
Aspartate Reduces Liver Inflammation and Fibrosis by Suppressing the NLRP3 Inflammasome Pathway via Upregulating NS3TP1 Expression

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Transfection
2.3. RNA Isolation and Quantitative Real-Time PCR (Real-Time qPCR)
2.4. Western Blotting
2.5. Cell Proliferation Assay
2.6. Cell Apoptosis Assay
2.7. Animal Studies
2.8. Hematoxylin–Eosin Staining, Masson Staining, Sirius Red Staining, and Oil Red O Staining
2.9. Immunohistochemistry
2.10. RNA-Seq and Analysis
2.11. Statistical Analysis
3. Results
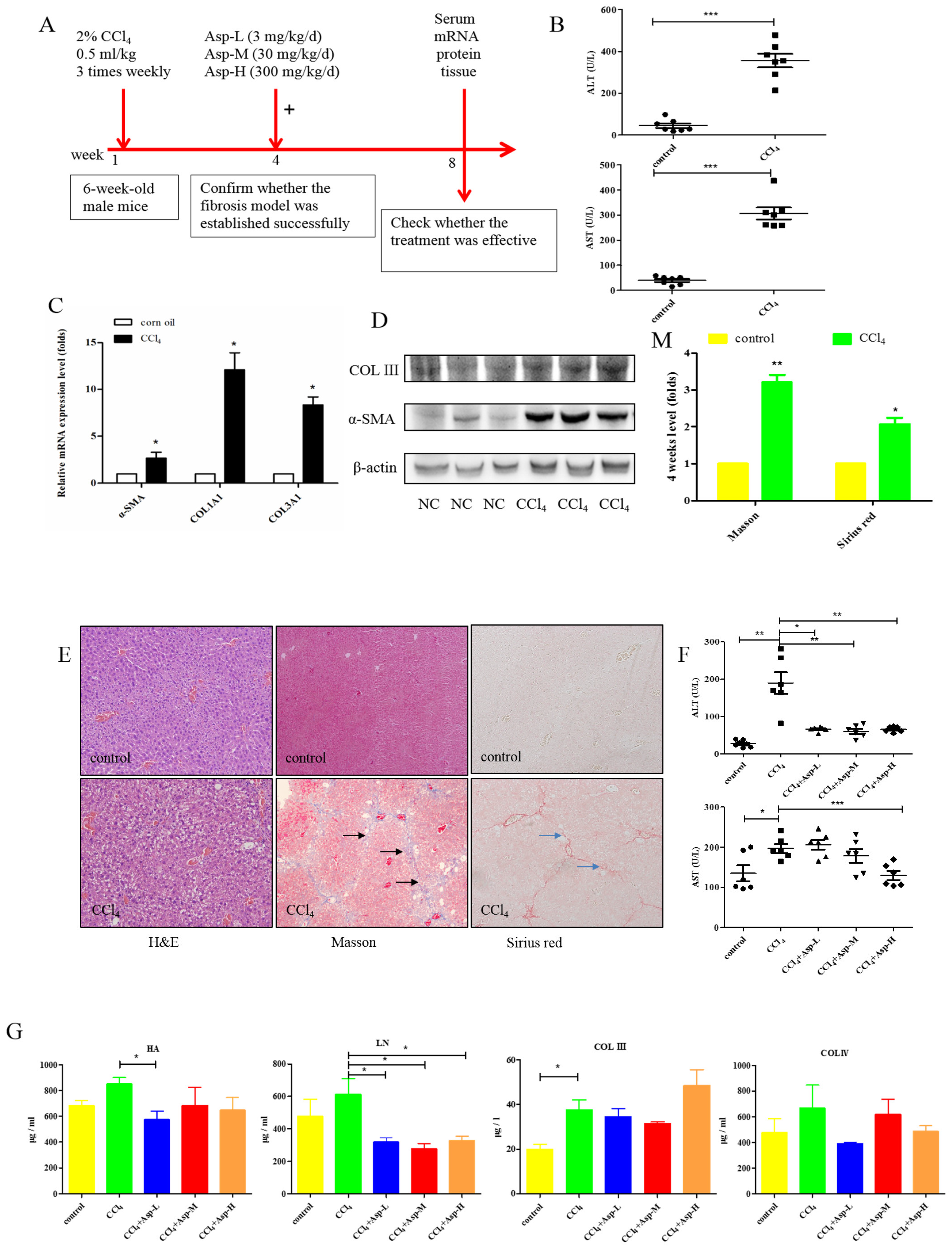
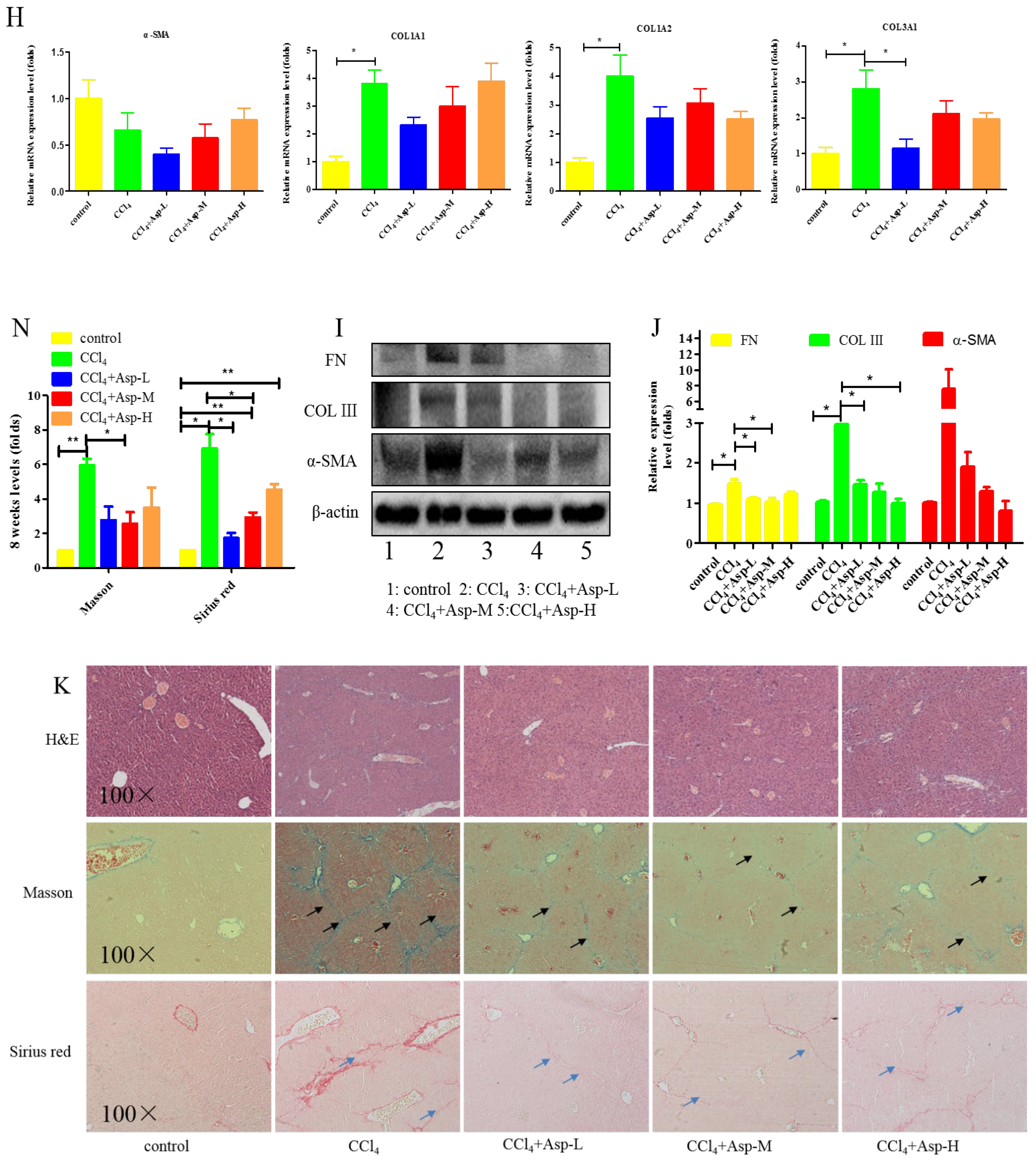
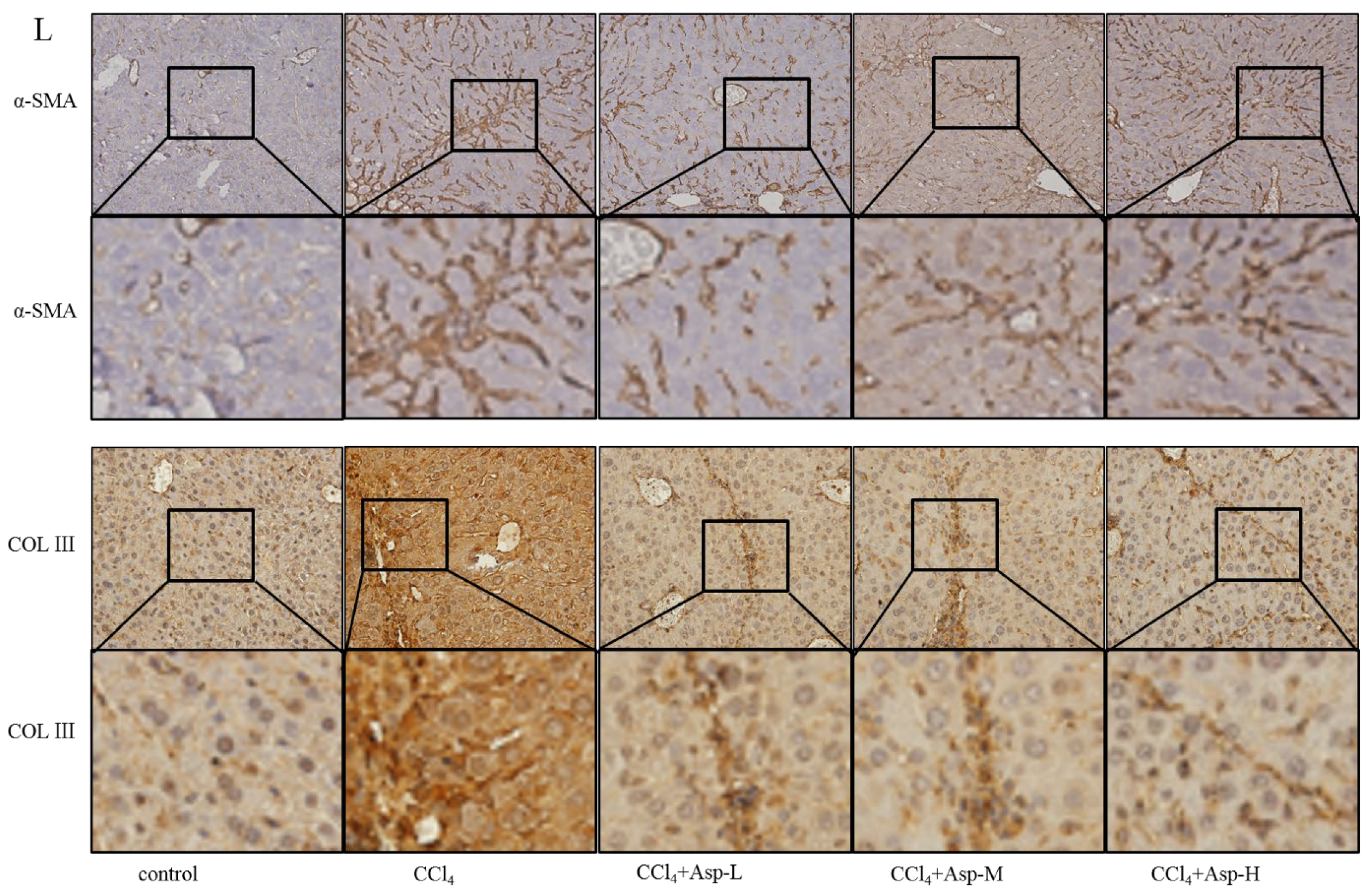
3.1. Asp Supplementation Protects against CCl4-Induced Liver Fibrosis in Mice
3.2. Inhibitory Effect of Asp on HSCs Activation In Vitro
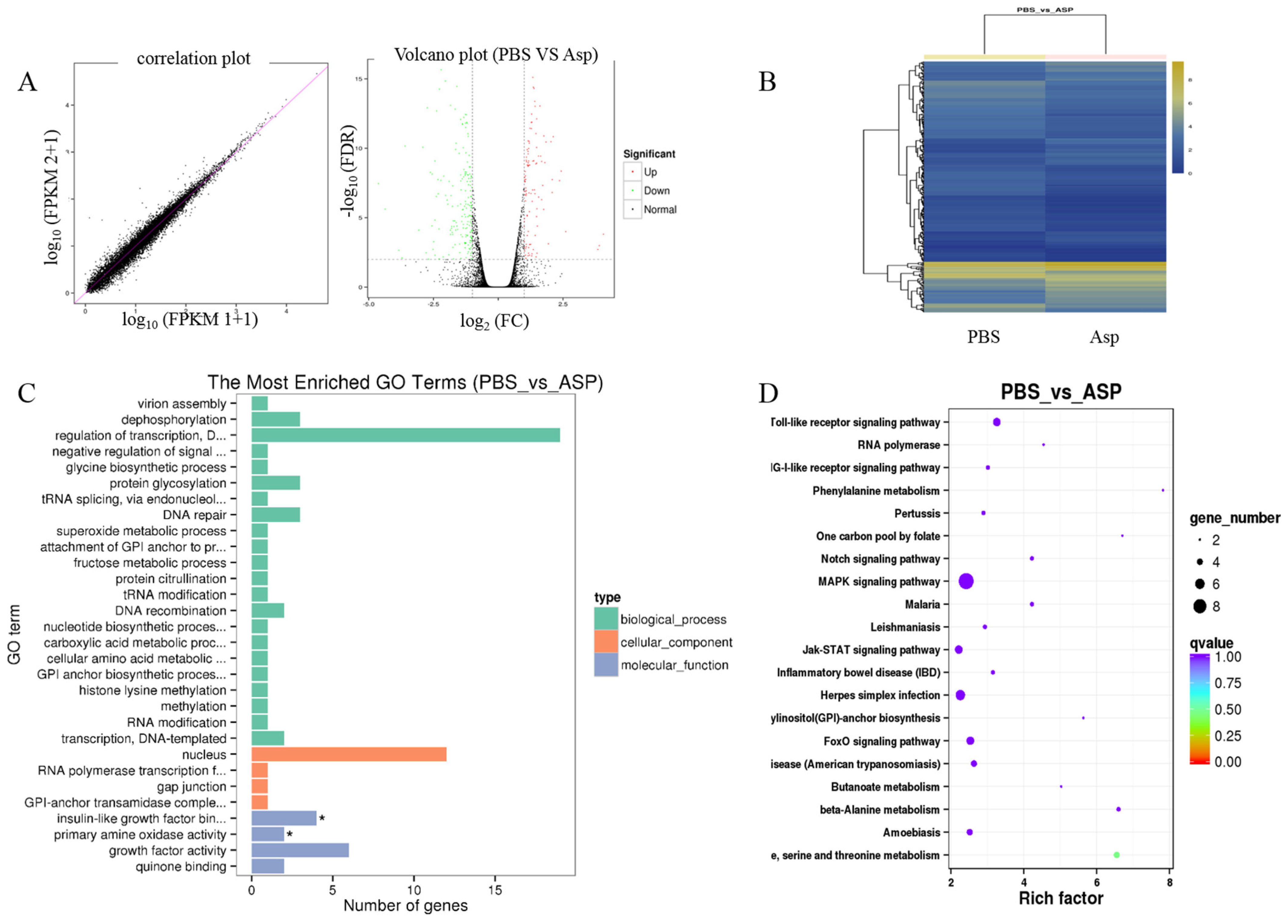
3.3. Gene Expression Profiling Using RNA-Seq
3.4. Effects of si-NLRP3 on Inflammation and Liver Fibrosis in LX-2 Cells
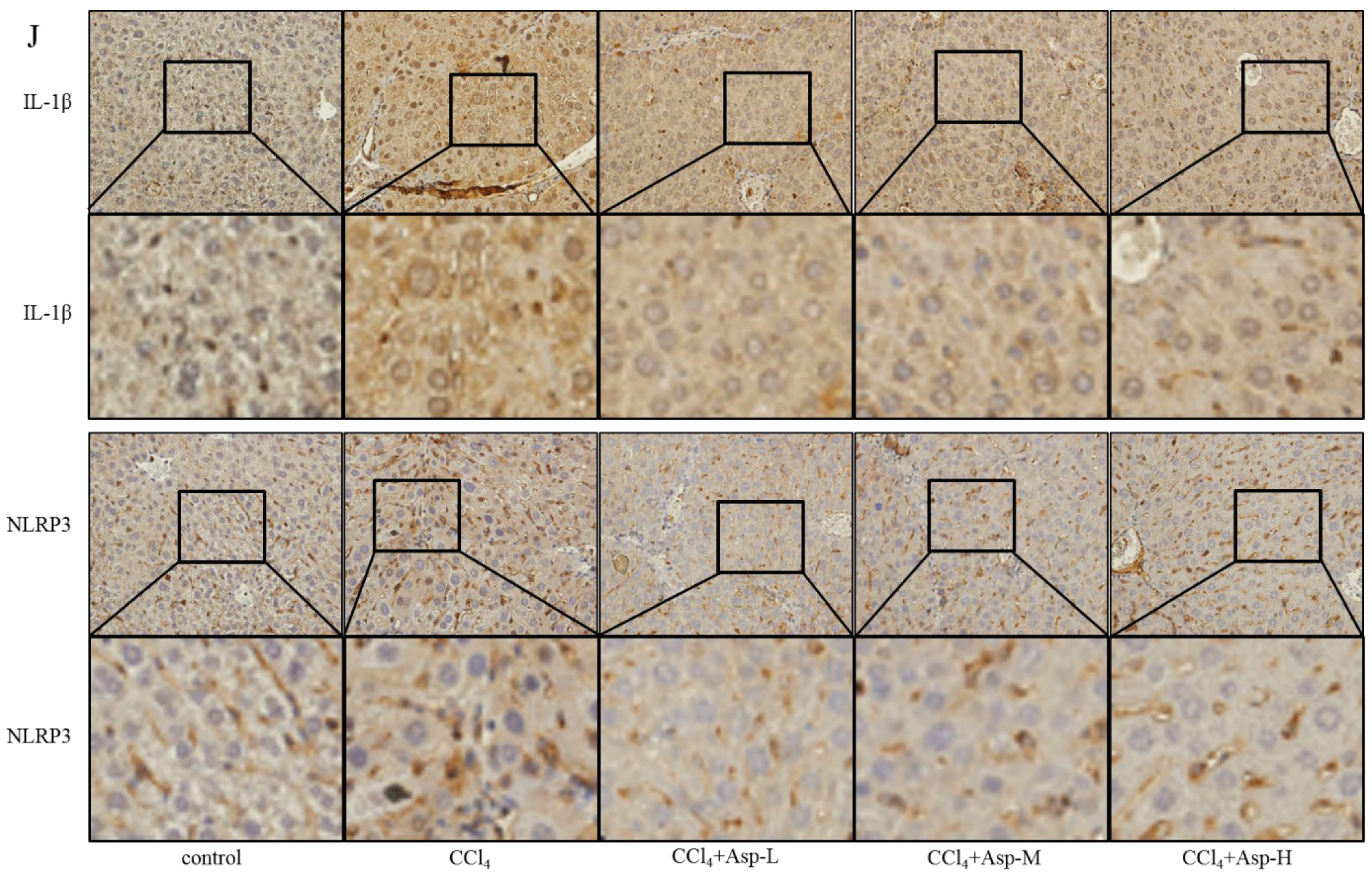
3.5. Treatment with Asp Blocked Hepatic NF-κB/NLRP3 Expression In Vitro and In Vivo
