Ultrasensitive ELISA Developed for Diagnosis


 ,
, 
Abstract
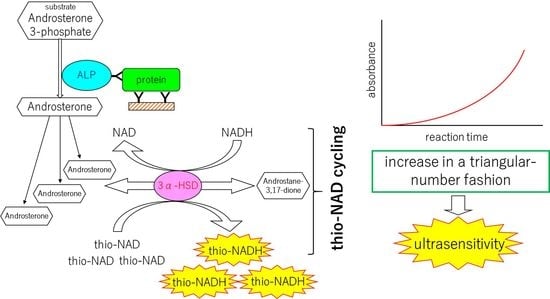
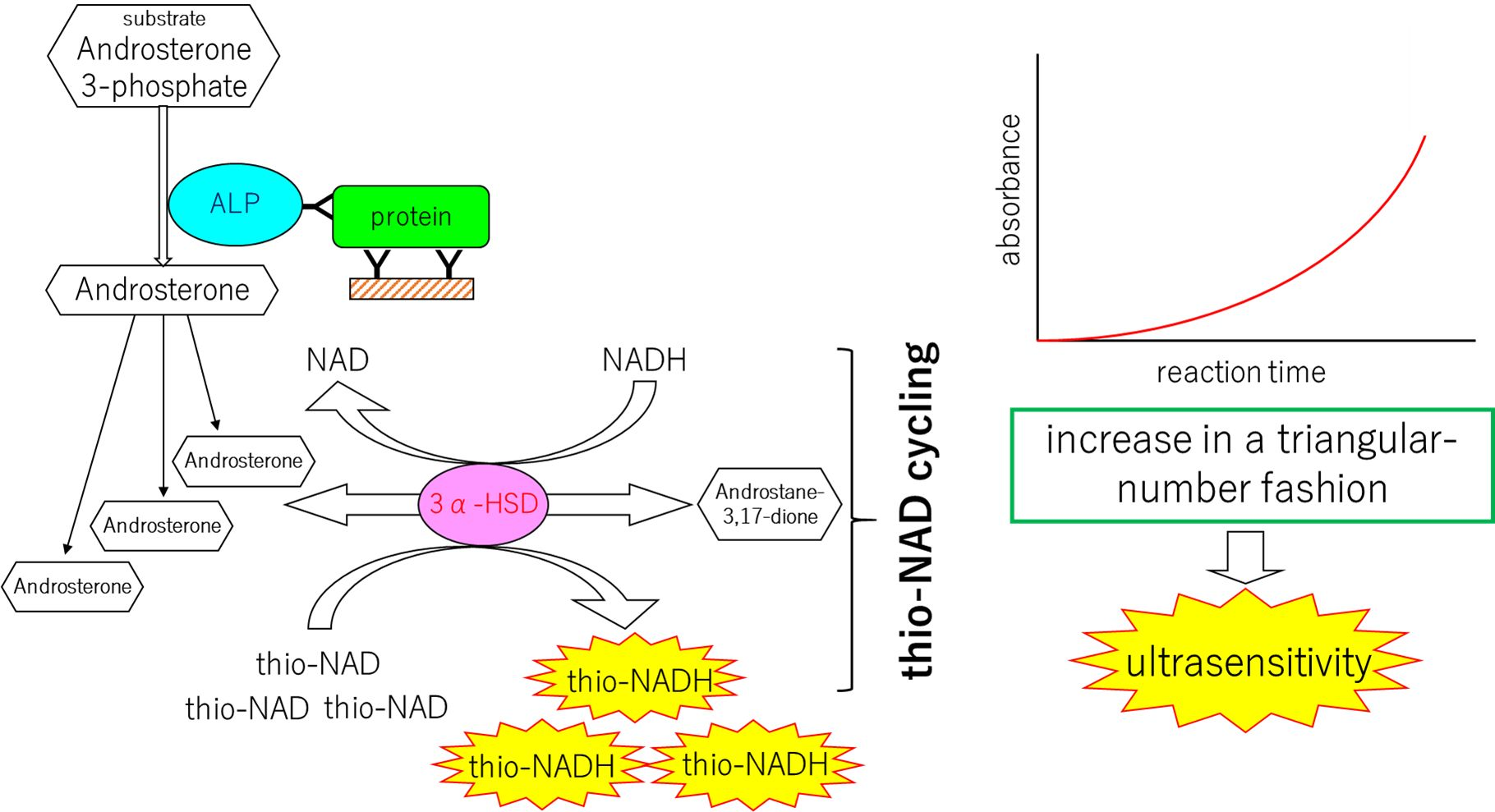{kind=link}
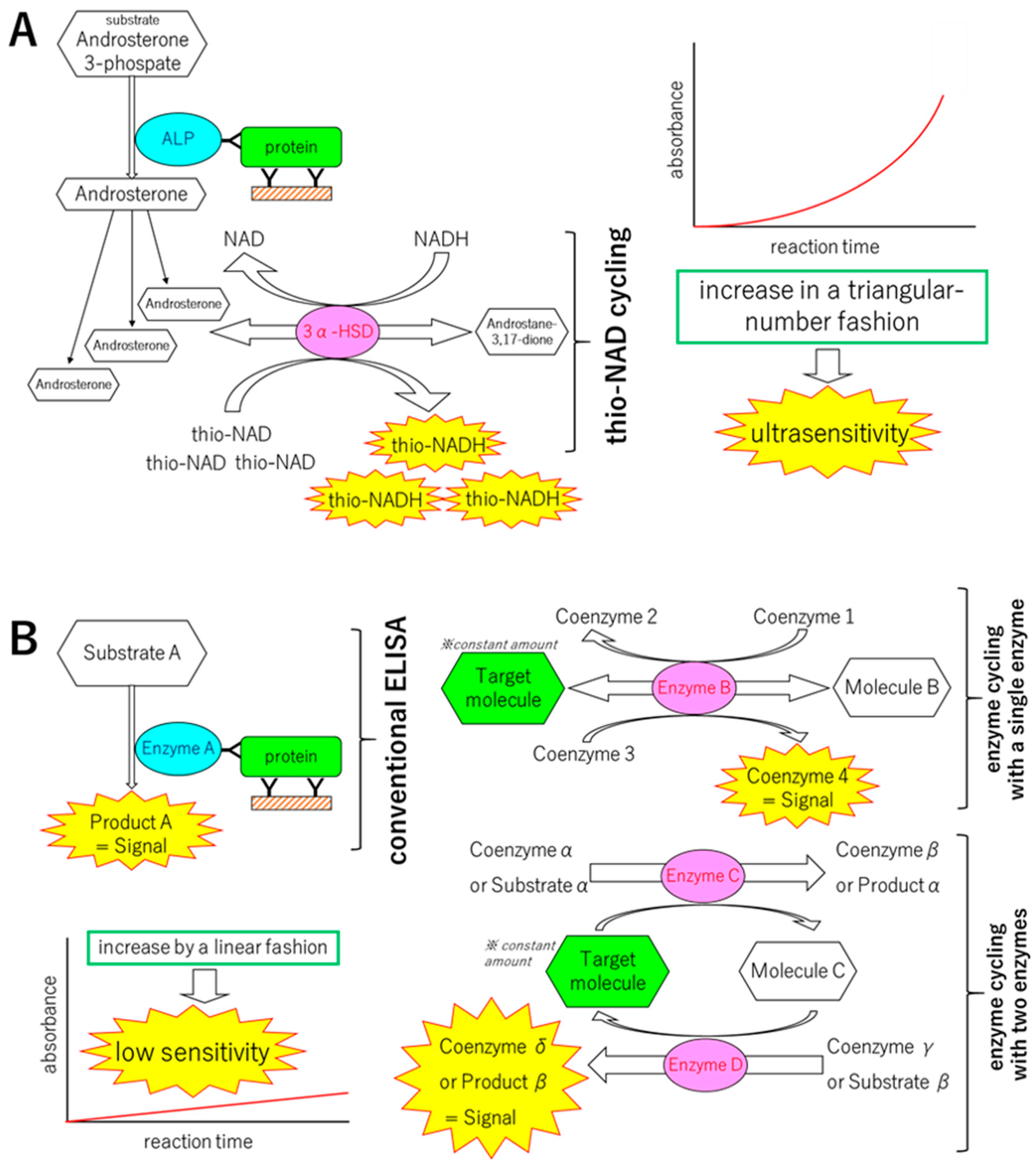{kind=link}
1. Introduction
2. Mechanisms of the Ultrasensitive ELISA
2.1. Principle of Protocol
2.2. Experimental Protocol
3. Detection of Trace Amount of Proteins
3.1. Detection of Proteins for an Infectious Disease
3.2. Detection of Proteins for a Lifestyle-Related Disease
4. Challenges: Expansion of the Ultrasensitive ELISA to a Non-Amplification Nucleic Acid Detection Method Using Hybridization and Thio-NAD Cycling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DM | diabetes mellitus |
| ELISA | enzyme-linked immunosorbent assay |
| FITC | fluorescein isothiocyanate |
| HIV | human immunodeficiency virus |
| LOD | limit of detection |
| LOQ | limit of quantification |
| NAD | nicotinamide adenine dinucleotide |
| NAT | nucleic acid test |
| PCR | polymerase chain reaction |
| WHO | World Health Organization |
References
- Chikamatsu, K.; Aono, A.; Yamada, H.; Sugamoto, T.; Kato, T.; Kazumi, Y.; Tamai, K.; Yanagisawa, H.; Mitarai, S. Comparative evaluation of three immunochromatographic identification tests for culture confirmation of Mycobacterium tuberculosis complex. BMC Infect. Dis. 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakaishi, K.; Watabe, S.; Kitagawa, T.; Ito, E. Immunochromatographic detection of MPB64 secreted from active BCG by heating: Toward same-day diagnosis of tuberculosis. BioTechniques 2019, 66, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Scherl, A. Clinical protein mass spectrometry. Methods 2015, 81, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Berger, S.J.; Carter, J.A.; Lowry, O.H. An enzymatic cycling method for nicotinamide-adenine dinucleotide with malic and alcohol dehydrogenases. Anal. Biochem. 1973, 53, 86–97. [Google Scholar] [CrossRef]
