Microvascular Density, Histoprognostic Factors and Survival. Pancreatology 2006, 6, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Johnbeck, C.B.; Knigge, U.; Kjær, A. PET tracers for somatostatin receptor imaging of neuroendocrine tumors: Current status and review of the literature. Future Oncol. 2014, 10, 2259–2277. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Knigge, U.; Mortensen, J.; Oturai, P.; Berthelsen, A.K.; Loft, A.; Binderup, T.; Rasmussen, P.; Elema, D.; Klausen, T.L.; et al. Clinical PET of Neuroendocrine Tumors Using 64Cu-DOTATATE: First-in-Humans Study. J. Nucl. Med. 2012, 53, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Binderup, T.; Knigge, U.; Mellon Mogensen, A.; Palnaes Hansen, C.; Kjær, A. Quantitative Gene Expression of Somatostatin Receptors and Noradrenaline Transporter Underlying Scintigraphic Results in Patients with Neuroendocrine Tumors. Neuroendocrinology 2008, 87, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Srirajaskanthan, R.; Watkins, J.; Marelli, L.; Khan, K.; Caplin, M.E. Expression of Somatostatin and Dopamine 2 Receptors in Neuroendocrine Tumours and the Potential Role for New Biotherapies. Neuroendocrinology 2009, 89, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Binderup, T.; Knigge, U.; Loft, A.; Mortensen, J.; Pfeifer, A.; Federspiel, B.; Hansen, C.P.; Højgaard, L.; Kjær, A. Functional Imaging of Neuroendocrine Tumors: A Head-to-Head Comparison of Somatostatin Receptor Scintigraphy, 123I-MIBG Scintigraphy, and 18F-FDG PET. J. Nucl. Med. 2010, 51, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Artale, S.; Giannetta, L.; Cerea, G.; Maggioni, D.; Pedrazzoli, P.; Schiavetto, I.; Napolitano, M.; Veronese, S.; Bramerio, E.; Gambacorta, M.; et al. Treatment of metastatic neuroendocrine carcinomas based on WHO classification. Anticancer Res. 2005, 25, 4463–4469. [Google Scholar] [PubMed]
- Welin, S.; Sorbye, H.; Sebjornsen, S.; Knappskog, S.; Busch, C.; Oberg, K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 2011, 117, 4617–4622. [Google Scholar] [CrossRef] [PubMed]
- Ezziddin, S.; Opitz, M.; Attassi, M.; Biermann, K.; Sabet, A.; Guhlke, S.; Brockmann, H.; Willinek, W.; Wardelmann, E.; Biersack, H.J.; et al. Impact of the Ki-67 proliferation index on response to peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Prasad, V.; Lee, D.S.; Baum, R.P. Effect of Peptide Receptor Radionuclide Therapy on Somatostatin Receptor Status and Glucose Metabolism in Neuroendocrine Tumors: Intraindividual Comparison of Ga-68 DOTANOC PET/CT and F-18 FDG PET/CT. Int. J. Mol. Imaging 2011, 2011, 524130. [Google Scholar] [CrossRef] [PubMed]
- Binderup, T.; Knigge, U.; Loft, A.; Federspiel, B.; Kjaer, A. 18F-Fluorodeoxyglucose Positron Emission Tomography Predicts Survival of Patients with Neuroendocrine Tumors. Clin. Cancer Res. 2010, 16, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Larghi, A.; Capurso, G.; Carnuccio, A.; Ricci, R.; Alfieri, S.; Galasso, D.; Lugli, F.; Bianchi, A.; Panzuto, F.; de Marinis, L.; et al. Ki-67 grading of nonfunctioning pancreatic neuroendocrine tumors on histologic samples obtained by EUS-guided fine-needle tissue acquisition: A prospective study. Gastrointest. Endosc. 2012, 76, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Öberg, K.; Knigge, U.; Kwekkeboom, D.; Perren, A.; on behalf of the ESMO Guidelines Working Group. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23 (Suppl. 7), vii124–vii130. [Google Scholar] [PubMed]
- Khasraw, M.; Yap, S.Y.; Ananda, S. Neuroendocrine neoplasms of the GI tract: The role of cytotoxic chemotherapy. Expert Rev. Anticancer Ther. 2013, 13, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Kvols, L.K.; O’Connell, M.J.; Rubin, J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 1991, 68, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Morizane, C.; Hiraoka, N.; Tsuda, H.; Tamura, T.; Shimada, Y.; Kaneko, S.; Kushima, R.; Ueno, H.; Kondo, S.; et al. Comparison of chemotherapeutic treatment outcomes of advanced extrapulmonary neuroendocrine carcinomas and advanced small-cell lung carcinoma. Neuroendocrinology 2012, 96, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Deutschbein, T.; Unger, N.; Yuece, A.; Eberhardt, W.; Gauler, T.; Lahner, H.; Mann, K.; Petersenn, S. Chemotherapy in patients with progressive, undifferentiated neuroendocrine tumors: A single-center experience. Horm. Metab. Res. 2011, 43, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.H.; Langer, S.W.; Jepsen, I.; Assens, M.; Federspiel, B.; Hasselby, J.P.; Hansen, C.P.; Kjær, A.; Knigge, U. First-line treatment of patients with disseminated poorly differentiated neuroendocrine carcinomas with carboplatin, etoposide, and vincristine: A single institution experience. Acta Oncol. 2012, 51, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Takahashi, S.; Yuasa, T.; Nishimura, N.; Mishima, Y.; Sakajiri, S.; Yokoyama, M.; Tsuyama, N.; Ishikawa, Y.; Hatake, K.; et al. Feasibility and efficacy of combined cisplatin and irinotecan chemotherapy for poorly differentiated neuroendocrine carcinomas. Jpn. J. Clin. Oncol. 2012, 42, 697–703. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, D.; Kuang, Q.; Liu, G.; Xu, W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: A meta-analysis in Caucasians. Pharm. J. 2014, 14, 120–129. [Google Scholar]
- Turner, N.C.; Strauss, S.J.; Sarker, D.; Gillmore, R.; Kirkwood, A.; Hackshaw, A.; Papadopoulou, A.; Bell, J.; Kayani, I.; Toumpanakis, C.; et al. Chemotherapy with 5-fluorouracil, cisplatin and streptozocin for neuroendocrine tumours. Br. J. Cancer 2010, 102, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Catena, L.; Procopio, G.; De, D.S.; Bichisao, E.; Ferrari, L.; Martinetti, A.; Platania, M.; Verzoni, E.; Formisano, B.; et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother. Pharmacol. 2007, 59, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotto, R.; Testa, L.; Riechelmann, R.P.; Sahade, M.; Siqueira, L.T.; Costa, F.P.; Hoff, P.M. Combination of capecitabine and oxaliplatin is an effective treatment option for advanced neuroendocrine tumors. Rare Tumors 2013, 5, 121–125. [Google Scholar] [CrossRef]
- Olsen, I.H.; Sorensen, J.B.; Federspiel, B.; Kjær, A.; Hansen, C.P.; Knigge, U.; Langer, S.W. Temozolomide as second or third line treatment of patients with neuroendocrine carcinomas. Sci. World J. 2012, 2012, 170496. [Google Scholar] [CrossRef]