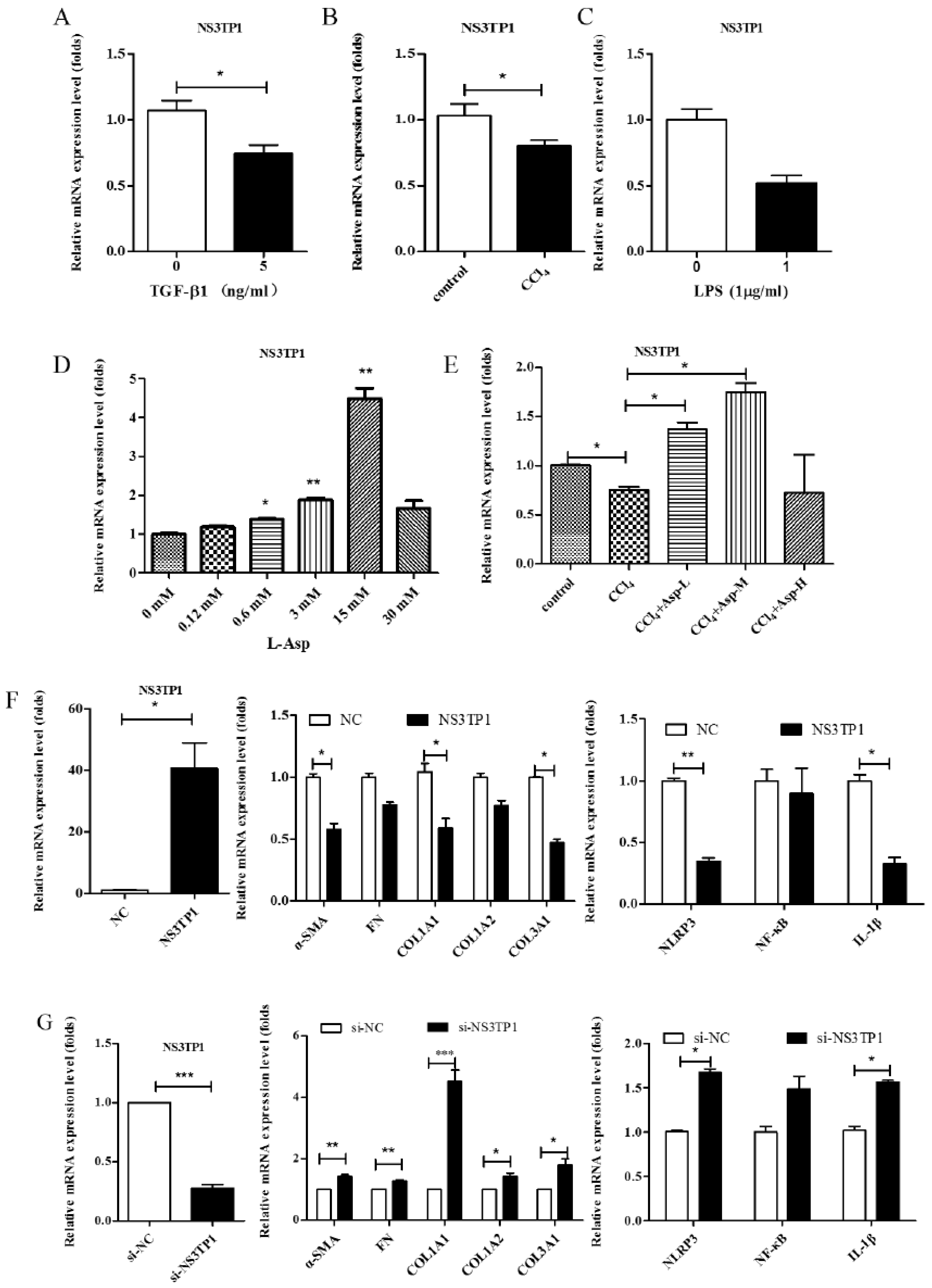
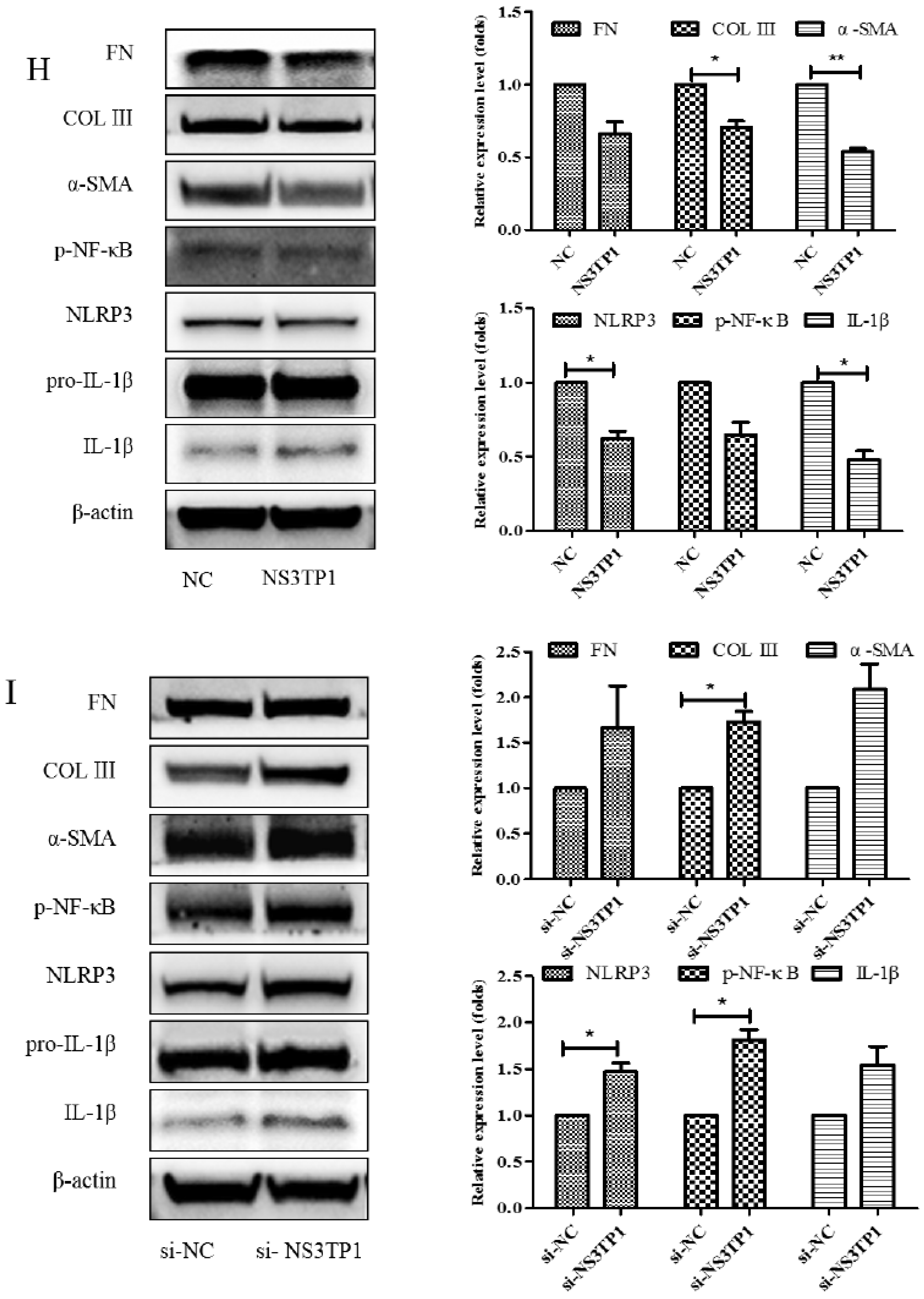
3.6. Asp Inhibited Liver Fibrosis by Upregulating NS3TP1 Expression and then Inhibiting the NF-κB/NLRP3 Signaling Pathway
3.7. Asp Supplementation Suppressed HSC Proliferation and Activation and Promoted Apoptosis In Vitro
3.8. D-Asp Had Effects Similar to Those of L-Asp
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.J.; Patel, K. Antifibrotic therapies: Will we ever get there. Curr. Gastroenterol. Rep. 2010, 12, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Parola, M. The role of redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2012, 5 (Suppl. S1), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Song, J.; Yang, J.; Zheng, J.; Yang, L.; Gao, J.; Tian, S.; Liu, Z.; Meng, X.; Wang, J.; et al. Tumor Necrosis Factor α-Induced Protein 8-Like 2 Alleviates Nonalcoholic Fatty Liver Disease Through Suppressing Transforming Growth Factor Beta-Activated Kinase 1 Activation. Hepatology 2021, 74, 1300–1318. