
Personalized Management of Sudden Death Risk in Primary Cardiomyopathies: From Clinical Evaluation and Multimodality Imaging to Ablation and Cardioverter-Defibrillator Implant
 , , ,
, , ,
Abstract
:1. Introduction
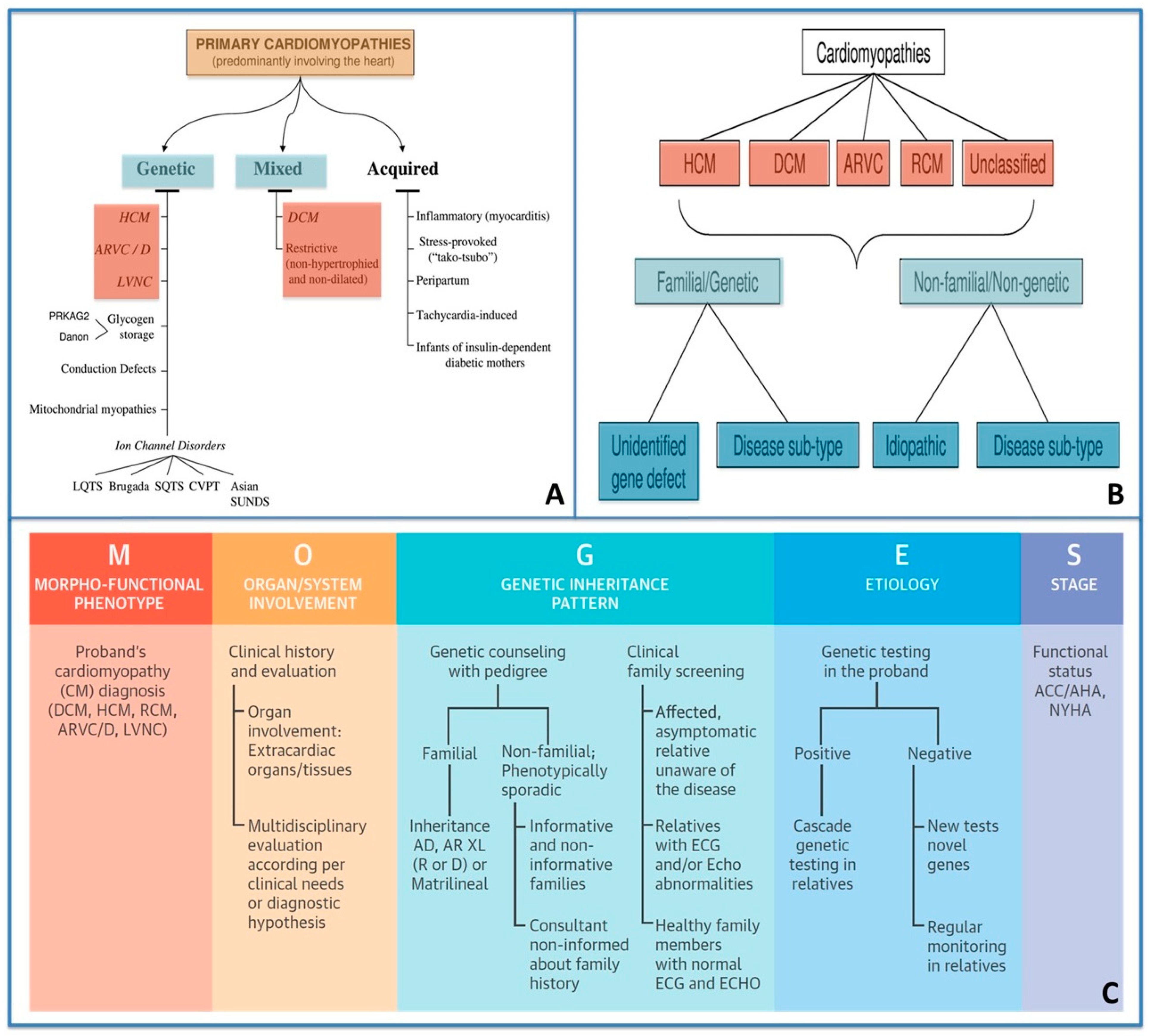
1.1. Classification of Cardiomyopathies from a Traditional Viewpoint to a Personalized Approach
- (1)
- HCM;
- (2)
- Dilated cardiomyopathy (DCM);
- (3)
- Arrhythmogenic cardiomyopathy (ACM);
- (4)
- Restrictive cardiomyopathy;
- (5)
- Unclassified cardiomyopathy.
1.2. Sudden Cardiac Death Risk in Cardiomyopathies: From a Traditional Viewpoint to a Personalized Approach
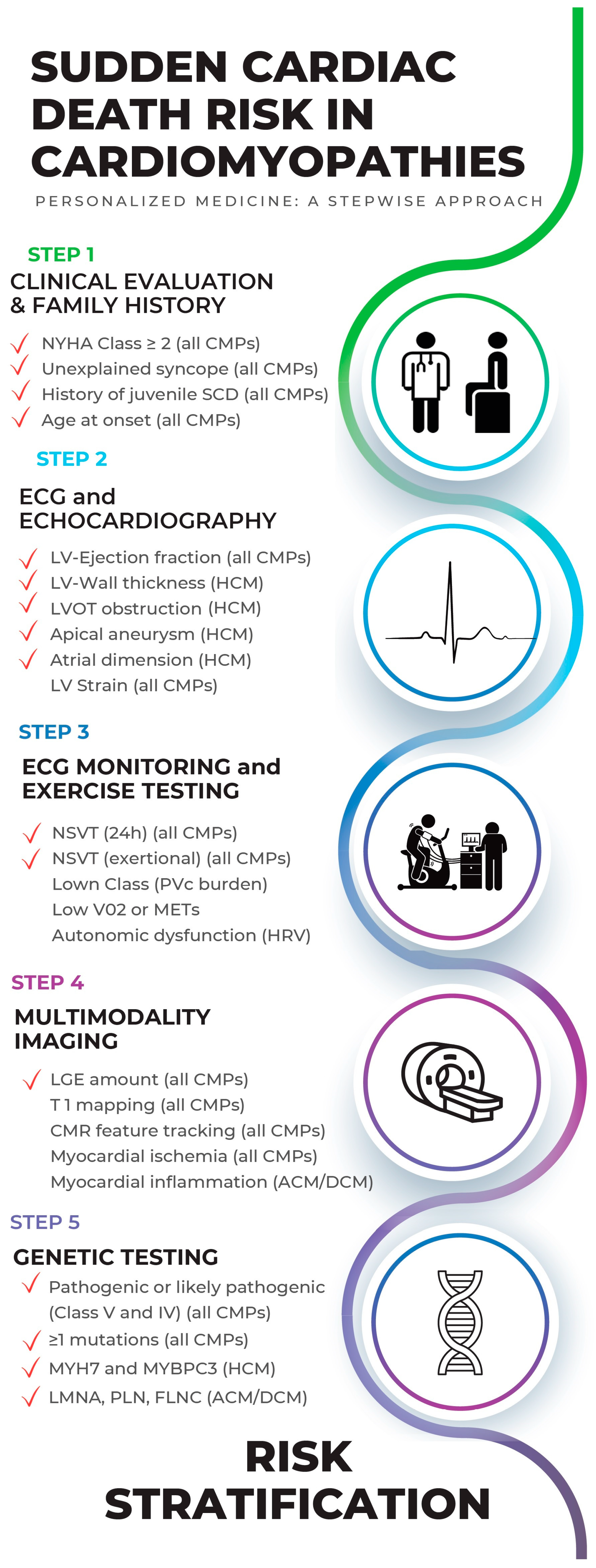
2. The Hierarchical and Multiparametric Approach of SCD Risk Estimation in Cardiomyopathies
2.1. Step 1: Clinical Evaluation and Family History (of CMPs and SCD)
2.2. Step 2: ECG and Echocardiography
2.2.1. Hypertrophic Cardiomyopathy
- (1)
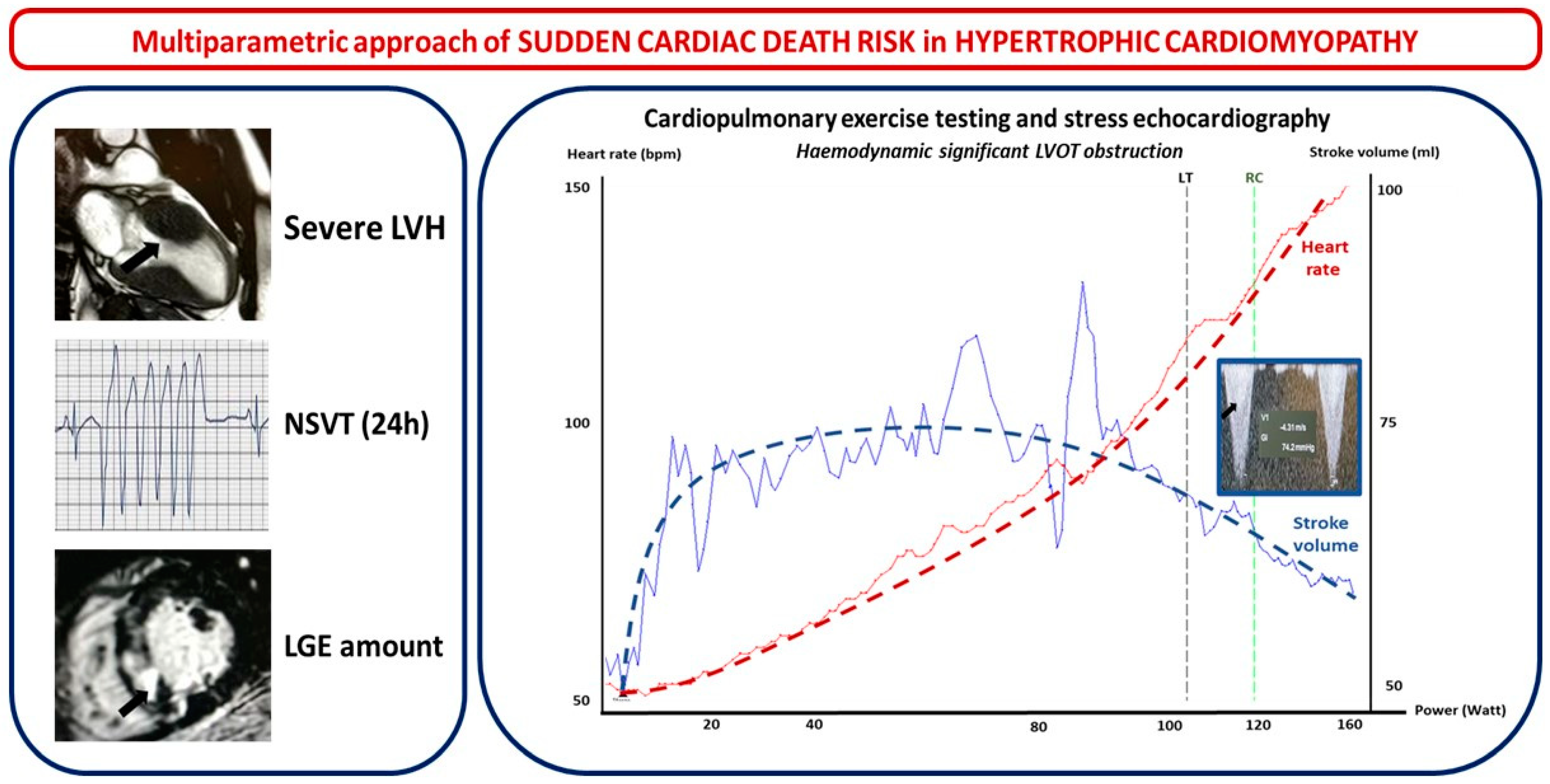
- Left ventricle wall thickness (LVWT), involved segments, maximal wall thickness, and septal morphology. LVWT represents the hallmark of HMC and, while listed as a major risk factor for SCD in both the 2014 ESC and 2020 US guidelines, the latter only considered massive LV hypertrophy ≥ 30 mm in any LV segment as a major risk factor for SCD. In the HCM Risk-SCD model proposed by ESC 2014 guidelines, maximal LVWT was considered as a continuous rather than a dichotomous variable [12,13]. Different hypertrophy patterns have been observed. Based on the location of LV hypertrophy, Maron et al. initially proposed a four-type classification: Type I hypertrophy involves the basal septum; type II involves the whole septum; type III involves the anterior and anterolateral walls of the septum; and type IV involves LV apex. A five-phenotype classification has recently been suggested. It includes the following: type A, predominant mid-septal convexity toward the LV cavity (reverse septum HCM); type B, septum concavity toward the LV cavity and a prominent basal septal bulge (sigmoid septum HCM); type C, an