- Ahn, H.K.; Choi, J.Y.; Kim, K.M.; Kim, H.; Choi, S.H.; Park, S.H.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Lee, J.; et al. Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br. J. Cancer 2013, 109, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.H.; Knigge, U.; Federspiel, B.; Hansen, C.P.; Skov, A.; Kjær, A.; Langer, S.W. Topotecan monotherapy in heavily pretreated patients with progressive advanced stage neuroendocrine carcinomas. J. Cancer 2014, 5, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.M.; Tang, L.M.; Hruban, R.H.M.; Adsay, V.M.; Yang, Z.M.; Krasinskas, A.M.M.; Vakiani, E.M.; la Rosa, S.M.; Jang, K.-T.M.P.; Frankel, W.L.M.; et al. Poorly Differentiated Neuroendocrine Carcinomas of the Pancreas: A Clinicopathologic Analysis of 44 Cases. Am. J. Surg. Pathol. 2014, 38, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Sauvanet, A.; Belghiti, J. Place of surgical resection in the treatment strategy of gastrointestinal neuroendocrine tumors. Target. Oncol. 2012, 7, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Reidy-Lagunes, D. The management of extrapulmonary poorly differentiated (high-grade) neuroendocrine carcinomas. Semin. Oncol. 2013, 40, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Frilling, A.; Li, J.; Malamutmann, E.; Schmid, K.W.; Bockisch, A.; Broelsch, C.E. Treatment of liver metastases from neuroendocrine tumours in relation to the extent of hepatic disease. Br. J. Surg. 2009, 96, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Chua, T.C.; Bester, L.; Kokandi, A.; Morris, D.L. Factors predicting response and survival after yttrium-90 radioembolization of unresectable neuroendocrine tumor liver metastases: A critical appraisal of 48 cases. Ann. Surg. 2010, 251, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Le Treut, Y.P.; Gregoire, E.; Belghiti, J.; Boillot, O.; Soubrane, O.; Mantion, G.; Cherqui, D.; Castaing, D.; Ruszniewski, P.; Wolf, P.; et al. Predictors of long-term survival after liver transplantation for metastatic endocrine tumors: An 85-case French multicentric report. Am. J. Transplant. 2008, 8, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Reidy-Lagunes, D.; Anthony, L.B.; Bertino, E.M.; Brendtro, K.; Chan, J.A.; Chen, H.; Jensen, R.T.; Kim, M.K.; Klimstra, D.S.; et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 2013, 42, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.V.; Chejfec, G.; Jao, W.; Gould, V.E. Neuroendocrine carcinomas of the esophagus. Ultrastruct. Pathol. 1980, 1, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Bardet, E.; Deguiral, P.; Douillard, J.Y. Small cell carcinoma of the esophagus: Analysis of 10 cases and review of the published data. Am. J. Clin. Oncol. 2000, 23, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.L.; Marshall, J.B.; Diaz-Arias, A.A.; Loy, T.S. Primary small-cell carcinoma of the esophagus. Report of 11 cases and review of the literature. J. Clin. Gastroenterol. 1991, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.C.; Ibrahim, N.B. Oat cell carcinomas of the oesophagus: A clinico-pathological study of 23 cases. Histopathology 1983, 7, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.B.; Yang, J.S.; Yang, W.P.; Weng, H.R.; Li, H.; Liu, D.T.; Chen, Y.P. Treatment and prognosis of limited disease primary small cell carcinoma of esophagus. Dis. Esophagus 2011, 24, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.R.; Carey, F.A.; Walker, W.S.; Cameron, E.W. Primary small-cell cancer of the esophagus. J. Thorac. Cardiovasc. Surg. 1995, 109, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ji, J.; Zhu, W.; Zhou, K.; Han, J.; Zhang, Y.; Yu, C.; Li, T.; Tao, G.; Ji, F.; et al. A retrospective study of different treatments of limited-stage small-cell esophageal carcinoma and associated prognostic factor analysis. Dis. Esophagus 2013, 26, 696–702. [Google Scholar] [PubMed]
- Doherty, M.A.; McIntyre, M.; Arnott, S.J. Oat cell carcinoma of esophagus: A report of six British patients with a review of the literature. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Wei, J.C.; Wu, J.X.; Wang, X.; Fu, J.H.; Lin, P.; Yang, H.X. Multidisciplinary Modalities Achieve Encouraging Long-Term Survival in Resectable Limited-Disease Esophageal Small Cell Carcinoma. PLoS ONE 2013, 8, e69259. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, H.; Nie, L.; Shi, J.; Lebenthal, A.; Chen, J.; Sun, Q.; Yang, J.; Huang, L.; Ye, Q.; et al. Primary high-grade neuroendocrine carcinoma of the esophagus: A clinicopathologic and immunohistochemical study of 42 resection cases. Am. J. Surg. Pathol. 2013, 37, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Huncharek, M.; Muscat, J. Small cell carcinoma of the esophagus. The Massachusetts General Hospital experience, 1978 to 1993. Chest 1995, 107, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Koide, N.; Saito, H.; Suzuki, A.; Sato, T.; Koiwai, K.; Nakamura, N.; Miyagawa, S. Clinicopathologic features and histochemical analyses of proliferative activity and angiogenesis in small cell carcinoma of the esophagus. J. Gastroenterol. 2007, 42, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Ku, G.Y.; Minsky, B.D.; Rusch, V.W.; Bains, M.; Kelsen, D.P.; Ilson, D.H. Small-cell carcinoma of the esophagus and gastroesophageal junction: Review of the Memorial Sloan-Kettering experience. Ann. Oncol. 2008, 19, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Kukar, M.; Groman, A.; Malhotra, U.; Warren, G.; Bogner, P.; Nwogu, C.; Demmy, T.; Yendamuri, S. Small Cell Carcinoma of the Esophagus: A SEER Database Analysis. Ann. Surg. Oncol. 2013, 20, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Hsieh, C.C.; Chan, M.L.; Li, A.F.; Huang, M.H.; Hsu, W.H.; Hsu, H.S. Small cell carcinoma of the esophagus: A report of 16 cases from a single institution and literature review. Ann. Thorac. Surg. 2011, 91, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.Y.; Law, S.; Tung, P.H.; Wong, J. Esophageal small cell carcinomas: Clinicopathologic parameters, p53 overexpression, proliferation marker, and their impact on pathogenesis. Arch. Pathol. Lab. Med. 2000, 124, 228–233. [Google Scholar] [PubMed]