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Tang, X.T.; Lin, M.; Yuan, J.; Peng, Y.J.; Yin, X.; Shang, G.; Ge, G.; Ren, Z.; Zhou, B.O. Perivenous Stellate Cells Are the Main Source of Myofibroblasts and Cancer-Associated Fibroblasts Formed After Chronic Liver Injuries. Hepatology 2021, 74, 1578–1594. [Google Scholar] [CrossRef]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; You, H.; Fan, X.; Jia, J. Hepatic macrophages in liver fibrosis: Pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. 2016, 3, e000079. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Peña, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Alegre, F.; Martí-Rodrigo, A.; Polo, M.; Ortiz-Masiá, D.; Bañuls, C.; Pinti, M.; Álvarez, Á.; Apostolova, N.; Esplugues, J.V.; Blas-García, A. Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz. Biomedicines 2022, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, D.M.; Chu, A.; Gaul, S.; Taghdiri, N.; Toomu, A.; Leszczynska, A.; Kaufmann, B.; Papouchado, B.; Wree, A.; Geisler, L.; et al. NOD-like receptor protein 3 activation causes spontaneous inflammation and fibrosis that mimics human NASH. Hepatology 2022, 10, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Meng, N.; Xia, M.; Lu, Y.-Q.; Wang, M.; Boini, K.M.; Li, P.-L.; Tang, W.-X. Activation of NLRP3 inflammasomes in mouse hepatic stellate cells during Schistosoma. J. Infect. Oncotarget 2016, 7, 39316–39331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.M.; Yang, R.Q.; Li, Y.; Ning, Z.W.; Zhang, L.L.; Zhou, G.S.; Luo, W.; Li, D.H.; Chen, Y.; Pan, M.X.; et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid. Redox Signal. 2016, 24, 795–812. [Google Scholar] [CrossRef]
- Ahmad, F.; Rafaz, H.; Ouyang, X.; Farooq, A.; Ghani, A.; Ahsan, K.; Guerra, M.; Mehal, W.Z. Activation of N-methyl-d-aspartate receptor downregulates inflammasome activity and liver inflammation via a β-arrestin-2 pathway. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, 107. [Google Scholar]
- Ji, D.; Cheng, J.; Wang, J.J.; Liu, Y.; Yang, Q.; Dang, X.-Y.; Wang, C.-H. Screening and cloning of the target genes transactivated by human gene 1 transactivated by hepatitis C virus NS3 protein using suppression subtractive hybridization. World J. Chin. Dig. 2004, 4, 93–96. [Google Scholar]