overall straight septum (neutral septum HCM); type D, predominant apical distribution of hypertrophy (apical HCM); and type E, predominant hypertrophy at the mid-ventricular level (mid-ventricular HCM). Interventricular septum (IVS) morphology has been correlated with the probability of a positive genetic test for sarcomeric mutations: accordingly, a reverse IVS is associated with a high probability of a positive genetic test, apical or neutral IVS with a moderate probability, and a “sigmoid” IVS with a low probability of a positive test [18]. The addition of contrast echocardiographic agents could be useful in order to diagnose other localized forms of HCM (such as apical or inferolateral).
- (2)
- LV apical aneurysm: US 2020 guidelines consider the presence of an LV apical aneurysm independent of its size as a major risk factor for SCD since it offers the substrate for re-entrant VT. Indeed, the prognosis of HCM patients with LV apical aneurysms is generally unfavorable, with an overall rate of life-threatening complications between 6% and 10% per year, mostly consisting of arrhythmic SCD and thromboembolic events [19,20].
- (3)
- The mitral valve, its apparatus, and left ventricular outflow tract obstruction. More than 50% of HCM patients have abnormal mitral leaflets, and more than 25% show abnormalities of the chordae and papillary muscles as a primary phenotypic expression of HCM that may have a pivotal role in left ventricular outflow tract (LVOT) obstruction [21]. One of the parameters of the HCM Risk-SCD model is the maximal LV outflow tract (LVOT) gradient, while US guidelines, although not considering LVOT gradient in SCD risk, include the exercise-induced hypotension that represents its hemodynamic consequence. LVOTO at rest is present in about one-third of HCM patients and is an independent determinant of adverse prognosis [22]. In another one-third of HCM patients, LVOTO is only seen after provocative maneuvers, and treadmill echocardiography (EE) is a key method in detecting an inducible obstruction in HCM [13].