- Lu, X.J.; Luo, J.D.; Ling, Y.; Kong, Y.Z.; Feng, L.L.; Zhou, J.; Wang, F. Management of small cell carcinoma of esophagus in China. J. Gastrointest. Surg. 2013, 17, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liang, J.; Wang, J.; Wang, L.; He, J.; Xiao, Z.; Yin, W. Primary small cell carcinoma of the esophagus. J. Thorac. Oncol. 2008, 3, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Maru, D.M.; Khurana, H.; Rashid, A.; Correa, A.M.; Anandasabapathy, S.; Krishnan, S.; Komaki, R.; Ajani, J.A.; Swisher, S.G.; Hofstetter, W.L.; et al. Retrospective study of clinicopathologic features and prognosis of high-grade neuroendocrine carcinoma of the esophagus. Am. J. Surg. Pathol. 2008, 32, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Medgyesy, D.C.; Wolff, R.A.; Putnam, J.B.; Ajani, J.A. Small cell carcinoma of the esophagus. Cancer 2000, 88, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.B.; Zaorsky, N.G.; Jiang, C.; Tian, L.J.; Wang, H.H.; Liu, C.L.; Wang, J.; Tao, Z.; Sun, Y.; Wang, J.; et al. Radiotherapy and chemotherapy are associated with improved outcomes over surgery and chemotherapy in the management of limited-stage small cell esophageal carcinoma. Radiother. Oncol. 2013, 106, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Kuwabara, Y.; Shinoda, N.; Sato, A.; Fujii, Y. Long-term survivors after the resection of limited esophageal small cell carcinoma. Dis. Esophagus 2000, 13, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Matsukuma, A.; Adachi, Y.; Miyagahara, T.; Matsuda, H.; Kuwano, H.; Sugimachi, K.; Enjoji, M. Small cell carcinoma of the esophagus. Cancer 1989, 63, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Zenda, S.; Minashi, K.; Yano, T.; Tahara, M.; Doi, T.; Onozawa, M.; Nihei, K.; Fujii, S.; Ohtsu, A.; et al. Non-surgical approach to small cell carcinoma of the esophagus: Does this rare disease have the same tumor behavior as SCLC? Int. J. Clin. Oncol. 2012, 17, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, K.; Zhao, H.J.; Goto, T.; Ogawa, Y.; Takai, Y.; Matsushita, H.; Takeda, K.; Takahashi, C.; Saito, H.; Yamada, S.; et al. Radiation therapy for limited-stage small-cell esophageal cancer. Am. J. Clin. Oncol. 2002, 25, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Nishimaki, T.; Suzuki, T.; Nakagawa, S.; Watanabe, K.; Aizawa, K.; Hatakeyama, K. Tumor spread and outcome of treatment in primary esophageal small cell carcinoma. J. Surg. Oncol. 1997, 64, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Takeno, S.; Kato, T.; Wada, S.; Noguchi, T.; Uchida, Y.; Kashima, K.; Yokoyama, S. Small cell carcinoma of the esophagus; clinicopathological and immunohistochemical analysis of six cases. Dis. Esophagus 2003, 16, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Osugi, H.; Takemura, M.; Morimura, K.; Kaneko, M.; Higashino, M.; Takada, N.; Lee, S.; Kinoshita, H. Clinicopathologic and immunohistochemical features of surgically resected small cell carcinoma of the esophagus. Oncol. Rep. 2002, 9, 1245–1249. [Google Scholar] [PubMed]
- Pantvaidya, G.H.; Pramesh, C.S.; Deshpande, M.S.; Jambhekar, N.A.; Sharma, S.; Deshpande, R.K. Small cell carcinoma of the esophagus: The Tata Memorial Hospital experience. Ann. Thorac. Surg. 2002, 74, 1924–1927. [Google Scholar] [CrossRef]
- Poynton, A.R.; Walsh, T.N.; Kelly, A.; Harney, M.; Stuart, R.; Daly, P.A.; Hennessy, T.P.J. Small cell carcinoma of the oesophagus. Eur. J. Surg. Oncol. 1997, 23, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Matono, S.; Nagano, T.; Nishimura, K.; Murata, K.; Yamana, H.; Shirouzu, K.; Fujita, H. Surgical management for small cell carcinoma of the esophagus. Dis. Esophagus 2010, 23, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Tennvall, J.; Johansson, L.; Albertsson, M. Small cell carcinoma of the oesophagus: A clinical and immunohistopathological review. Eur. J. Surg. Oncol. 1990, 16, 109–115. [Google Scholar] [PubMed]
- Terada, T. Small cell neuroendocrine carcinoma of the esophagus: Report of 6 cases with immunohistochemical and molecular genetic analysis of KIT and PDGFRA. Int. J. Clin. Exp. Pathol. 2013, 6, 485–491. [Google Scholar] [PubMed]
- Wang, S.Y.; Mao, W.M.; Du, X.H.; Xu, Y.P.; Zhang, S.Z. The 2002 AJCC TNM classification is a better predictor of primary small cell esophageal carcinoma outcome than the VALSG staging system. Chin. J. Cancer 2013, 32, 342–352. [Google Scholar] [PubMed]
- Wu, Z.; Ma, J.Y.; Yang, J.J.; Zhao, Y.F.; Zhang, S.F. Primary small cell carcinoma of esophagus: Report of 9 cases and review of literature. World J. Gastroenterol. 2004, 10, 3680–3682. [Google Scholar] [PubMed]
- Yamashita, H.; Nakagawa, K.; Asari, T.; Murakami, N.; Igaki, H.; Okuma, K.; Ohtomo, K. Concurrent chemoradiation alone with curative intent for limited-disease small-cell esophageal cancer in nine Japanese patients. Dis. Esophagus 2009, 22, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.K.; Siu, W.T.; Wong, D.C.T.; Chau, C.H.; Li, A.C.N.; Law, B.K.B.; Li, M.K.W. Non-operative management of small cell carcinoma of esophagus. Dis. Esophagus 2007, 20, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Zhimin, Z.; Hualiang, X.; Fei, X.; Hui, Z.; Chuan, C.; He, X.; Zhenzhou, Y.; Dong, W.; Zengpeng, L.; Ge, W.; et al. High-incidence of PTEN mutations in Chinese patients with primary small cell carcinoma of the esophagus. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Wang, F.; Zhang, D.S.; Luo, H.Y.; Wang, Z.Q.; Wang, F.H.; Qiu, M.Z.; Ren, C.; Wei, X.L.; Wu, W.J.; et al. Primary small cell carcinoma of the esophagus: Clinicopathological study of 44 cases. BMC Cancer 2014, 14, 222. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; Baba, S.; Hosaka, H.; Ishiyama, A.; Mizunuma, N.; Shinozaki, E.; Suenaga, M.; Kozuka, T.; Seto, Y.; Yamamoto, N.; et al. Irinotecan plus cisplatin for therapy of small-cell carcinoma of the esophagus: Report of 12 cases from single institution experience. Jpn. J. Clin. Oncol. 2008, 38, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, A.; Shimada, Y.; Matsumura, Y.; Yamada, Y.; Muro, K.; Hamaguchi, T.; Igaki, H.; Tachimori, Y.; Kato, H.; Shirao, K.; et al. Small cell carcinoma of the esophagus. Analysis of 14 cases and literature review. Hepatogastroenterology 2005, 52, 1738–1741. [Google Scholar] [PubMed]
