
The First Eight Mitogenomes of Leaf-Mining Dactylispa Beetles (Coleoptera: Chrysomelidae: Cassidinae) Shed New Light on Subgenus Relationships

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Genomic Sequencing, Assembly, and Annotation
2.3. Bioinformatic Analysis
2.4. Phylogenetic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subgenus | Species | GenBank No. | Size (bp) | Total A + T % | AT% of All PCGs | References |
|---|---|---|---|---|---|---|
| Dactylispa s. str. | Dactylispa approximata | MN016958 | 18,896 | 75.6 | 75.5 | This study |
| Dactylispa s. str. | Dactylispa albopilosa | MN016964 | 18,774 | 70.9 | 70.5 | This study |
| Dactylispa s. str. | Dactylispa longispina | MN016959 | 20,363 | 71.2 | 70.6 | This study |
| Triplispa | Dactylispa paucispina | MN016963 | 18,116 | 74.9 | 74.7 | This study |
| Triplispa | Dactylispa nigrodiscalis | MN016961 | 17,189 | 75.3 | 75.1 | This study |
| Triplispa | Dactylispa xanthopus | MN016966 | 19,581 | 75 | 74.8 | This study |
| Platypriella | Dactylispa planispina | MN016960 | 18,530 | 73.9 | 73.5 | This study |
| Platypriella | Dactylispa latispina | MN016962 | 17,963 | 74.9 | 74.5 | This study |
| OUTGROUP | Agnoita chinensis | MF351622 | 17,866 | 79.1 | 78.4 | [41] |
| OUTGROUP | Downesia tarsata | MW176089 | 18,557 | 78.6 | 76.3 | [42] |