- Ji, D.; Cheng, J.; Liu, Y.; Wang, J.; Guo, J. Screening of differentially expressed genes in NS3TP1-transfected cells by gene expression profiling chip technology. World J. Chin. Dig. 2004, 7, 201–205. [Google Scholar]
- Bansal, R.; Frelin, L.; Brenndörfer, E.D.; Storm, G.; Prakash, J.; Sällberg, M. Hepatitis C Virus Nonstructural 3/4A Protein Dampens Inflammation and Contributes to Slow Fibrosis Progression during Chronic Fibrosis In Vivo. PLoS ONE 2015, 10, e0128466. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, S.; Han, M.; Ma, Y.; Feng, S.; Zhao, J.; Lu, H.; Yuan, X.; Cheng, J. miR-185 Inhibits Fibrogenic Activation of Hepatic Stellate Cells and Prevents Liver Fibrosis. Mol. Nucleic Acids 2018, 10, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Domenicali, M.; Caraceni, P.; Giannone, F.; Baldassarre, M.; Lucchetti, G.; Quarta, C.; Patti, C.; Catani, L.; Nanni, C.; Lemoli, R.M.; et al. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J. Hepatol. 2009, 51, 991–999. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S73–S78. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Sun, H.; Xue, T.; Gan, C.; Liu, H.; Xie, Y.; Yao, Y.; Ye, T. Liver Fibrosis: Therapeutic Targets and Advances in Drug Therapy. Front. Cell Dev. Biol. 2021, 9, 730176. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Pinzani, M.; Marra, F. Cytokine receptors and signaling in hepatic stellate cells. Semin. Liver Dis. 2001, 21, 397–416. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Gallego, P.; Castejón-Vega, B.; Del Campo, J.A.; Cordero, M.D. The Absence of NLRP3-inflammasome Modulates Hepatic Fibrosis Progression, Lipid Metabolism, and Inflammation in KO NLRP3 Mice during Aging. Cells 2020, 9, 2148. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Zhao, J.; Han, M.; Zhou, L.; Liang, P.; Wang, Y.; Feng, S.; Lu, H.; Yuan, X.; Han, K.; Chen, X.; et al. TAF and TDF attenuate liver fibrosis through NS5ATP9, TGFβ1_Smad3, and NF-κB_NLRP3 inflammasome signaling pathways. Hepatol. Int. 2019, 14, 145–160. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med. 2011, 17, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENES | SPECIES | SENSE (5′-3′) | ANTISENSE (5′-3′) |
|---|---|---|---|
| β-actin | Hum | CATCCGCAAAGAC CTG TAC GC | AGTACTTGCGCTCAGGAGGAG |
| α-SMA | Hum | GGGAATGGGACAAAAAGACA | CTTCAGGGGCAACACGAA |
| COLL1A1 | Hum | GGGATTCCCTGGACCTAAAG | GGAACACCTCGCTCTCCA |
| COLL1A2 | Hum | CTGGAGAGGCTGGTACTGCT | AGCACCAAGAAGACCCTGAG |
| COLL3A1 | Hum | CTGGACCCCAGGGTCTTC | GACCATCTGATCCAGGGTTTC |
| ARRB2 | Hum | GGAAACTCAAGCACGAGGAC | CTTGTTGGCACCCTCCTTC |
| NLRP3 | Hum | CACCTGTTGTGCAATCTGAAG | GCAAGATCCTGACAACATGC |
| IL-1β | Hum | TCGCCAGTGAAATGATGGCT | TGGAAGGAGCACTTCATCTGT |
| β-actin | Mus | CTAAGGCCAACCGTGAAAAG | ACCAGAGGCATACAGGGACA |
| COLL1A1 | Mus | TTCTCCTGGCAAAGACGGAC | CGGCCACCATCTTGAGACTT |
| COLL1A2 | Mus | TAGCCAACCGTGCTTCTCAG | TCTCCTCATCCAGGTACGCA |
| COLL3A1 | Mus | AAGGCTGCAAGATGGATGCT | GTGCTTACGTGGGACAGTCA |
| α-SMA | Mus | GAGACTCTCTTCCAGCCATCTT | TGATCTCCTTCTGCATCCTGTC |