- (4)
- LV systolic function: US 2020 guidelines consider decreased LV systolic function (ejection fraction < 50%) as a major risk factor for SCD in HCM patients. However, the limitations of LVEF are well known when LVH is present. EF, mostly reflecting radial wall thickening, is often preserved in HCM, compensating for the reduced longitudinal function seen in this disease. Doppler myocardial imaging (DMI) and 2D speckle-tracking echocardiography (2D-STE) overcome some of these pitfalls [23]. As early signs of LV systolic dysfunction, HCM patients show a decrease in regional and global longitudinal strain (LS) before the impairment of LVEF. A decreased septal and regional LS (>10%) has been related to susceptibility to ventricular arrhythmias in HCM [24]. LV-GLS is significantly related to an increased risk of SCD events [25] and is an independent predictor of appropriate ICD therapy [26]. A 3D echo provides potential further insights on LV function in HCM, showing good correlations with CMR. Three-dimensional strain echocardiography also represents a promising tool, although it is not yet defined as useful in HCM risk stratification [27].
- (5)
- LV diastolic function and left atrial volume: HCM is classically defined as a “diastolic disease” and the hallmark of diastolic HF [28]. Nevertheless, no single non-invasive echo Doppler parameter has been validated to be completely accurate in the assessment of LV filling pressures (LV-FPs) in HCM. Although the ESC risk score does not consider diastolic function, left atrial (LA) diameter, which represents one of its main structural consequences, is included. On the other hand, in the US SCD risk evaluation, diastolic dysfunction was not considered. The 2D echo LA volume indexed to body surface area (LAVI, mL/m2, in the four-chamber view) is a simple and mandatory parameter for assessing diastolic function in HCM patients. Moreover, strain analysis of the LA may represent a novel promising tool in HCM risk stratification; in fact, atrial LS was correlated with HF symptoms [29].
2.2.2. Arrhythmogenic Cardiomyopathy
2.2.3. Dilated Cardiomyopathy
2.3. Step 3: ECG Monitoring and Exercise Testing in SCD Risk Evaluation
2.4. Step 4: Multimodality Imaging in SCD Risk Evaluation
2.4.1. Hypertrophic Cardiomyopathy
2.4.2. Arrhythmogenic Cardiomyopathy
2.4.3. Dilated Cardiomyopathy
2.5. Step 5: Genetic Testing in SCD Risk Evaluation
- Class V: pathogenic;
- Class IV: likely pathogenic;
- Class III: variant of uncertain significance;
- Class II: likely benign;
- Class I: benign [69].
3. The World of ACM and DCM: How “Complex Imaging” (Electroanatomic Mapping) Directs Diagnosis and Invasive Management
3.1. Arrhythmogenic Cardiomyopathy
3.2. Dilated Cardiomyopathy
3.3. ICD Implant in Cardiomyopathies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braunwald, E. Hypertrophic Cardiomyopathy: The First Century 1869–1969. Glob. Cardiol. Sci. Pract. 2012, 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A Beta Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Pera, L.L.; Carletti, R.; Cerbelli, B.; Pignataro, M.G.; Pernazza, A.; Ferre, F.; Lombardi, M.; Lazzeroni, D.; Olivotto, I.; et al. RNA-Seq Profiling Reveals Different Pathways between Remodeled Vessels and Myocardium in Hypertrophic Cardiomyopathy. Microcirculation 2022, 29, e12790. [Google Scholar] [CrossRef] [PubMed]
- Foglieni, C.; Lombardi, M.; Lazzeroni, D.; Zerboni, R.; Lazzarini, E.; Bertoli, G.; Pisano, A.; Girolami, F.; Andolfo, A.; Magagnotti, C.; et al. Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy. Biomedicines 2022, 10, 2180. [Google Scholar] [CrossRef]
- Horizon 2020 Advisory Group. Available online: https://Eur-Lex.Europa.Eu/Legal-Content/EN/TXT/?Uri=uriserv%3AOJ.C_.2015.421.01.0002.01.ENG&toc=OJ%3AC%3A2015%3A421%3AFULL (accessed on 5 April 2023).
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Arbustini, E.; Narula, N.; Tavazzi, L.; Serio, A.; Grasso, M.; Favalli, V.; Bellazzi, R.; Tajik, J.A.; Bonow, R.D.; Fuster, V.; et al. The MOGE(S) Classification of Cardiomyopathy for Clinicians. J. Am. Coll. Cardiol. 2014, 64, 304–318. [Google Scholar] [CrossRef]
- European Heart Rhythm Association; Heart Rhythm Society; Zipes, D.P.; Camm, A.J.; Borggrefe, M.; Buxton, A.E.; Chaitman, B.; Fromer, M.; Gregoratos, G.; Klein, G.; et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J. Am. Coll. Cardiol. 2006, 48, e247–e346. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Authors/Task Force members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G. 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
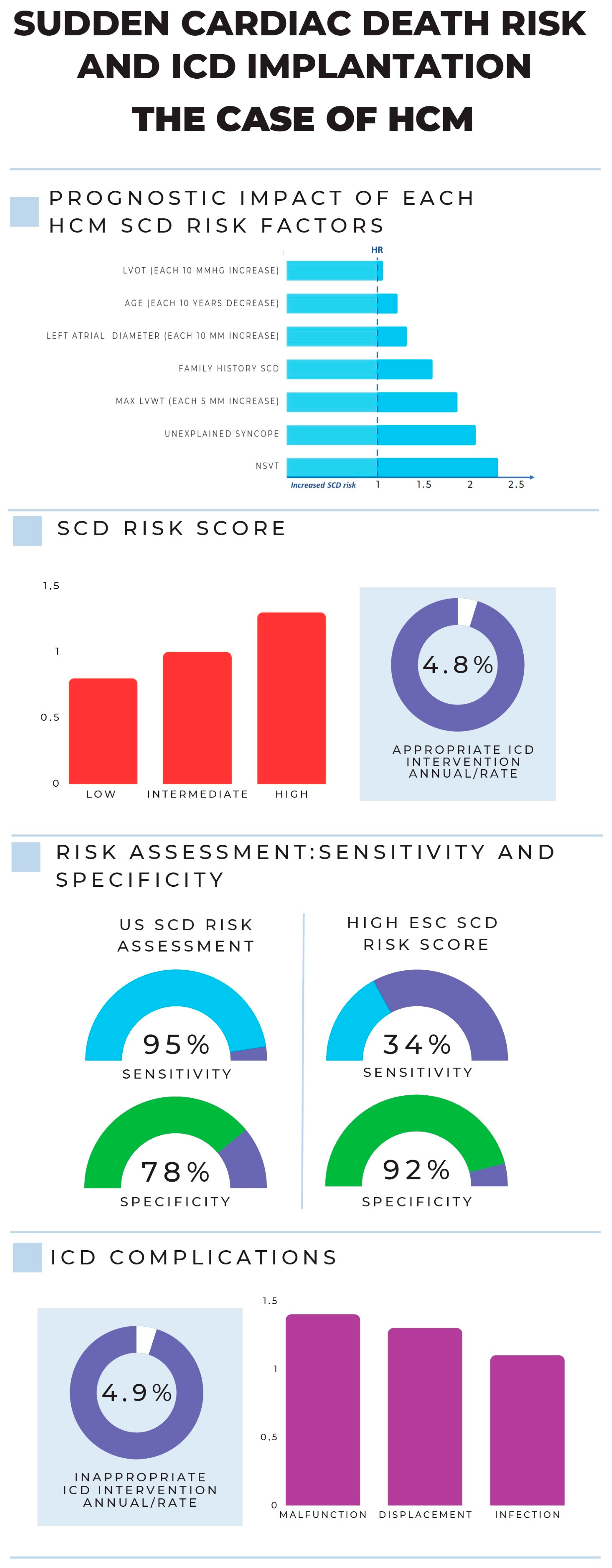
- Francia, P.; Olivotto, I.; Lambiase, P.D.; Autore, C. Implantable Cardioverter-Defibrillators for Hypertrophic Cardiomyopathy: The Times They Are a-Changin’. Europace 2022, 24, 1384–1394. [Google Scholar] [CrossRef]
- Norrish, G.; Ding, T.; Field, E.; McLeod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; et al. A Validation Study of the European Society of Cardiology Guidelines for Risk Stratification of Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy. Europace 2019, 21, 1559–1565. [Google Scholar] [CrossRef]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic Work-up in Cardiomyopathies: Bridging the Gap between Clinical Phenotypes and Final Diagnosis. A Position Statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef]