- Lowry, O.H. Amplification by enzymatic cycling. Mol. Cell. Biochem. 1980, 32, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Watabe, S.; Kodama, H.; Kaneda, M.; Morikawa, M.; Nakaishi, K.; Yoshimura, T.; Iwai, A.; Miura, T.; Ito, E. Ultrasensitive enzyme-linked immunosorbent assay (ELISA) of proteins by combination with the thio-NAD cycling method. Biophysics 2014, 10, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Watabe, S.; Morikawa, M.; Kaneda, M.; Nakaishi, K.; Nakatsuma, A.; Ninomiya, M.; Yoshimura, T.; Miura, T.; Ito, E. Ultrasensitive detection of proteins and sugars at single-cell level. Commun. Integr. Biol. 2016, 9, e1124201. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuma, A.; Kaneda, M.; Kodama, H.; Morikawa, M.; Watabe, S.; Nakaishi, K.; Yamashita, M.; Yoshimura, T.; Miura, T.; Ninomiya, M.; et al. Detection of HIV-1 p24 at attomole level by ultrasensitive ELISA with thio-NAD cycling. PLoS ONE 2015, 10, e0131319. [Google Scholar] [CrossRef][Green Version]
- Ito, E.; Kaneda, M.; Kodama, H.; Morikawa, M.; Tai, M.; Aoki, K.; Watabe, S.; Nakaishi, K.; Hashida, S.; Tada, S.; et al. Immunoreactive insulin in diabetes mellitus patient sera detected by ultrasensitive ELISA with thio-NAD cycling. BioTechniques 2015, 59, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Naito, R.; Mita, K.; Watabe, S.; Nakaishi, K.; Yoshimura, T.; Miura, T.; Hashida, S.; Ito, E. Subattomole detection of adiponectin in urine by ultrasensitive ELISA coupled with thio-NAD cycling. Biophys. Physicobiol. 2015, 12, 79–86. [Google Scholar] [CrossRef][Green Version]
- Yamakado, S.; Cho, H.; Inada, M.; Morikawa, M.; Jiang, Y.-H.; Saito, K.; Nakaishi, K.; Watabe, S.; Takagi, H.; Kaneda, M.; et al. Urinary adiponectin as a new diagnostic index for chronic kidney disease due to diabetic nephropathy. BMJ Open Diabetes Res. Care 2019, 7, e000661. [Google Scholar] [CrossRef] [PubMed]