| IL-6 | Mus | AGACAAAGCCAGAGTCCTTCAG | GCCACTCCTTCTGTGACTCCAG |
| NLRP3 | Mus | CCACATCTGATTGTGTTAATGGCT | GGGCTTAGGTCCACACAGAA |
| IL-1β | Mus | GCCACCTTTTGACAGTGATGAG | GACAGCCCAGGTCAAAGGTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Zhao, J.; Han, M.; Ma, A.; Yang, S.; Zeng, Y.; Cheng, J. Aspartate Reduces Liver Inflammation and Fibrosis by Suppressing the NLRP3 Inflammasome Pathway via Upregulating NS3TP1 Expression. J. Pers. Med. 2023, 13, 386. https://doi.org/10.3390/jpm13030386
Zhou L, Zhao J, Han M, Ma A, Yang S, Zeng Y, Cheng J. Aspartate Reduces Liver Inflammation and Fibrosis by Suppressing the NLRP3 Inflammasome Pathway via Upregulating NS3TP1 Expression. Journal of Personalized Medicine. 2023; 13(3):386. https://doi.org/10.3390/jpm13030386
Chicago/Turabian StyleZhou, Li, Jing Zhao, Ming Han, Anlin Ma, Song Yang, Yilan Zeng, and Jun Cheng. 2023. "Aspartate Reduces Liver Inflammation and Fibrosis by Suppressing the NLRP3 Inflammasome Pathway via Upregulating NS3TP1 Expression" Journal of Personalized Medicine 13, no. 3: 386. https://doi.org/10.3390/jpm13030386