- Lazzeroni, D.; Stefano Centorbi, C. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Diagnosis, Clinical Course and Therapy. In Cardiomyopathy—Disease of the Heart Muscle; IntechOpen: Uppsala, Sweden, 2021. [Google Scholar] [CrossRef]
- Bos, J.M.; Towbin, J.A.; Ackerman, M.J. Diagnostic, Prognostic, and Therapeutic Implications of Genetic Testing for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 201–211. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, B.J.; Haas, T.S.; Garberich, R.F.; Wang, W.; Link, M.S.; Maron, M.S. Hypertrophic Cardiomyopathy with Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J. Am. Coll. Cardiol. 2017, 69, 761–773. [Google Scholar] [CrossRef]
- Elsheshtawy, M.O.; Mahmoud, A.N.; Abdelghany, M.; Suen, I.H.; Sadiq, A.; Shani, J. Left Ventricular Aneurysms in Hypertrophic Cardiomyopathy with Midventricular Obstruction: A Systematic Review of Literature. Pacing Clin. Electrophysiol. 2018, 41, 854–865. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Harrigan, C.; Appelbaum, E.; Gibson, C.M.; Lesser, J.R.; Haas, T.S.; Udelson, J.E.; Manning, W.J.; Maron, B.J. Mitral Valve Abnormalities Identified by Cardiovascular Magnetic Resonance Represent a Primary Phenotypic Expression of Hypertrophic Cardiomyopathy. Circulation 2011, 124, 40–47. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of Left Ventricular Outflow Tract Obstruction on Clinical Outcome in Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef]
- Bayrak, F.; Kahveci, G.; Mutlu, B.; Sonmez, K.; Degertekin, M. Tissue Doppler Imaging to Predict Clinical Course of Patients with Hypertrophic Cardiomyopathy. Eur. J. Echocardiogr. 2008, 9, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Thaman, R.; Gimeno, J.R.; Murphy, R.T.; Kubo, T.; Sachdev, B.; Mogensen, J.; Elliott, P.M.; McKenna, W.J. Prevalence and Clinical Significance of Systolic Impairment in Hypertrophic Cardiomyopathy. Heart 2005, 91, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, H.K.; Lee, S.C.; Kim, J.; Park, J.B.; Hwang, I.C.; Choi, Y.J.; Lee, S.P.; Chang, S.A.; Lee, W.; et al. Supplementary Role of Left Ventricular Global Longitudinal Strain for Predicting Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 1108–1116. [Google Scholar] [CrossRef]
- Candan, O.; Gecmen, C.; Bayam, E.; Guner, A.; Celik, M.; Doğan, C. Mechanical Dispersion and Global Longitudinal Strain by Speckle Tracking Echocardiography: Predictors of Appropriate Implantable Cardioverter Defibrillator Therapy in Hypertrophic Cardiomyopathy. Echocardiography 2017, 34, 835–842. [Google Scholar] [CrossRef] [PubMed]
- de Gregorio, C.; Recupero, A.; Grimaldi, P.; Coglitore, S. Can Transthoracic Live 3-Dimensional Echocardiography Improve the Recognition of Midventricular Obliteration in Hypertrophic Obstructive Cardiomyopathy? J. Am. Soc. Echocardiogr. 2006, 19, e1–e4. [Google Scholar] [CrossRef]
- Rakowski, H.; Carasso, S. Quantifying Diastolic Function in Hypertrophic Cardiomyopathy: The Ongoing Search for the Holy Grail. Circulation 2007, 116, 2662–2665. [Google Scholar] [CrossRef]
- Roşca, M.; Popescu, B.A.; Beladan, C.C.; Călin, A.; Muraru, D.; Popa, E.C.; Lancellotti, P.; Enache, R.; Coman, I.M.; Jurcuţ, R.; et al. Left Atrial Dysfunction as a Correlate of Heart Failure Symptoms in Hypertrophic Cardiomyopathy. J. Am. Soc. Echocardiogr. 2010, 23, 1090–1098. [Google Scholar] [CrossRef]
- Monda, E.; Rubino, M.; Palmiero, G.; Verrillo, F.; Lioncino, M.; Diana, G.; Cirillo, A.; Fusco, A.; Dongiglio, F.; Caiazza, M.; et al. Multimodality Imaging in Arrhythmogenic Left Ventricular Cardiomyopathy. J. Clin. Med. 2023, 12, 1568. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic Cardiomyopathies (ACs): Diagnosis, Risk Stratification and Management. Heart 2019, 105, 1117–1128. [Google Scholar] [CrossRef]
- Peretto, G.; Di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular Features of Patients with LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458–463. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; Benedetti, S.; Di Resta, C.; Gigli, L.; Ferrari, M.; Della Bella, P. Updated Clinical Overview on Cardiac Laminopathies: An Electrical and Mechanical Disease. Nucleus 2018, 9, 380–391. [Google Scholar] [CrossRef]
- Raafs, A.G.; Boscutti, A.; Henkens, M.T.H.M.; van den Broek, W.W.A.; Verdonschot, J.A.J.; Weerts, J.; Stolfo, D.; Nuzzi, V.; Manca, P.; Hazebroek, M.R.; et al. Global Longitudinal Strain Is Incremental to Left Ventricular Ejection Fraction for the Prediction of Outcome in Optimally Treated Dilated Cardiomyopathy Patients. J. Am. Heart Assoc. 2022, 11, e024505. [Google Scholar] [CrossRef]
- Mignot, A.; Donal, E.; Zaroui, A.; Reant, P.; Salem, A.; Hamon, C.; Monzy, S.; Roudaut, R.; Habib, G.; Lafitte, S. Global Longitudinal Strain as a Major Predictor of Cardiac Events in Patients with Depressed Left Ventricular Function: A Multicenter Study. J. Am. Soc. Echocardiogr. 2010, 23, 1019–1024. [Google Scholar] [CrossRef]
- Popescu, B.A.; Beladan, C.C.; Calin, A.; Muraru, D.; Deleanu, D.; Rosca, M.; Ginghina, C. Left Ventricular Remodelling and Torsional Dynamics in Dilated Cardiomyopathy: Reversed Apical Rotation as a Marker of Disease Severity. Eur. J. Heart Fail 2009, 11, 945–951. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving Concepts in Dilated Cardiomyopathy. Eur. J. Heart Fail 2018, 20, 228–239. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, D.H.; Choi, W.S.; Kim, K.H.; Park, S.H.; Bae, M.H.; Lee, J.H.; Park, H.S.; Cho, Y.; Chae, S.C.; et al. Prediction of Improvement in Cardiac Function by High Dose Dobutamine Stress Echocardiography in Patients with Recent Onset Idiopathic Dilated Cardiomyopathy. Int. J. Cardiol. 2013, 167, 1649–1650. [Google Scholar] [CrossRef]