- Alexander, T.S. Human immunodeficiency virus diagnostic testing: 30 years of evolution. Clin. Vaccine Immunol. 2016, 23, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Miedouge, M.; Grèze, M.; Bailly, A.; Izopet, J. Analytical sensitivity of four HIV combined antigen/antibody assays using the p24 WHO standard. J. Clin. Virol. 2011, 50, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.D.; Plantier, J.C.; Leballais, L.; Gonzalo, S.; Lemée, V.; Laperche, S. The variable sensitivity of HIV Ag/Ab combination assays in the detection of p24Ag according to genotype could compromise the diagnosis of early HIV infection. J. Clin. Virol. 2012, 55, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Sokoll, L.; Yip, P.; Elliott, D.J.; Dua, R.; Mohr, P.; Wang, X.Y.; Spencer, M.; Swanson, P.; Dawson, G.J.; et al. Comparative evaluation of three FDA-approved HIV Ag/Ab combination tests using a genetically diverse HIV panel and diagnostic specimens. J. Clin. Virol. 2017, 92, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.M.; Edelman, D.C.; Constantine, N.T. Lowering the detection limits of HIV-1 viral load using real-time immuno-PCR for HIV-1 p24 antigen. Am. J. Clin. Pathol. 2004, 122, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Wagatsuma, A.; Sadamoto, H.; Kitahashi, T.; Lukowiak, K.; Urano, A.; Ito, E. Determination of the exact copy numbers of particular mRNAs in a single cell by quantitative real-time RT-PCR. J. Exp. Biol. 2005, 208, 2389–2398. [Google Scholar] [CrossRef][Green Version]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. Molecular mechanism of metabolic syndrome X: Contribution of adipocytokines adipocyte-derived bioactive substances. Ann. N. Y. Acad. Sci. 1999, 892, 146–154. [Google Scholar] [CrossRef]
- Nishida, M.; Funahashi, T.; Shimomura, I. Pathophysiological significance of adiponectin. Med. Mol. Morphol. 2007, 40, 55–67. [Google Scholar] [CrossRef]
- Koshimura, J.; Fujita, H.; Narita, T.; Shimotomai, T.; Hosoba, M.; Yoshioka, N.; Kakei, M.; Fujishima, S.; Ito, S. Urinary adiponectin excretion is increased in patients with overt diabetic nephropathy. Biochem. Biophys. Res. Commun. 2004, 316, 165–169. [Google Scholar] [CrossRef]