- Sadanaga, N.; Morita, M.; Masuda, T.; Okada, S.; Sugiyama, M.; Ando, K.; Kakeji, Y.; Matsuura, H.; Maehara, Y. Clinical features of primary small cell carcinoma of the thoracic esophagus: A retrospective analysis of 12 surgically resected cases. Esophagus 2009, 6, 161–165. [Google Scholar] [CrossRef]
- Atsumi, K.; Shioyama, Y.; Nomoto, S.; Ohga, S.; Toba, T.; Sasaki, T.; Kunitake, N.; Yoshitake, T.; Nakamura, K.; Honda, H.; et al. Chemoradiation for small cell esophageal carcinoma: Report of 11 cases from multi-institution experience. J. Radiat. Res. 2010, 51, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Vos, B.; Rozema, T.; Miller, R.C.; Hendlisz, A.; van Laethem, J.L.; Khanfir, K.; Weber, D.C.; el Nakadi, I.; van Houtte, P. Small Cell Carcinoma of the Esophagus: A Multicentre Rare Cancer Network Study. Dis. Esophagus 2011, 24, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.; Powell, J.; Mukherjee, S.; Crosby, T.D.; Brewster, A.E.; Maughan, T.S.; Bailey, H.; Lester, J.F. Small cell oesophageal carcinoma: An institutional experience and review of the literature. Br. J. Cancer 2007, 96, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xue, L.Y.; Lu, N.; Zou, S.M.; Liu, X.Y.; Wen, P. Superficial primary small cell carcinoma of the esophagus: Clinicopathological and immunohistochemical analysis of 15 cases. Dis. Esophagus 2010, 23, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qiu, B.; Liu, H.; Li, Q.; Xiao, W.; Hu, Y.; Liu, M. Primary small cell carcinoma of the esophagus: Review of 64 cases from a single institution. Dis. Esophagus 2014, 27, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Wang, F.; Chen, S.; Wang, L.; Ren, C.; Luo, H.Y.; Wang, F.H.; Li, Y.H.; Zhang, D.S.; Xu, R.H.; et al. Detailed Analysis of Prognostic Factors in Primary Esophageal Small Cell Carcinoma. Ann. Thorac. Surg. 2014, 97, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.P.; Zhang, M.F.; Hou, J.H.; Tian, Q.H.; Fu, J.; Liang, X.M.; Wu, Q.L.; Rong, T.H. Primary small cell carcinoma of the esophagus: Clinicopathological and immunohistochemical features of 21 cases. BMC Cancer 2007, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Al, M.S.; Ziske, C.; Schmidt-Wolf, I.G. Primary small cell carcinoma of the esophagus: Patient data metaanalysis and review of the literature. Ger. Med. Sci. 2013, 11. [Google Scholar] [CrossRef]
- Chow, V.; Law, S.; Lam, K.Y.; Luk, J.M.; Wong, J. Telomerase activity in small cell esophageal carcinoma. Dis. Esophagus 2001, 14, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Gollard, R.; Ellis, C.; VanderHarten, C. Small cell/neuroendocrine tumors of the esophagus: Presentation of two cases and review of the literature. Tumori 2010, 96, 780–783. [Google Scholar] [PubMed]
- Okuma, H.; Iwasa, S.; Shoji, H.; Takashima, A.; Okita, N.; Honma, Y.; Kato, K.; Hamaguchi, T.; Yamada, Y.; Shimada, Y.; et al. Irinotecan plus Cisplatin in Patients with Extensive-Disease Poorly Differentiated Neuroendocrine Carcinoma of the Esophagus. Anticancer Res. 2014, 34, 5037–5041. [Google Scholar] [PubMed]
- Usami, S.; Motoyama, S.; Maruyama, K.; Sato, Y.; Shibuya, K.H.; Nakatsu, T.; Saito, H.; Minamiya, Y.; Ogawa, J. Small Cell Carcinoma of the Esophagus Treated with Esophagectomy and following Chemotherapy: Case Report with Review of the Literature. Eur. Surg. Res. 2010, 45, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.; Rice, T.W.; Rajeswaran, J.; Zhong, J.; Blackstone, E.H. Esophageal small-cell cancer: Study of a rare disease. Dis. Esophagus 2013, 26, 690–695. [Google Scholar] [PubMed]
- Leichman, L.; Rosser, R. Uncommon Tumors of the Esophagus. In Textbook of Uncommon Cancer; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 373–388. [Google Scholar]
- Boo, Y.J.; Park, S.S.; Kim, J.H.; Mok, Y.J.; Kim, S.J.; Kim, C.S. Gastric neuroendocrine carcinoma: Clinicopathologic review and immunohistochemical study of E-cadherin and Ki-67 as prognostic markers. J. Surg. Oncol. 2007, 95, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, Y.; Zhao, X.; Zhang, H.; Yuan, X.; Wang, J. Primary small cell carcinoma of the stomach: An experience of two decades (1990–2011) in a Chinese cancer institute. J. Surg. Oncol. 2012, 106, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Sekine, S.; Fukagawa, T.; Ohashi, M.; Morita, S.; Taniguchi, H.; Katai, H.; Tsuda, H.; Kushima, R. Neuroendocrine carcinoma of the stomach: Morphologic and immunohistochemical characteristics and prognosis. Am. J. Surg. Pathol. 2013, 37, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.X.; Mikami, T.; Umezawa, A.; Saegusa, M.; Kameya, T.; Okayasu, I. Gastric large cell neuroendocrine carcinomas: A distinct clinicopathologic entity. Am. J. Surg. Pathol. 2006, 30, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Oh, S.T.; Yook, J.H.; Kim, K.C.; Kim, M.G.; Jeong, J.W.; Kim, B.S. Typical carcinoids and neuroendocrine carcinomas of the stomach: Differing clinical courses and prognoses. Am. J. Surg. 2010, 200, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Ohyama, S.; Hiki, N.; Nunobe, S.; Yamamoto, N.; Yamaguchi, T. Endocrine carcinoma of the stomach: Clinicopathological analysis of 27 surgically treated cases in a single institute. Gastric Cancer 2012, 15, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Jin, X.M.; Kitagawa, M.; Miwa, A. Clinicopathologic features of neuroendocrine carcinomas of the stomach: Appraisal of small cell and large cell variants. Arch. Pathol. Lab. Med. 1998, 122, 1010–1017. [Google Scholar] [PubMed]
- Namikawa, T.; Oki, T.; Kitagawa, H.; Okabayashi, T.; Kobayashi, M.; Hanazaki, K. Neuroendocrine carcinoma of the stomach: Clinicopathological and immunohistochemical evaluation. Med. Mol. Morphol. 2013, 46, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K.; Watanabe, H.; Iwafuchi, M.; Fujiwara, T.; Kojima, K.; Ajioka, Y. Carcinogenesis of gastric endocrine cell carcinoma: Analysis of histopathology and p53 gene alteration. Gastric Cancer 2003, 6, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Borch, K.M.; Ahren, B.M.; Ahlman, H.M.; Falkmer, S.M.; Granerus, G.M.; Grimelius, L.M. Gastric Carcinoids: Biologic Behavior and Prognosis After Differentiated Treatment in Relation to Type. Ann. Surg. 2005, 242, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Luinetti, O.; Cornaggia, M.; Capella, C.; Solcia, E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: A clinicopathologic study. Gastroenterology 1993, 104, 994–1006. [Google Scholar] [PubMed]