- Bhonsale, A.; James, C.A.; Tichnell, C.; Murray, B.; Gagarin, D.; Philips, B.; Dalal, D.; Tedford, R.; Russell, S.D.; Abraham, T.; et al. Incidence and Predictors of Implantable Cardioverter-Defibrillator Therapy in Patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Undergoing Implantable Cardioverter-Defibrillator Implantation for Primary Prevention. J. Am. Coll. Cardiol. 2011, 58, 1485–1496. [Google Scholar] [CrossRef]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, Ø.H.; Saguner, A.M.; et al. A New Prediction Model for Ventricular Arrhythmias in Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858. [Google Scholar] [CrossRef]
- Goldberger, J.J.; Cain, M.E.; Hohnloser, S.H.; Kadish, A.H.; Knight, B.P.; Lauer, M.S.; Maron, B.J.; Page, R.L.; Passman, R.S.; Siscovick, D.; et al. American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society Scientific Statement on Noninvasive Risk Stratification Techniques for Identifying Patients at Risk for Sudden Cardiac Death. A Scientific Statement from the American Heart Association Council on Clinical Cardiology Committee on Electrocardiography and Arrhythmias and Council on Epidemiology and Prevention. J. Am. Coll. Cardiol. 2008, 52, 1179–1199. [Google Scholar] [CrossRef]
- Solbiati, M.; Casazza, G.; Dipaola, F.; Barbic, F.; Caldato, M.; Montano, N.; Furlan, R.; Sheldon, R.S.; Costantino, G. The Diagnostic Yield of Implantable Loop Recorders in Unexplained Syncope: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2017, 231, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Exercise Testing Oversights Underlie Missed and Delayed Diagnosis of Catecholaminergic Polymorphic Ventricular Tachycardia in Young Sudden Cardiac Arrest Survivors. Heart Rhythm 2019, 16, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, J.R.; Tomé-Esteban, M.; Lofiego, C.; Hurtado, J.; Pantazis, A.; Mist, B.; Lambiase, P.; McKenna, W.J.; Elliott, P.M. Exercise-Induced Ventricular Arrhythmias and Risk of Sudden Cardiac Death in Patients with Hypertrophic Cardiomyopathy. Eur. Heart J. 2009, 30, 2599–2605. [Google Scholar] [CrossRef]
- Bayonas-Ruiz, A.; Muñoz-Franco, F.M.; Ferrer, V.; Pérez-Caballero, C.; Sabater-Molina, M.; Tomé-Esteban, M.T.; Bonacasa, B. Cardiopulmonary Exercise Test in Patients with Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 2312. [Google Scholar] [CrossRef] [PubMed]
- McLELLAN, A.J.A.; Ellims, A.H.; Prabhu, S.; Voskoboinik, A.; Iles, L.M.; Hare, J.L.; Kaye, D.M.; Macciocca, I.; Mariani, J.A.; Kalman, J.M.; et al. Diffuse Ventricular Fibrosis on Cardiac Magnetic Resonance Imaging Associates with Ventricular Tachycardia in Patients with Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2016, 27, 571–580. [Google Scholar] [CrossRef]
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Pencina, M.J.; Assenza, G.E.; Haas, T.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; et al. Prognostic Value of Quantitative Contrast-Enhanced Cardiovascular Magnetic Resonance for the Evaluation of Sudden Death Risk in Patients with Hypertrophic Cardiomyopathy. Circulation 2014, 130, 484–495. [Google Scholar] [CrossRef]
- Negri, F.; Muser, D.; Driussi, M.; Sanna, G.D.; Masè, M.; Cittar, M.; Poli, S.; De Bellis, A.; Fabris, E.; Puppato, M.; et al. Prognostic Role of Global Longitudinal Strain by Feature Tracking in Patients with Hypertrophic Cardiomyopathy: The STRAIN-HCM Study. Int. J. Cardiol. 2021, 345, 61–67. [Google Scholar] [CrossRef]
- Xu, J.; Zhuang, B.; Sirajuddin, A.; Li, S.; Huang, J.; Yin, G.; Song, L.; Jiang, Y.; Zhao, S.; Lu, M. MRI T1 Mapping in Hypertrophic Cardiomyopathy: Evaluation in Patients without Late Gadolinium Enhancement and Hemodynamic Obstruction. Radiology 2020, 294, 275–286. [Google Scholar] [CrossRef]
- O’Hara, R.P.; Binka, E.; Prakosa, A.; Zimmerman, S.L.; Cartoski, M.J.; Abraham, M.R.; Lu, D.-Y.; Boyle, P.M.; Trayanova, N.A. Personalized Computational Heart Models with T1-Mapped Fibrotic Remodeling Predict Sudden Death Risk in Patients with Hypertrophic Cardiomyopathy. eLife 2022, 11, e73325. [Google Scholar] [CrossRef]
- Di Marco, A.; Brown, P.F.; Bradley, J.; Nucifora, G.; Anguera, I.; Miller, C.A.; Schmitt, M. Extracellular Volume Fraction Improves Risk-Stratification for Ventricular Arrhythmias and Sudden Death in Non-Ischaemic Cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2023, 24, 512–521. [Google Scholar] [CrossRef]
- Androulakis, A.F.A.; Zeppenfeld, K.; Paiman, E.H.M.; Piers, S.R.D.; Wijnmaalen, A.P.; Siebelink, H.-M.J.; Sramko, M.; Lamb, H.J.; van der Geest, R.J.; de Riva, M.; et al. Entropy as a Novel Measure of Myocardial Tissue Heterogeneity for Prediction of Ventricular Arrhythmias and Mortality in Post-Infarct Patients. JACC Clin. Electrophysiol. 2019, 5, 480–489. [Google Scholar] [CrossRef]
- Ye, Y.; Ji, Z.; Zhou, W.; Pu, C.; Li, Y.; Zhou, C.; Hu, X.; Chen, C.; Sun, Y.; Huang, Q.; et al. Mean Scar Entropy by Late Gadolinium Enhancement Cardiac Magnetic Resonance Is Associated with Ventricular Arrhythmias Events in Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 758635. [Google Scholar] [CrossRef]