- Panduru, N.M.; Saraheimo, M.; Forsblom, C.; Thorn, L.M.; Gordin, D.; Wadén, J.; Tolonen, N.; Bierhaus, A.; Humpert, P.M.; Per-Henrik Groop on behalf of the FinnDiane Study Group. Urinary adiponectin is an independent predictor of progression to end-stage renal disease in patients with type 1 diabetes and diabetic nephropathy. Diabetes Care 2015, 38, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Song, H.; Hebert, L.A.; Nadasdy, T.; Nadasdy, G.; Birmingham, D.J.; Yu, C.Y.; Nagaraja, H.N. Plasma, urine, and renal expression of adiponectin in human systemic lupus erythematosus. Kidney Int. 2005, 68, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- WHO Reference Regent Insulin. Available online: http://www.nibsc.org/documents/ifu/66-304.pdf (accessed on 18 July 2019).
- Miller, W.G.; Thienpont, L.M.; Van Uytfanghe, K.; Clark, P.M.; Lindstedt, P.; Nilsson, G.; Steffes, M.W.; Insulin Standardization Work Group. Toward standardization of insulin immunoassays. Clin. Chem. 2009, 55, 1011–1018. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iha, K.; Inada, M.; Kawada, N.; Nakaishi, K.; Watabe, S.; Tan, Y.H.; Shen, C.; Ke, L.-Y.; Yoshimura, T.; Ito, E. Ultrasensitive ELISA Developed for Diagnosis. Diagnostics 2019, 9, 78. https://doi.org/10.3390/diagnostics9030078
Iha K, Inada M, Kawada N, Nakaishi K, Watabe S, Tan YH, Shen C, Ke L-Y, Yoshimura T, Ito E. Ultrasensitive ELISA Developed for Diagnosis. Diagnostics. 2019; 9(3):78. https://doi.org/10.3390/diagnostics9030078
Chicago/Turabian StyleIha, Kanako, Mikio Inada, Naoki Kawada, Kazunari Nakaishi, Satoshi Watabe, Yong Hong Tan, Chieh Shen, Liang-Yin Ke, Teruki Yoshimura, and Etsuro Ito. 2019. "Ultrasensitive ELISA Developed for Diagnosis" Diagnostics 9, no. 3: 78. https://doi.org/10.3390/diagnostics9030078
APA StyleIha, K., Inada, M., Kawada, N., Nakaishi, K., Watabe, S., Tan, Y. H., Shen, C., Ke, L.-Y., Yoshimura, T., & Ito, E. (2019). Ultrasensitive ELISA Developed for Diagnosis. Diagnostics, 9(3), 78. https://doi.org/10.3390/diagnostics9030078