- Rindi, G.; Bordi, C.; Rappel, S.; la Rosa, S.; Stolte, M.; Solcia, E. Gastric Carcinoids and Neuroendocrine Carcinomas: Pathogenesis, Pathology, and Behavior. World J. Surg. 1996, 20, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.; Kidd, M.; Svejda, B.; Modlin, I. A clinical perspective on gastric neuroendocrine neoplasia. Curr. Gastroenterol. Rep. 2011, 13, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Popa, E.; Schnoll-Sussman, F.; Jesudian, A.; Nandakumar, G.; Shah, M.A. Uncommon Cancers of the Stomach. In Textbook of Uncommon Cancer; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 389–408. [Google Scholar]
- Richards, D.; Davis, D.; Yan, P.; Guha, S. Unusual case of small cell gastric carcinoma: Case report and literature review. Dig. Dis. Sci. 2011, 56, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Scherübl, H.; Cadiot, G.; Jensen, R.; Rösch, T.; Stölzel, U.; Klöppel, G. Neuroendocrine tumors of the stomach (gastric carcinoids) are on the rise: Small tumors, small problems? Endoscopy 2010, 42, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Klöppel, G.; Alhman, H.; Caplin, M.; Couvelard, A.; de Herder, W.W.; Erikssson, B.; Falchetti, A.; Falconi, M.; Komminoth, P.; et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch. 2006, 449, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.; Brock, G.; Scoggins, C.; McMasters, K.; Martin, R. A Proposed Staging System for Gastric Carcinoid Tumors Based on an Analysis of 1543 Patients. Ann. Surg. Oncol. 2009, 16, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Konishi, K.; Kaji, M.; Maeda, K.; Yabushita, K.; Tsuji, M.; Miwa, A. Highly aggressive behavior and poor prognosis of small cell carcinoma in the stomach: Flow cytometric and immunohistochemical analysis. Oncol. Rep. 1999, 6, 767–772. [Google Scholar] [PubMed]
- Okita, N.T.; Kato, K.; Takahari, D.; Hirashima, Y.; Nakajima, T.E.; Matsubara, J.; Hamaguchi, T.; Yamada, Y.; Shimada, Y.; Taniguchi, H.; et al. Neuroendocrine tumors of the stomach: Chemotherapy with cisplatin plus irinotecan is effective for gastric poorly-differentiated neuroendocrine carcinoma. Gastric Cancer 2011, 14, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.V.; Wang, T. Undifferentiated small cell carcinoma of the pancreas: A report of five cases. Cancer 1981, 47, 2500–2502. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Narang, A.K.; Mansfield, A.S.; Herman, J.M.; Cameron, J.L.; Laheru, D.; Eckhauser, F.E.; Olson, M.T.; Hruban, R.H.; Miller, R.C.; et al. Resectable pancreatic small cell carcinoma. Rare Tumors 2011, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Arvold, N.D.; Willett, C.G.; Fernandez-del, C.C.; Ryan, D.P.; Ferrone, C.R.; Clark, J.W.; Blaszkowsky, L.S.; Deshpande, V.; Niemierko, A.; Allen, J.N.; et al. Pancreatic neuroendocrine tumors with involved surgical margins: Prognostic factors and the role of adjuvant radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e337–e343. [Google Scholar] [CrossRef] [PubMed]
- Bertani, E.; Fazio, N.; Botteri, E.; Chiappa, A.; Falconi, M.; Grana, C.; Bodei, L.; Papis, D.; Spada, F.; Bazolli, B.; et al. Resection of the primary pancreatic neuroendocrine tumor in patients with unresectable liver metastases: Possible indications for a multimodal approach. Surgery 2014, 155, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.Y.; Talamonti, M.S.; Tomlinson, J.S.; Stewart, A.K.; Winchester, D.P.; Ko, C.Y.; Bentrem, D.J. Prognostic score predicting survival after resection of pancreatic neuroendocrine tumors: Analysis of 3851 patients. Ann. Surg. 2008, 247, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Casadei, R.; Ricci, C.; Pezzilli, R.; Campana, D.; Tomassetti, P.; Calculli, L.; Santini, D.; Antonacci, N.; Minni, F. Value of Both WHO and TNM Classification Systems for Patients with Pancreatic Endocrine Tumors: Results of a Single-Center Series. World J. Surg. 2009, 33, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Cherenfant, J.; Stocker, S.J.; Gage, M.K.; Du, H.; Thurow, T.A.; Odeleye, M.; Schimpke, S.W.; Kaul, K.L.; Hall, C.R.; Lamzabi, I.; et al. Predicting aggressive behavior in nonfunctioning pancreatic neuroendocrine tumors. Surgery 2013, 154, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Partelli, S.; Zamboni, G.; Scarpa, A.; Tamburrino, D.; Bassi, C.; Pederzoli, P.; Falconi, M. Incidental diagnosis as prognostic factor in different tumor stages of nonfunctioning pancreatic endocrine tumors. Surgery 2014, 155, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ekeblad, S.; Skogseid, B.; Dunder, K.; Öberg, K.; Eriksson, B. Prognostic Factors and Survival in 324 Patients with Pancreatic Endocrine Tumor Treated at a Single Institution. Clin. Cancer Res. 2008, 14, 7798–7803. [Google Scholar] [CrossRef]
- Ellison, T.A.; Wolfgang, C.L.; Shi, C.; Cameron, J.L.; Murakami, P.; Mun, L.J.; Singhi, A.D.; Cornish, T.C.; Olino, K.; Meriden, Z.; et al. A single institution’s 26-year experience with nonfunctional pancreatic neuroendocrine tumors: A validation of current staging systems and a new prognostic nomogram. Ann. Surg. 2014, 259, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, F.A.F.; Giovannini, M.; Monges, G.; Bories, E.; Pesenti, C.; Caillol, F.; Delpero, J.R. EUS-FNA predicts 5-year survival in pancreatic endocrine tumors. Gastrointest. Endosc. 2009, 70, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Kleeff, J.; Esposito, I.; Hinz, U.; Zimmermann, A.; Friess, H.; Buchler, M.W. Clinical outcome and long-term survival in 118 consecutive patients with neuroendocrine tumours of the pancreas. Br. J. Surg. 2008, 95, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.A.; Liu, T.C.; Cavatiao, A.; Mawad, K.; Chen, L.; Strasberg, S.S.; Linehan, D.C.; Cao, D.; Hawkins, W.G. Ki-67 predicts disease recurrence and poor prognosis in pancreatic neuroendocrine neoplasms. Surgery 2012, 152, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kaifi, J.T.; Zinnkann, U.; Yekebas, E.F.; Schurr, P.G.; Reichelt, U.; Wachowiak, R.; Fiegel, H.C.; Petri, S.; Schachner, M.; Izbicki, J.R.; et al. L1 is a potential marker for poorly-differentiated pancreatic neuroendocrine carcinoma. World J. Gastroenterol. 2006, 12, 94–98. [Google Scholar] [PubMed]