- Lazzeroni, D.; Rimoldi, O.; Camici, P.G. From Left Ventricular Hypertrophy to Dysfunction and Failure. Circ. J. 2016, 80, 555–564. [Google Scholar] [CrossRef]
- Ciampi, Q.; Olivotto, I.; Gardini, C.; Mori, F.; Peteiro, J.; Monserrat, L.; Fernandez, X.; Cortigiani, L.; Rigo, F.; Lopes, L.R.; et al. Prognostic Role of Stress Echocardiography in Hypertrophic Cardiomyopathy: The International Stress Echo Registry. Int. J. Cardiol. 2016, 219, 331–338. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Chen, X.; Ji, K.; Lu, M.; Hauer, R.; Chen, L.; Zhao, S. Left Ventricular Longitudinal Dyssynchrony by CMR Feature Tracking Is Related to Adverse Prognosis in Advanced Arrhythmogenic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 712832. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical Profile of Four Families with Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Dominant Desmoplakin Mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Tessier, R.; Marteau, L.; Vivien, M.; Guyomarch, B.; Thollet, A.; Fellah, I.; Jamet, B.; Sébille, J.C.; Eugene, T.; Serfaty, J.M.; et al. 18F-Fluorodeoxyglucose Positron Emission Tomography for the Detection of Myocardial Inflammation in Arrhythmogenic Left Ventricular Cardiomyopathy. Circ. Cardiovasc. Imaging 2022, 15, e014065. [Google Scholar] [CrossRef]
- Peretto, G.; Sommariva, E.; Di Resta, C.; Rabino, M.; Villatore, A.; Lazzeroni, D.; Sala, S.; Pompilio, G.; Cooper, L.T. Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench. Biomolecules 2023, 13, 646. [Google Scholar] [CrossRef]
- Palmisano, A.; Vignale, D.; Peretto, G.; Busnardo, E.; Calcagno, C.; Campochiaro, C.; De Luca, G.; Sala, S.; Ferro, P.; Basso, C.; et al. Hybrid FDG-PET/MR or FDG-PET/CT to Detect Disease Activity in Patients with Persisting Arrhythmias After Myocarditis. JACC Cardiovasc. Imaging 2021, 14, 288–292. [Google Scholar] [CrossRef]
- Becker, M.A.J.; Cornel, J.H.; van de Ven, P.M.; van Rossum, A.C.; Allaart, C.P.; Germans, T. The Prognostic Value of Late Gadolinium-Enhanced Cardiac Magnetic Resonance Imaging in Nonischemic Dilated Cardiomyopathy: A Review and Meta-Analysis. JACC Cardiovasc. Imaging 2018, 11, 1274–1284. [Google Scholar] [CrossRef]
- Alba, A.C.; Gaztañaga, J.; Foroutan, F.; Thavendiranathan, P.; Merlo, M.; Alonso-Rodriguez, D.; Vallejo-García, V.; Vidal-Perez, R.; Corros-Vicente, C.; Barreiro-Pérez, M.; et al. Prognostic Value of Late Gadolinium Enhancement for the Prediction of Cardiovascular Outcomes in Dilated Cardiomyopathy: An International, Multi-Institutional Study of the MINICOR Group. Circ. Cardiovasc. Imaging 2020, 13, e010105. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated Cardiomyopathy and Arrhythmogenic Left Ventricular Cardiomyopathy: A Comprehensive Genotype-Imaging Phenotype Study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carr-White, G.; Jabbour, A.; Yu, C.-Y.; Gebker, R.; Kelle, S.; Hinojar, R.; Doltra, A.; Varma, N.; Child, N.; et al. T1-Mapping and Outcome in Nonischemic Cardiomyopathy: All-Cause Mortality and Heart Failure. JACC Cardiovasc. Imaging 2016, 9, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Judd, R.M.; Kim, R.J.; Kim, H.W.; Klem, I.; Heitner, J.F.; Shah, D.J.; Jue, J.; White, B.E.; Indorkar, R.; et al. Feature-Tracking Global Longitudinal Strain Predicts Death in a Multicenter Population of Patients with Ischemic and Nonischemic Dilated Cardiomyopathy Incremental to Ejection Fraction and Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2018, 11, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Gerson, M.; Thomas, G.S.; Cerqueira, M.D.; Jacobson, A.F. 123I-MIBG Imaging for Prediction of Mortality and Potentially Fatal Events in Heart Failure: The ADMIRE-HFX Study. J. Nucl. Med. 2015, 56, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hong, Y.; Su, W.W.; Li, X. Risk Factors of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Curr. Opin. Cardiol. 2022, 37, 15–21. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Paldino, A.; De Angelis, G.; Merlo, M.; Gigli, M.; Dal Ferro, M.; Severini, G.M.; Mestroni, L.; Sinagra, G. Genetics of Dilated Cardiomyopathy: Clinical Implications. Curr Cardiol. Rep. 2018, 20, 83. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-Phenotype Associations in Dilated Cardiomyopathy: Meta-Analysis on More than 8000 Individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.W.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin a/c Mutation Carriers a European Cohort Study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
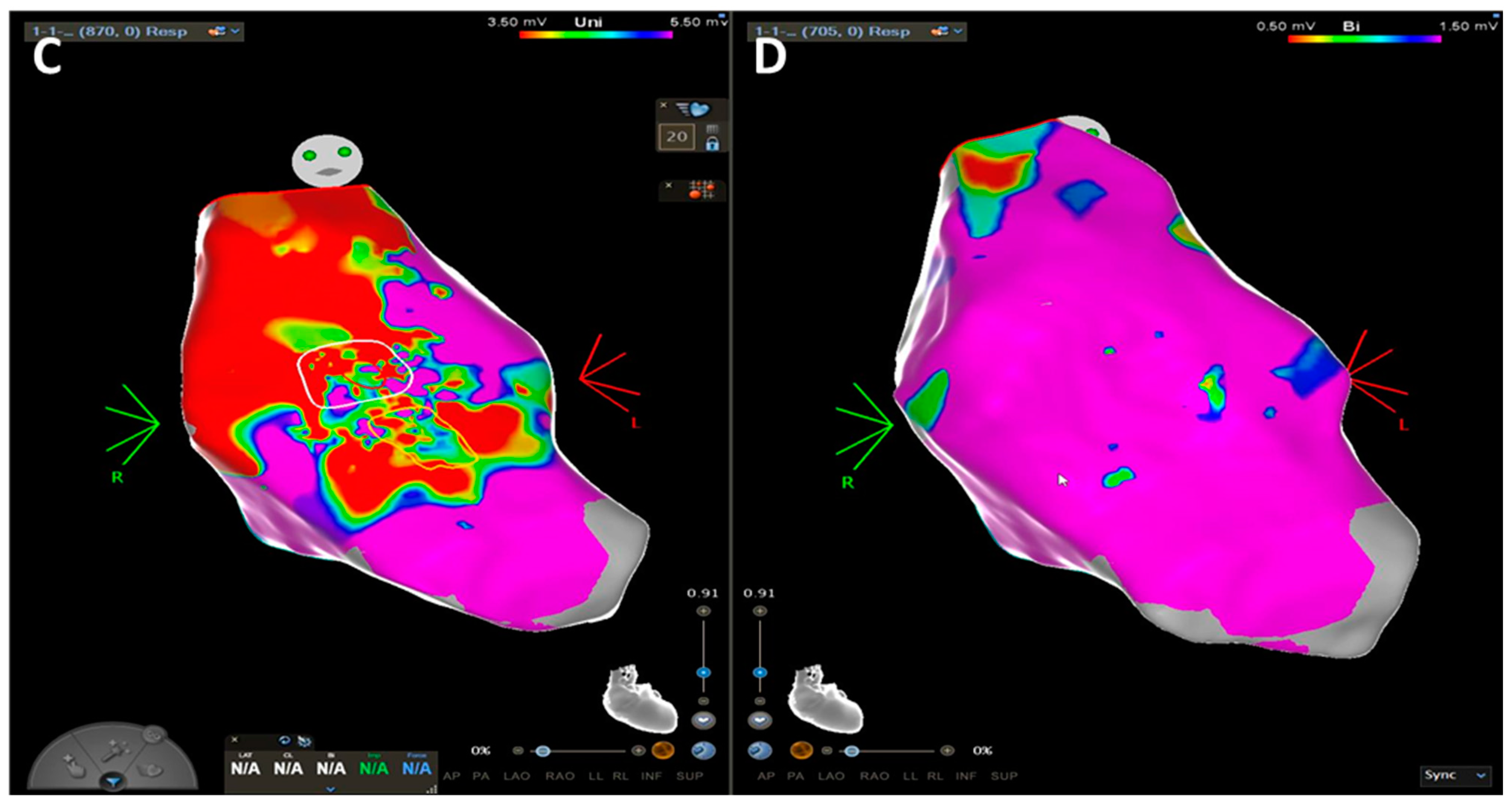
- Boulos, M.; Lashevsky, I.; Reisner, S.; Gepstein, L. Electroanatomic Mapping of Arrhythmogenic Right Ventricular Dysplasia. J. Am. Coll. Cardiol. 2001, 38, 2020–2027. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Leoni, L.; Tokajuk, B.; Bauce, B.; Frigo, G.; Tarantini, G.; Napodano, M.; Turrini, P.; Ramondo, A.; et al. Three-Dimensional Electroanatomic Voltage Mapping Increases Accuracy of Diagnosing Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation 2005, 111, 3042–3050. [Google Scholar] [CrossRef]
- Santangeli, P.; Zado, E.S.; Supple, G.E.; Haqqani, H.M.; Garcia, F.C.; Tschabrunn, C.M.; Callans, D.J.; Lin, D.; Dixit, S.; Hutchinson, M.D.; et al. Long-Term Outcome with Catheter Ablation of Ventricular Tachycardia in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Arrhythm Electrophysiol. 2015, 8, 1413–1421. [Google Scholar] [CrossRef]