- La Rosa, S.; Klersy, C.; Uccella, S.; Dainese, L.; Albarello, L.; Sonzogni, A.; Doglioni, C.; Capella, C.; Solcia, E. Improved histologic and clinicopathologic criteria for prognostic evaluation of pancreatic endocrine tumors. Hum. Pathol. 2009, 40, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Liszka, L.; Pajak, J.; Mrowiec, S.; Zielinska-Pajak, E.; Golka, D.; Lampe, P. Discrepancies between two alternative staging systems (European Neuroendocrine Tumor Society 2006 and American Joint Committee on Cancer/Union for International Cancer Control 2010) of neuroendocrine neoplasms of the pancreas. A study of 50 cases. Pathol. Res. Pract. 2011, 207, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tang, L.H.; Liu, Z.; Mei, M.; Yu, R.; Dhall, D.; Qiao, X.W.; Zhang, T.P.; Zhao, Y.P.; Liu, T.H.; et al. a-Internexin: A Novel Biomarker for Pancreatic Neuroendocrine Tumor Aggressiveness. J. Clin. Endocrinol. Metab. 2014, 99, E786–E795. [Google Scholar] [CrossRef] [PubMed]
- Madeira, I.; Terris, B.; Voss, M.; Denys, A.; Sauvanet, A.; Flejou, J.F.; Vilgrain, V.; Belghiti, J.; Bernades, P.; Ruszniewski, P.; et al. Prognostic factors in patients with endocrine tumours of the duodenopancreatic area. Gut 1998, 43, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Martin-Perez, E.; Capdevila, J.; Castellano, D.; Jimenez-Fonseca, P.; Salazar, R.; Beguiristain-Gomez, A.; Alonso-Orduña, V.; Martinez del Prado, P.; Villabona-Artero, C.; Diaz-Perez, J.A.; et al. Prognostic Factors and Long-Term Outcome of Pancreatic Neuroendocrine Neoplasms: Ki-67 Index Shows a Greater Impact on Survival than Disease Stage. The Large Experience of the Spanish National Tumor Registry (RGETNE). Neuroendocrinology 2013, 98, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.C.; Kooby, D.A.; Weber, S.M.; Merchant, N.B.; Parikh, A.A.; Cho, C.S.; Ahmad, S.A.; Kim, H.J.; Hawkins, W.; Scoggins, C.R.; et al. Analysis of 6,747 pancreatic neuroendocrine tumors for a proposed staging system. J. Gastrointest. Surg. 2011, 15, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Pape, U.F.; Jann, H.; Muller-Nordhorn, J.; Bockelbrink, A.; Berndt, U.; Willich, S.N.; Koch, M.; Rocken, C.; Rindi, G.; Wiedenmann, B.; et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer 2008, 113, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Sellner, F.; Thalhammer, S.; Stattner, S.; Karner, J.; Klimpfinger, M. TNM stage and grade in predicting the prognosis of operated, non-functioning neuroendocrine carcinoma of the pancreas—A single-institution experience. J. Surg. Oncol. 2011, 104, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Cheema, A.; Weber, J.; Han, G.; Coppola, D.; Kvols, L.K. Prognostic validity of a novel American Joint Committee on Cancer Staging Classification for pancreatic neuroendocrine tumors. J. Clin. Oncol. 2011, 29, 3044–3049. [Google Scholar] [CrossRef] [PubMed]
- Toste, P.; Kadera, B.; Tatishchev, S.; Dawson, D.; Clerkin, B.; Muthusamy, R.; Watson, R.; Tomlinson, J.; Hines, O.; Reber, H.; et al. Nonfunctional Pancreatic Neuroendocrine Tumors <2 cm on Preoperative Imaging are Associated with a Low Incidence of Nodal Metastasis and an Excellent Overall Survival. J. Gastrointest. Surg. 2013; 17, 2105–2113. [Google Scholar]
- You, D.D.; Lee, H.G.; Paik, K.Y.; Heo, J.S.; Choi, S.H.; Choi, D.W. The outcomes after surgical resection in pancreatic endocrine tumors: An institutional experience. Eur. J. Surg. Oncol. 2009, 35, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Sellner, F.; Sobhian, B.; de Santis, M.; Pont, J.; Staettner, S.; Sellner, S.; Karner, J.; Klimpfinger, M. Well or poorly differentiated nonfunctioning neuroendocrine carcinoma of the pancreas: A single institution experience with 17 cases. Eur. J. Surg. Oncol. 2008, 34, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, G.; Bresaola, E.; Bogina, G.; Pasini, F.; Rodella, S.; Castelli, P.; Iacono, C.; Serio, G.; Zamboni, G. Endocrine tumors of the pancreas: Ki-67 immunoreactivity on paraffin sections is an independent predictor for malignancy: A comparative study with proliferating-cell nuclear antigen and progesterone receptor protein immunostaining, mitotic index, and other clinicopathologic variables. Hum. Pathol. 1996, 27, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Zerbi, A.; Falconi, M.; Rindi, G.; Fave, G.D.; Tomassetti, P.; Pasquali, C.; Capitanio, V.; Boninsegna, L.; di Carlo, V. Clinicopathological Features of Pancreatic Endocrine Tumors: A Prospective Multicenter Study in Italy of 297 Sporadic Cases. Am. J. Gastroenterol. 2010, 105, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Hashim, Y.M.; Trinkaus, K.M.; Linehan, D.C.; Strasberg, S.S.; Fields, R.C.; Cao, D.; Hawkins, W.G. Regional lymphadenectomy is indicated in the surgical treatment of pancreatic neuroendocrine tumors (PNETs). Ann. Surg. 2014, 259, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.; Tomlinson, J.; Merkow, R.; Stewart, A.; Ko, C.; Talamonti, M.; Bentrem, D. Clinicopathologic Features and Treatment Trends of Pancreatic Neuroendocrine Tumors: Analysis of 9821 Patients. J. Gastrointest. Surg. 2007, 11, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Bartsch, D.K.; Eriksson, B.; Kloppel, G.; Lopes, J.M.; O’Connor, J.M.; Salazar, R.; Taal, B.G.; Vullierme, M.P.; O’Toole, D.; et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: Well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012, 95, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Couvelard, A.; O’Toole, D.; Turley, H.; Leek, R.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.L.; Gatter, K.; et al. Microvascular density and hypoxia-inducible factor pathway in pancreatic endocrine tumours: Negative correlation of microvascular density and VEGF expression with tumour progression. Br. J. Cancer 2005, 92, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Sobin, L.H.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours, 7th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009. [Google Scholar]