- Berg, J.; Villatore, A.; Sivilotti, F.; Pili, G.; Sala, S.; Della Bella, P.; Peretto, G. Diagnostic Value of Electroanatomical Mapping in Patients with Suspected Arrhythmogenic Cardiomyopathy. Eur. Heart J. 2022, 43 (Suppl. S2), ehac544.661. [Google Scholar] [CrossRef]
- Chung, F.P.; Lin, C.Y.; Lin, Y.J.; Chang, S.L.; Lo, L.W.; Hu, Y.F.; Tuan, T.C.; Chao, T.F.; Liao, J.N.; Chang, T.Y.; et al. Catheter Ablation of Ventricular Tachycardia in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Korean Circ. J. 2018, 48, 890–905. [Google Scholar] [CrossRef]
- Wijnmaalen, A.P.; van der Geest, R.J.; van Huls van Taxis, C.F.B.; Siebelink, H.-M.J.; Kroft, L.J.M.; Bax, J.J.; Reiber, J.H.C.; Schalij, M.J.; Zeppenfeld, K. Head-to-Head Comparison of Contrast-Enhanced Magnetic Resonance Imaging and Electroanatomical Voltage Mapping to Assess Post-Infarct Scar Characteristics in Patients with Ventricular Tachycardias: Real-Time Image Integration and Reversed Registration. Eur. Heart J. 2011, 32, 104–114. [Google Scholar] [CrossRef]
- Mahida, S.; Venlet, J.; Saguner, A.M.; Kumar, S.; Baldinger, S.H.; AbdelWahab, A.; Tedrow, U.B.; Castelletti, S.; Pantazis, A.; John, R.M.; et al. Ablation Compared with Drug Therapy for Recurrent Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy: Results from a Multicenter Study. Heart Rhythm 2019, 16, 536–543. [Google Scholar] [CrossRef]
- Muser, D.; Santangeli, P.; Castro, S.A.; Pathak, R.K.; Liang, J.J.; Hayashi, T.; Magnani, S.; Garcia, F.C.; Hutchinson, M.D.; Supple, G.G.; et al. Long-Term Outcome after Catheter Ablation of Ventricular Tachycardia in Patients with Nonischemic Dilated Cardiomyopathy. Circ. Arrhythm Electrophysiol. 2016, 9, e004328. [Google Scholar] [CrossRef]
- Soejima, K.; Stevenson, W.G.; Sapp, J.L.; Selwyn, A.P.; Couper, G.; Epstein, L.M. Endocardial and Epicardial Radiofrequency Ablation of Ventricular Tachycardia Associated with Dilated Cardiomyopathy: The Importance of Low-Voltage Scars. J. Am. Coll. Cardiol. 2004, 43, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Hsia, H.H.; Callans, D.J.; Marchlinski, F.E. Characterization of Endocardial Electrophysiological Substrate in Patients with Nonischemic Cardiomyopathy and Monomorphic Ventricular Tachycardia. Circulation 2003, 108, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K. Ventricular Tachycardia Ablation in Nonischemic Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, D.M.; Vassallo, J.A.; Miller, J.M.; Poll, D.S.; Buxton, A.E.; Marchlinski, F.E.; Josephson, M.E. Endocardial Catheter Mapping in Patients in Sinus Rhythm: Relationship to Underlying Heart Disease and Ventricular Arrhythmias. Circulation 1986, 73, 645–652. [Google Scholar] [CrossRef]
- Nakahara, S.; Tung, R.; Ramirez, R.J.; Michowitz, Y.; Vaseghi, M.; Buch, E.; Gima, J.; Wiener, I.; Mahajan, A.; Boyle, N.G.; et al. Characterization of the Arrhythmogenic Substrate in Ischemic and Nonischemic Cardiomyopathy Implications for Catheter Ablation of Hemodynamically Unstable Ventricular Tachycardia. J. Am. Coll. Cardiol. 2010, 55, 2355–2365. [Google Scholar] [CrossRef]
- Hutchinson, M.D.; Gerstenfeld, E.P.; Desjardins, B.; Bala, R.; Riley, M.P.; Garcia, F.C.; Dixit, S.; Lin, D.; Tzou, W.S.; Cooper, J.M.; et al. Endocardial Unipolar Voltage Mapping to Detect Epicardial Ventricular Tachycardia Substrate in Patients with Nonischemic Left Ventricular Cardiomyopathy. Circ. Arrhythm Electrophysiol. 2011, 4, 49–55. [Google Scholar] [CrossRef]
- Oloriz, T.; Silberbauer, J.; Maccabelli, G.; Mizuno, H.; Baratto, F.; Kirubakaran, S.; Vergara, P.; Bisceglia, C.; Santagostino, G.; Marzi, A.; et al. Catheter Ablation of Ventricular Arrhythmia in Nonischemic Cardiomyopathy: Anteroseptal versus Inferolateral Scar Sub-Types. Circ. Arrhythm Electrophysiol. 2014, 7, 414–423. [Google Scholar] [CrossRef]
- Kumar, S.; Androulakis, A.F.A.; Sellal, J.-M.; Maury, P.; Gandjbakhch, E.; Waintraub, X.; Rollin, A.; Richard, P.; Charron, P.; Baldinger, S.H.; et al. Multicenter Experience with Catheter Ablation for Ventricular Tachycardia in Lamin A/C Cardiomyopathy. Circ. Arrhythm Electrophysiol. 2016, 9, e004357. [Google Scholar] [CrossRef]
- Cano, O.; Hutchinson, M.; Lin, D.; Garcia, F.; Zado, E.; Bala, R.; Riley, M.; Cooper, J.; Dixit, S.; Gerstenfeld, E.; et al. Electroanatomic Substrate and Ablation Outcome for Suspected Epicardial Ventricular Tachycardia in Left Ventricular Nonischemic Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 799–808. [Google Scholar] [CrossRef]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. Hypertrophic Cardiomyopathy Outcomes Investigators. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Maron, M.S.; Rowin, E.J.; Wessler, B.S.; Mooney, P.J.; Fatima, A.; Patel, P.; Koethe, B.C.; Romashko, M.; Link, M.S.; Maron, B.J. Enhanced American College of Cardiology/American Heart Association Strategy for Prevention of Sudden Cardiac Death in High-Risk Patients with Hypertrophic Cardiomyopathy. JAMA Cardiol. 2019, 4, 644–657. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Pinamonti, B.; Dragos, A.M.; Pyxaras, S.A.; Merlo, M.; Pivetta, A.; Barbati, G.; Di Lenarda, A.; Morgera, T.; Mestroni, L.; Sinagra, G. Prognostic Predictors in Arrhythmogenic Right Ventricular Cardiomyopathy: Results from a 10-Year Registry. Eur. Heart J. 2011, 32, 1105–1113. [Google Scholar] [CrossRef]