- Scarpa, A.; Mantovani, W.; Capelli, P.; Beghelli, S.; Boninsegna, L.; Bettini, R.; Panzuto, F.; Pederzoli, P.; Fave, G.D.; Falconi, M.; et al. Pancreatic endocrine tumors: Improved TNM staging and histopathological grading permit a clinically efficient prognostic stratification of patients. Mod. Pathol. 2010, 23, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Falconi, M.; Klersy, C.; Albarello, L.; Boninsegna, L.; Buchler, M.W.; Capella, C.; Caplin, M.; Couvelard, A.; Doglioni, C.; et al. TNM staging of neoplasms of the endocrine pancreas: Results from a large international cohort study. J. Natl. Cancer Inst. 2012, 104, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Panzuto, F.; Boninsegna, L.; Fazio, N.; Campana, D.; Pia, B.M.; Capurso, G.; Scarpa, A.; de Braud, F.; Dogliotti, L.; Tomassetti, P.; et al. Metastatic and locally advanced pancreatic endocrine carcinomas: Analysis of factors associated with disease progression. J. Clin. Oncol. 2011, 29, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Hamilton, N.; Hawkins, W.; Gao, F.; Cao, D. Comparison of WHO Classifications (2004, 2010), the Hochwald grading system, and AJCC and ENETS staging systems in predicting prognosis in locoregional well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2013, 37, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Amato, E.; Barbi, S.; Malpeli, G.; Bersani, S.; Pelosi, G.; Capelli, P.; Scarpa, A. Chromosome 3p alterations in pancreatic endocrine neoplasia. Virchows Arch. 2011, 458, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Jensen, R.T. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): Recent insights and advances. J. Gastroenterol. 2012, 47, 941–960. [Google Scholar] [CrossRef] [PubMed]
- Bettini, R.; Boninsegna, L.; Mantovani, W.; Capelli, P.; Bassi, C.; Pederzoli, P.; Delle Fave, G.F.; Panzuto, F.; Scarpa, A.; Falconi, M.; et al. Prognostic factors at diagnosis and value of WHO classification in a mono-institutional series of 180 non-functioning pancreatic endocrine tumours. Ann. Oncol. 2008, 19, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Franko, J.; Feng, W.; Yip, L.; Genovese, E.; Moser, A.J. Non-functional Neuroendocrine Carcinoma of the Pancreas: Incidence, Tumor Biology, and Outcomes in 2,158 Patients. J. Gastrointest. Surg. 2010, 14, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bettini, R.; Mantovani, W.; Boninsegna, L.; Crippa, S.; Capelli, P.; Bassi, C.; Scarpa, A.; Pederzoli, P.; Falconi, M. Primary tumour resection in metastatic nonfunctioning pancreatic endocrine carcinomas. Dig. Liver Dis. 2009, 41, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Rong, Y.; Wu, W.; Jin, D. Primary small cell carcinoma of the pancreas: Rare type of pancreatic cancer and review of the literatures. World J. Surg. Oncol. 2012, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Morizane, C.; Okusaka, T.; Ueno, H.; Ikeda, M.; Kondo, S.; Tanaka, T.; Nakachi, K.; Mitsunaga, S.; Kojima, Y.; et al. Cisplatin and etoposide as first-line chemotherapy for poorly differentiated neuroendocrine carcinoma of the hepatobiliary tract and pancreas. Jpn. J. Clin. Oncol. 2010, 40, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Weber, J.M.; Feldman, M.; Coppola, D.; Meredith, K.; Kvols, L.K. Prognostic validity of the American Joint Committee on Cancer staging classification for midgut neuroendocrine tumors. J. Clin. Oncol. 2013, 31, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Albores-Saavedra, J.; Soriano, J.; Larraza-Hernandez, O.; Aguirre, J.; Henson, D.E. Oat cell carcinoma of the gallbladder. Hum. Pathol. 1984, 15, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Albores-Saavedra, J.; Batich, K.; Hossain, S.; Henson, D.E.; Schwartz, A.M. Carcinoid tumors and small-cell carcinomas of the gallbladder and extrahepatic bile ducts: A comparative study based on 221 cases from the Surveillance, Epidemiology, and End Results Program. Ann. Diagn. Pathol. 2009, 13, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.M.D.; Tascilar, M.M.D.; Hruban, R.H.M.; Offerhaus, G.J.; Albores-Saavedra, J.M.D. Small Cell Carcinoma of the Gallbladder: A Clinicopathologic, Immunohistochemical, and Molecular Pathology Study of 12 Cases. Am. J. Surg. Pathol. 2001, 25, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Moskal, T.L.; Zhang, P.J.; Nava, H.R. Small cell carcinoma of the gallbladder. J. Surg. Oncol. 1999, 70, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, K.; Tsuneyoshi, M. Small-cell carcinoma of the gallbladder—A clinicopathological, immunohistochemical and flow cytometric study of 15 cases. Int. J. Oncol. 1993, 3, 901–908. [Google Scholar] [PubMed]
- Albores-Saavedra, J.; Cruz-Ortiz, H.; Alcantara-Vazques, A.; Henson, D.E. Unusual types of gallbladder carcinoma. A report of 16 cases. Arch. Pathol. Lab. Med. 1981, 105, 287–293. [Google Scholar] [PubMed]
- Eltawil, K.M.M.; Gustafsson, B.I.M.; Kidd, M.P.; Modlin, I.M.M. Neuroendocrine Tumors of the Gallbladder: An Evaluation and Reassessment of Management Strategy. J. Clin. Gastroenterol. 2010, 44, 687–695. [Google Scholar] [PubMed]
- Nassar, H.; Albores-Saavedra, J.; Klimstra, D.S. High-grade neuroendocrine carcinoma of the ampulla of vater: A clinicopathologic and immunohistochemical analysis of 14 cases. Am. J. Surg. Pathol. 2005, 29, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascu, T.; Dima, S.; Herlea, V.; Tomulescu, V.; Ionescu, M.; Popescu, I. Neuroendocrine tumours of the ampulla of Vater: Clinico-pathological features, surgical approach and assessment of prognosis. Langenbecks Arch. Surg. 2012, 397, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Randle, R.; Ahmed, S.; Newman, N.; Clark, C. Clinical Outcomes for Neuroendocrine Tumors of the Duodenum and Ampulla of Vater:A Population-Based Study. J. Gastrointest. Surg. 2014, 18, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Albores-Saavedra, J.; Hart, A.; Chablé-Montero, F.; Henson, D.E. Carcinoids and High-Grade Neuroendocrine Carcinomas of the Ampulla of Vater: A Comparative Analysis of 139 Cases from the Surveillance, Epidemiology, and End Results Program—A Population Based Study. Arch. Pathol. Lab. Med. 2010, 134, 1692–1696. [Google Scholar] [PubMed]
- Rindi, G.; Klöppel, G.; Couvelard, A.; Komminoth, P.; Körner, M.; Lopes, J.M.; McNicol, A.M.; Nilsson, O.; Perren, A.; Scarpa, A.; et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: A consensus proposal including a grading system. Virchows Arch. 2007, 451, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.S.; Brock, G.; Scoggins, C.R.; McMasters, K.M.; Martin, R.C. A proposed staging system for small bowel carcinoid tumors based on an analysis of 6380 patients. Am. J. Surg. 2008, 196, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.S.; Woodall, C.; Scoggins, C.R.; McMasters, K.M.; Martin, R.G., II. Analysis of 900 appendiceal carcinoid tumors for a proposed predictive staging system. Arch. Surg. 2008, 143, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Aytac, E.; Ozdemir, Y.; Ozuner, G. Long term outcomes of neuroendocrine carcinomas (high-grade neuroendocrine tumors) of the colon, rectum, and anal canal. J. Visc. Surg. 2014, 151, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Staren, E.D.; Gould, V.E.; Warren, W.H.; Wool, N.L.; Bines, S.; Baker, J.; Bonomi, P.; Roseman, D.L.; Economou, S.G. Neuroendocrine carcinomas of the colon and rectum: A clinicopathologic evaluation. Surgery 1988, 104, 1080–1089. [Google Scholar] [PubMed]
- Bernick, P.E.; Klimstra, D.S.; Shia, J.; Minsky, B.; Saltz, L.; Shi, W.; Thaler, H.; Guillem, J.; Paty, P.; Cohen, A.M.; et al. Neuroendocrine carcinomas of the colon and rectum. Dis. Colon Rectum 2004, 47, 163–169. [Google Scholar] [CrossRef]
- Akintola-Ogunremi, O.; Pfeifer, J.D.; Tan, B.R.; Yan, Y.; Zhu, X.; Hart, J.; Goldblum, J.R.; Burgart, L.; Lauwers, G.Y.; Montgomery, E.; et al. Analysis of protein expression and gene mutation of c-kit in colorectal neuroendocrine carcinomas. Am. J. Surg. Pathol. 2003, 27, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.N.; Nagasaka, T.; Goel, A.; Scharf, I.; Grabowski, P.; Sosnowski, A.; Schmitt-Graff, A.; Boland, C.R.; Arnold, R.; Blum, H.E.; et al. Molecular characteristics and predictors of survival in patients with malignant neuroendocrine tumors. Int. J. Cancer 2008, 123, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.B.; Shekitka, K.M.; Sobin, L.H. Small cell carcinomas of the large intestine. Am. J. Clin. Pathol. 1991, 95, 315–321. [Google Scholar] [PubMed]
- Chagpar, R.; Chiang, Y.J.; Xing, Y.; Cormier, J.N.; Feig, B.W.; Rashid, A.; Chang, G.J.; You, Y.N. Neuroendocrine tumors of the colon and rectum: Prognostic relevance and comparative performance of current staging systems. Ann. Surg. Oncol. 2013, 20, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Gaffey, M.J.; Mills, S.E.; Lack, E.E. Neuroendocrine carcinoma of the colon and rectum. A clinicopathologic, ultrastructural, and immunohistochemical study of 24 cases. Am. J. Surg. Pathol. 1990, 14, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- La, R.S.; Marando, A.; Furlan, D.; Sahnane, N.; Capella, C. Colorectal poorly differentiated neuroendocrine carcinomas and mixed adenoneuroendocrine carcinomas: Insights into the diagnostic immunophenotype, assessment of methylation profile, and search for prognostic markers. Am. J. Surg. Pathol. 2012, 36, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Patta, A.; Fakih, M. First-line cisplatin plus etoposide in high-grade metastatic neuroendocrine tumors of colon and rectum (MCRC NET): Review of 8 cases. Anticancer Res. 2011, 31, 975–978. [Google Scholar] [PubMed]
- Smith, J.; Reidy, D.; Goodman, K.; Shia, J.; Nash, G. A Retrospective Review of 126 High-Grade Neuroendocrine Carcinomas of the Colon and Rectum. Ann. Surg. Oncol. 2014, 21, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Spread, C.; Berkel, H.; Jewell, L.; Jenkins, H.; Yakimets, W. Colon carcinoid tumors. Dis. Colon Rectum 1994, 37, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Vortmeyer, A.O.; Lubensky, I.A.; Merino, M.J.; Wang, C.Y.; Pham, T.; Furth, E.E.; Zhuang, Z. Concordance of genetic alterations in poorly differentiated colorectal neuroendocrine carcinomas and associated adenocarcinomas. J. Natl. Cancer Inst. 1997, 89, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Wick, M.R.; Weatherby, R.P.; Weiland, L.H. Small cell neuroendocrine carcinoma of the colon and rectum: Clinical, histologic, and ultrastructural study and immunohistochemical comparison with cloacogenic carcinoma. Hum. Pathol. 1987, 18, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.E.; Allen, M.S., Jr.; Cohen, A.R. Small-cell undifferentiated carcinoma of the colon. A clinicopathological study of five cases and their association with colonic adenomas. Am. J. Surg. Pathol. 1983, 7, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.; Sundin, A.; Nillson, O.; Baum, R.P.; Klose, K.J.; Kelestimur, F.; Plockinger, U.; Papotti, M.; Salazar, R.; Pascher, A.; et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: Colorectal neuroendocrine neoplasms. Neuroendocrinology 2012, 95, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Mandair, D.; Caplin, M.E. Colonic and rectal NET’s. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.S.; Brock, G.; Scoggins, C.R.; McMasters, K.M.; Martin, R.C.G., II. Proposed Staging System for Colon Carcinoid Tumors Based on an Analysis of 2459 Patients. J. Am. Coll. Surg. 2008, 207, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.S.; Brock, G.; Scoggins, C.R.; McMasters, K.M.; Martin, R.C.G., II. A proposed staging system for rectal carcinoid tumors based on an analysis of 4701 patients. Surgery 2008, 144, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Stelow, E.B.M.; Moskaluk, C.A.M.; Mills, S.E.M. The Mismatch Repair Protein Status of Colorectal Small Cell Neuroendocrine Carcinomas. Am. J. Surg. Pathol. 2006, 30, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Furlan, D.; Sahnane, N.; Mazzoni, M.; Pastorino, R.; Carnevali, I.; Stefanoli, M.; Ferretti, A.; Chiaravalli, A.M.; La, R.S.; Capella, C.; et al. Diagnostic utility of MS-MLPA in DNA methylation profiling of adenocarcinomas and neuroendocrine carcinomas of the colon-rectum. Virchows Arch. 2013, 462, 47–56. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilett, E.E.; Langer, S.W.; Olsen, I.H.; Federspiel, B.; Kjær, A.; Knigge, U. Neuroendocrine Carcinomas of the Gastroenteropancreatic System: A Comprehensive Review. Diagnostics 2015, 5, 119-176. https://doi.org/10.3390/diagnostics5020119
Ilett EE, Langer SW, Olsen IH, Federspiel B, Kjær A, Knigge U. Neuroendocrine Carcinomas of the Gastroenteropancreatic System: A Comprehensive Review. Diagnostics. 2015; 5(2):119-176. https://doi.org/10.3390/diagnostics5020119
Chicago/Turabian StyleIlett, Emma Elizabeth, Seppo W. Langer, Ingrid Holst Olsen, Birgitte Federspiel, Andreas Kjær, and Ulrich Knigge. 2015. "Neuroendocrine Carcinomas of the Gastroenteropancreatic System: A Comprehensive Review" Diagnostics 5, no. 2: 119-176. https://doi.org/10.3390/diagnostics5020119