- Bosman, L.P.; Sammani, A.; James, C.A.; Cadrin-Tourigny, J.; Calkins, H.; van Tintelen, J.P.; Hauer, R.N.W.; Asselbergs, F.W.; Te Riele, A.S.J.M. Predicting Arrhythmic Risk in Arrhythmogenic Right Ventricular Cardiomyopathy: A Systematic Review and Meta-Analysis. Heart Rhythm 2018, 15, 1097–1107. [Google Scholar] [CrossRef]
- Saguner, A.M.; Vecchiati, A.; Baldinger, S.H.; Rüeger, S.; Medeiros-Domingo, A.; Mueller-Burri, A.S.; Haegeli, L.M.; Biaggi, P.; Manka, R.; Lüscher, T.F.; et al. Different Prognostic Value of Functional Right Ventricular Parameters in Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circ. Cardiovasc. Imaging 2014, 7, 230–239. [Google Scholar] [CrossRef]
- Link, M.S.; Laidlaw, D.; Polonsky, B.; Zareba, W.; McNitt, S.; Gear, K.; Marcus, F.; Estes, N.A.M. Ventricular Arrhythmias in the North American Multidisciplinary Study of ARVC: Predictors, Characteristics, and Treatment. J. Am. Coll. Cardiol. 2014, 64, 119–125. [Google Scholar] [CrossRef]
- Sharif, Z.I.; Lubitz, S.A. Ventricular Arrhythmia Management in Patients with Genetic Cardiomyopathies. Heart Rhythm O2 2021, 2, 819–831. [Google Scholar] [CrossRef]
- Hodgkinson, K.A.; Connors, S.P.; Merner, N.; Haywood, A.; Young, T.-L.; McKenna, W.J.; Gallagher, B.; Curtis, F.; Bassett, A.S.; Parfrey, P.S. The Natural History of a Genetic Subtype of Arrhythmogenic Right Ventricular Cardiomyopathy Caused by a p.S358L Mutation in TMEM43. Clin. Genet. 2013, 83, 321–331. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inheritance | Signs or Symptoms of Multiorgan Involvement | ECG Abnormalities beyond LVH Criteria | Routine Laboratory Tests | Echocardiography | CMR (LGE) | |
|---|---|---|---|---|---|---|
| Athlete’s heart | None | Uncommon | Isolated LVH | Not specific | LVH symmetrical or eccentric (mild-to-moderate); Normal systolic and diastolic function | Negative |
| Hypertensive heart | None | Uncommon | ST and T abnormalities | Not specific | LVH usually concentric (mild-to-moderate) | Mild degree; no specific pattern |
| HCM | AD | Uncommon | High LVH; ST and T abnormalities; Giant T wave inversion; Q waves | Not specific | Moderate-to-severe LVH (asymmetrical and septal, potentially found at any location); diastolic dysfunction, LVOT obstruction, mitral valve abnormalities (mitral SAM, leaflets and chordal elongation, dysplasia, prolapse, hypermobility); atrial enlargement; apical aneurysm | Frequent; RV insertion points and intramural; potentially found at any location |
| Anderson–Fabry disease | X-linked | Visual impairment; sensorineural deafness; paresthesiae and sensory abnormalities; angiokeratoma | Short P-R/preexcitation; AV block | Proteinuria with or without glomerular filtration rate | Concentric LVH; increased atrioventricular valve and RV free wall thickness; global hypokinesia (with/without LV dilatation) | Frequent; posterolateral in concentric LVH |
| Familial amyloidosis | AD | Visual impairment; paresthesiae and sensory abnormalities; carpal tunnel syndrome (bilateral) | Low QRS voltage; AV block | Proteinuria with or without glomerular filtration rate | Increased interatrial septum, atrioventricular valve, and RV free wall thickness; pericardial effusion; myocardium’s ground-glass appearance; global hypokinesia (with/without LV dilatation) | Frequent; diffuse subendocardial “zebra” pattern; intense myocardial “avidity” for gadolinium |
| Danon disease | X-linked | Learning difficulties, mental retardation; visual impairment | Short P-R/preexcitation; AV block; extreme LVH (Sokolow > 100) | ↑Creatine kinase ↑Transaminase | Extreme concentric LVH; global hypokinesia (with/without LV dilatation) | Frequent; large amount subendocardial or transmural |
| Mitochondrial CMP | X-linked or matrilinear | Sensorineural deafness; learning difficulties, mental retardation; visual impairment; muscle weakness | Short P-R/preexcitation | ↑Creatine kinase ↑Transaminase Lactic acidosis | Global hypokinesia (with/without LV dilatation) | Frequent; large amount nonischemic intramural pattern mostly basal LV inferolateral wall |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzeroni, D.; Crocamo, A.; Ziveri, V.; Notarangelo, M.F.; Rizzello, D.; Spoladori, M.; Donelli, D.; Cacciola, G.; Ardissino, D.; Niccoli, G.; et al. Personalized Management of Sudden Death Risk in Primary Cardiomyopathies: From Clinical Evaluation and Multimodality Imaging to Ablation and Cardioverter-Defibrillator Implant. J. Pers. Med. 2023, 13, 877. https://doi.org/10.3390/jpm13050877
Lazzeroni D, Crocamo A, Ziveri V, Notarangelo MF, Rizzello D, Spoladori M, Donelli D, Cacciola G, Ardissino D, Niccoli G, et al. Personalized Management of Sudden Death Risk in Primary Cardiomyopathies: From Clinical Evaluation and Multimodality Imaging to Ablation and Cardioverter-Defibrillator Implant. Journal of Personalized Medicine. 2023; 13(5):877. https://doi.org/10.3390/jpm13050877
Chicago/Turabian StyleLazzeroni, Davide, Antonio Crocamo, Valentina Ziveri, Maria Francesca Notarangelo, Davide Rizzello, Matteo Spoladori, Davide Donelli, Giovanna Cacciola, Diego Ardissino, Giampaolo Niccoli, and et al. 2023. "Personalized Management of Sudden Death Risk in Primary Cardiomyopathies: From Clinical Evaluation and Multimodality Imaging to Ablation and Cardioverter-Defibrillator Implant" Journal of Personalized Medicine 13, no. 5: 877. https://doi.org/10.3390/jpm13050877