3. Results
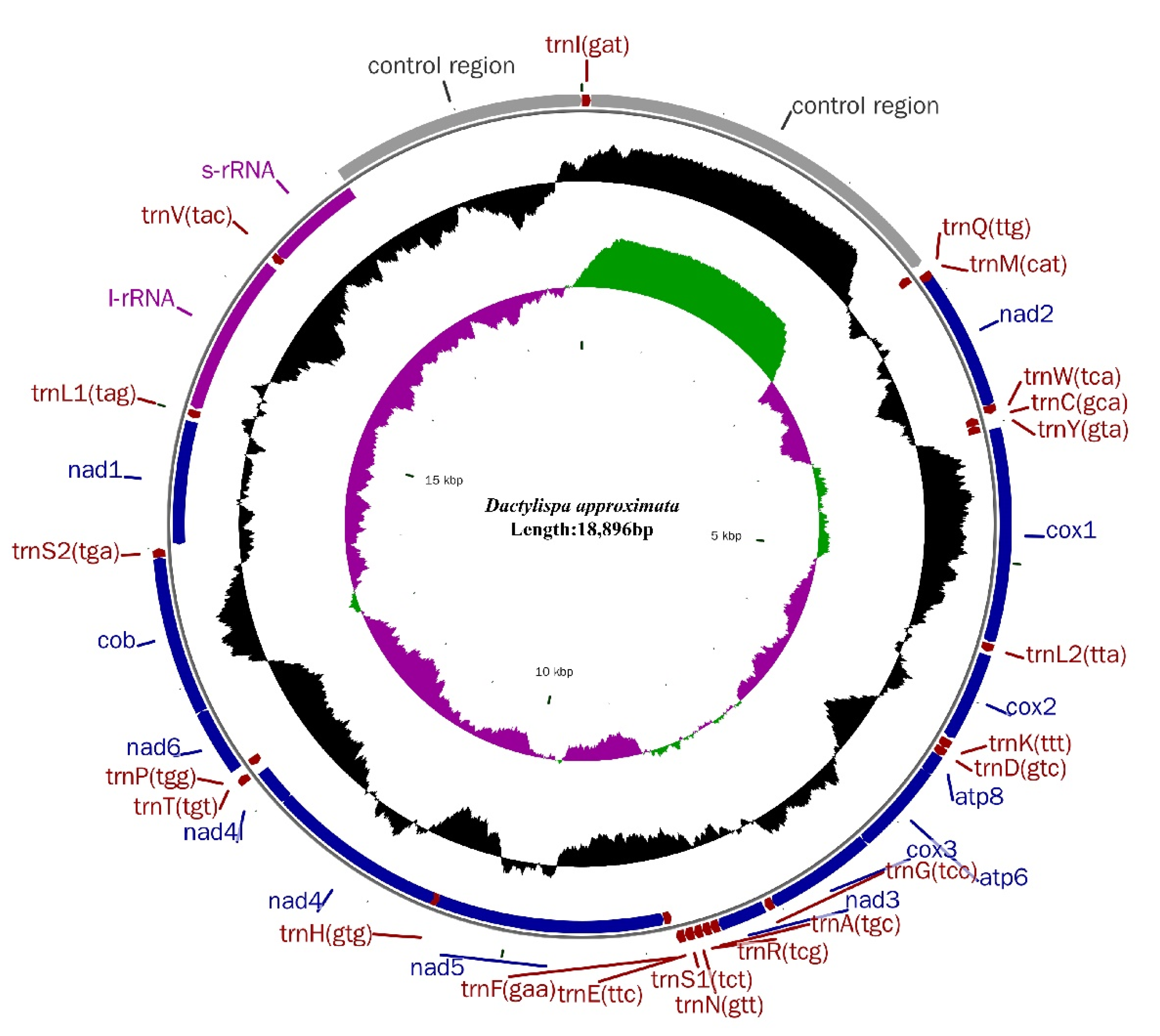
3.1. Genome Organization and Nucleotide Composition
3.2. Protein-Coding Genes
3.3. Codon Usage
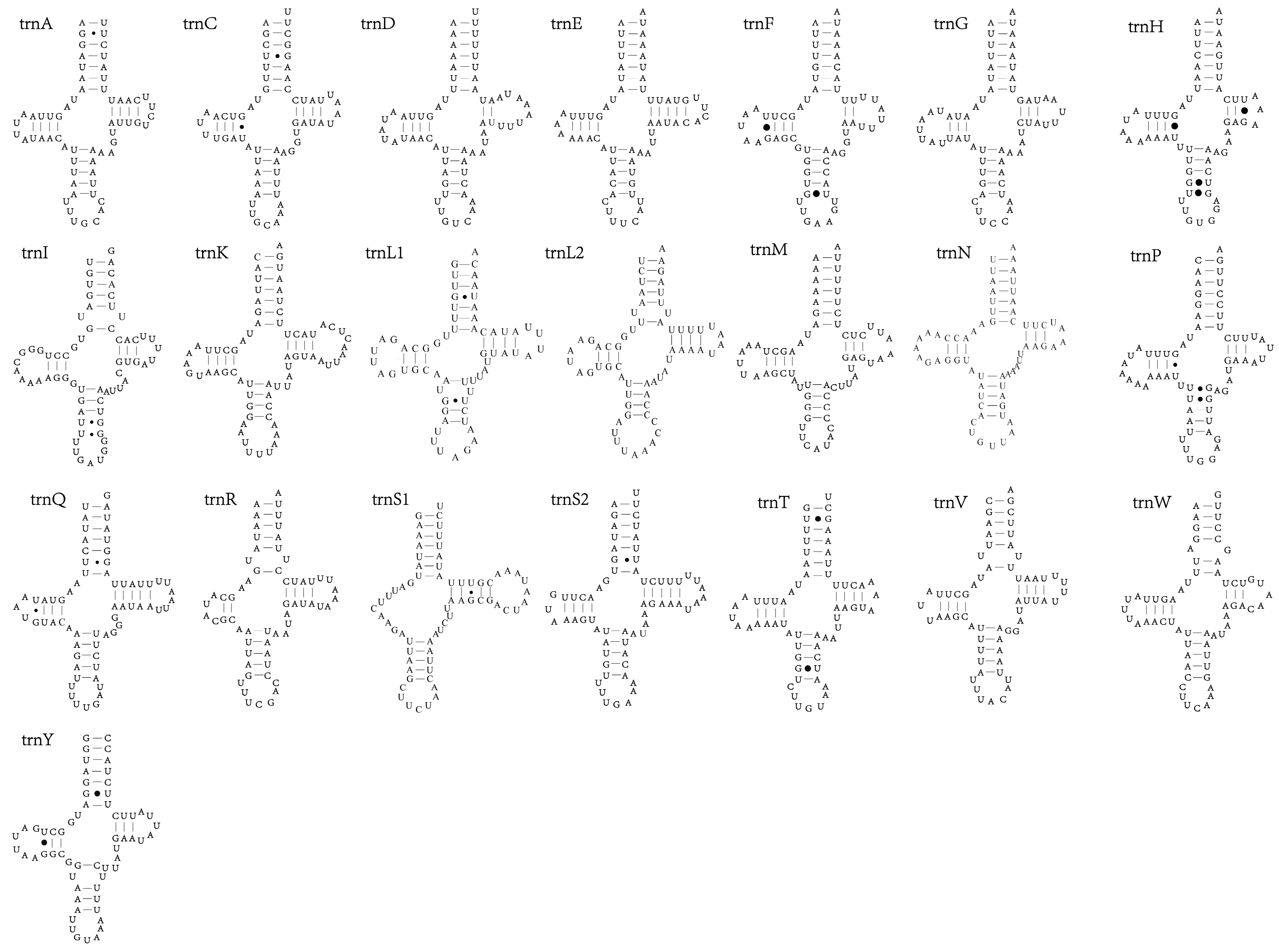
3.4. tRNAs and rRNAs
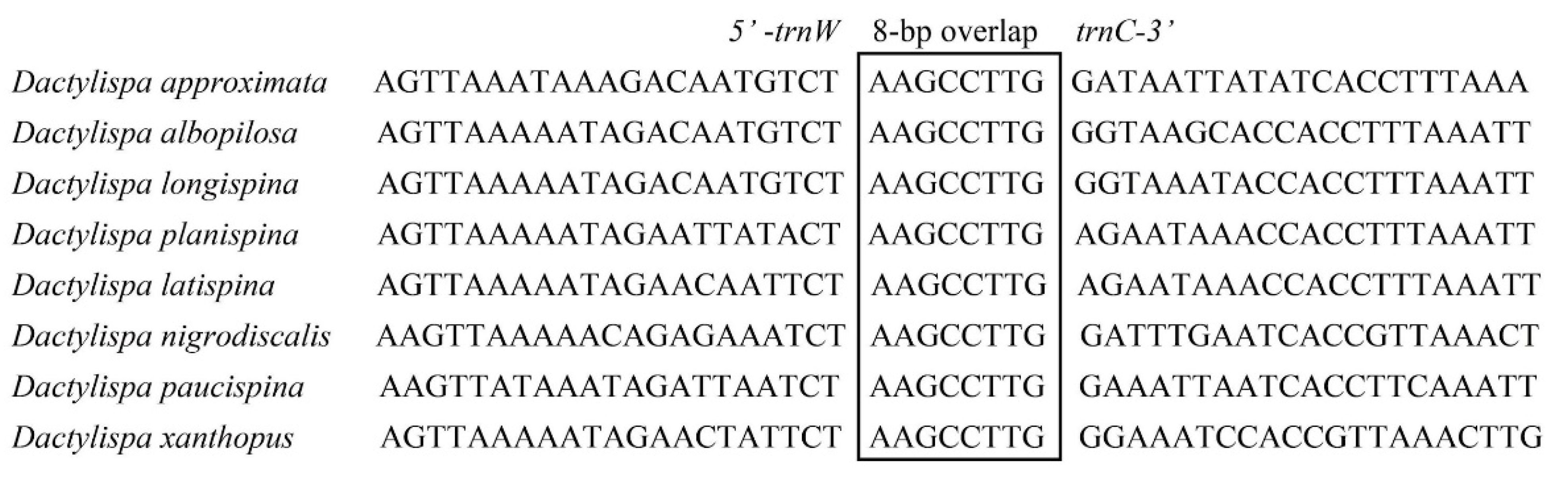
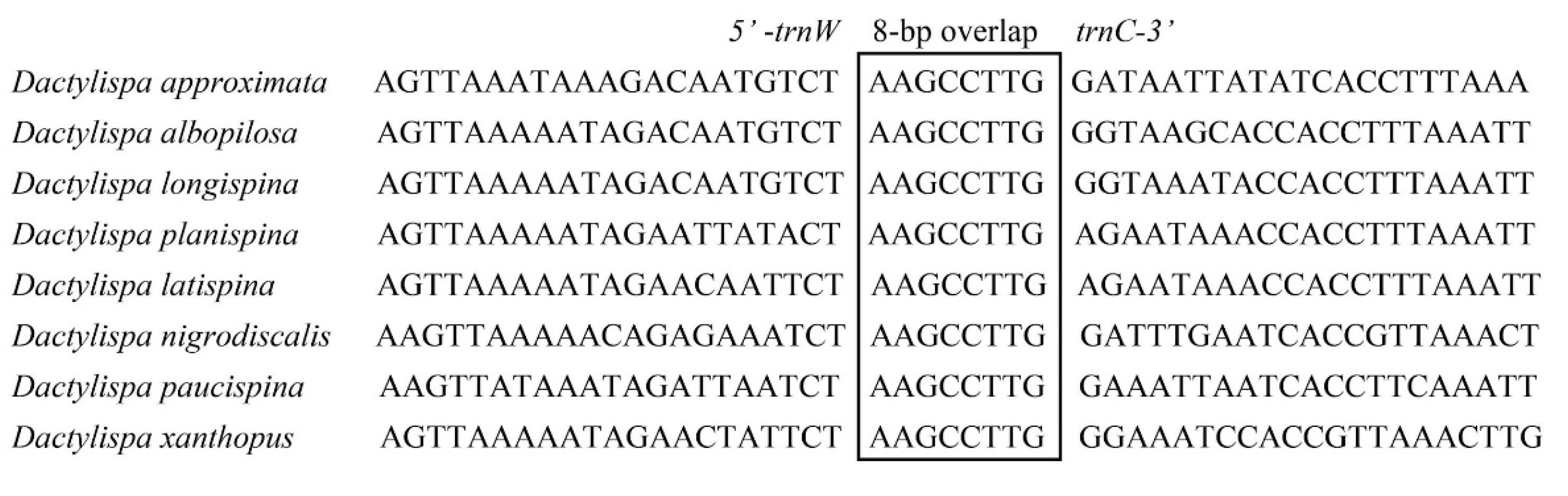
3.5. Gene Overlap, Intergenic Spacers and Noncoding A + T-Rich Regions
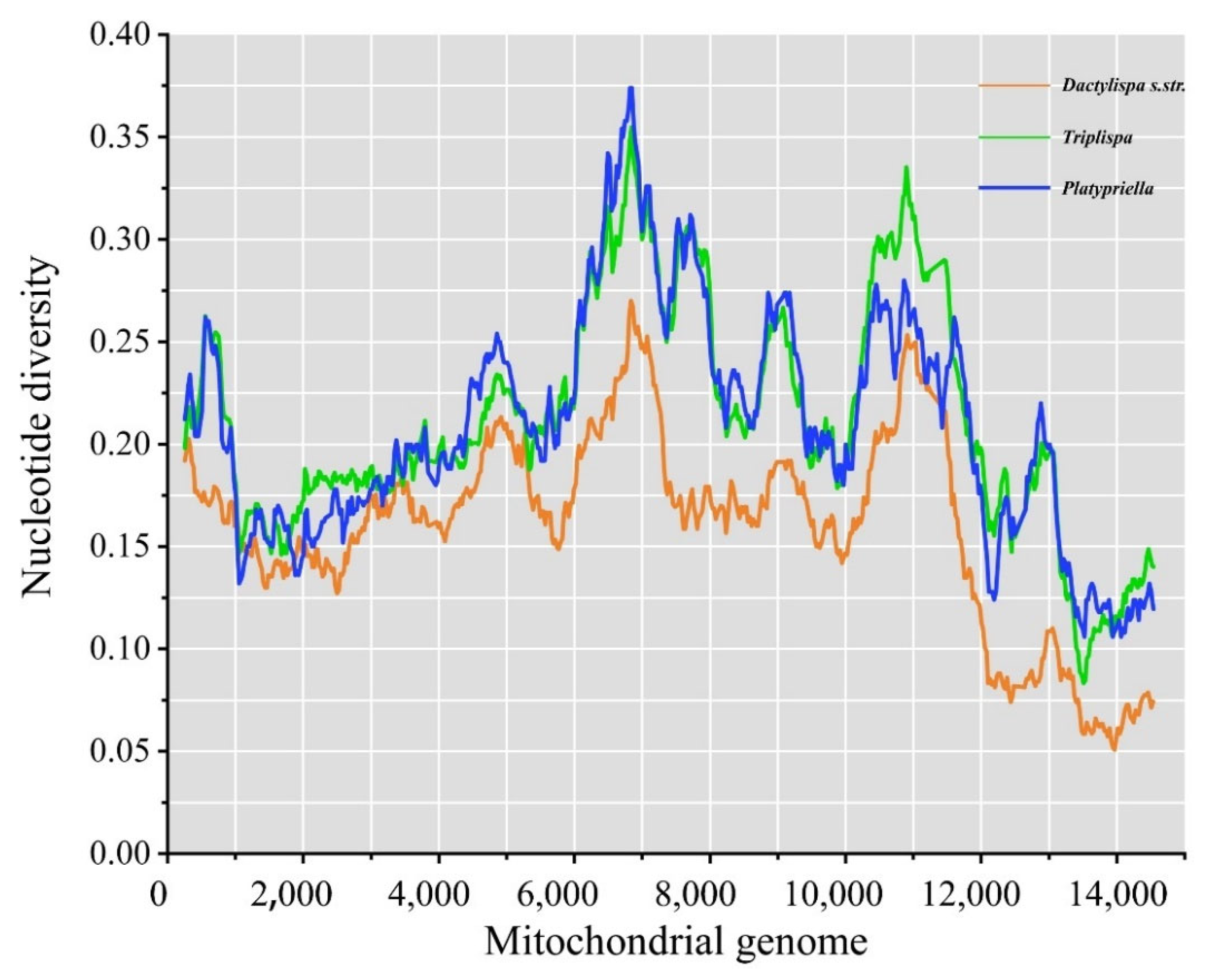
3.6. Genetic Diversity among Mitogenomes
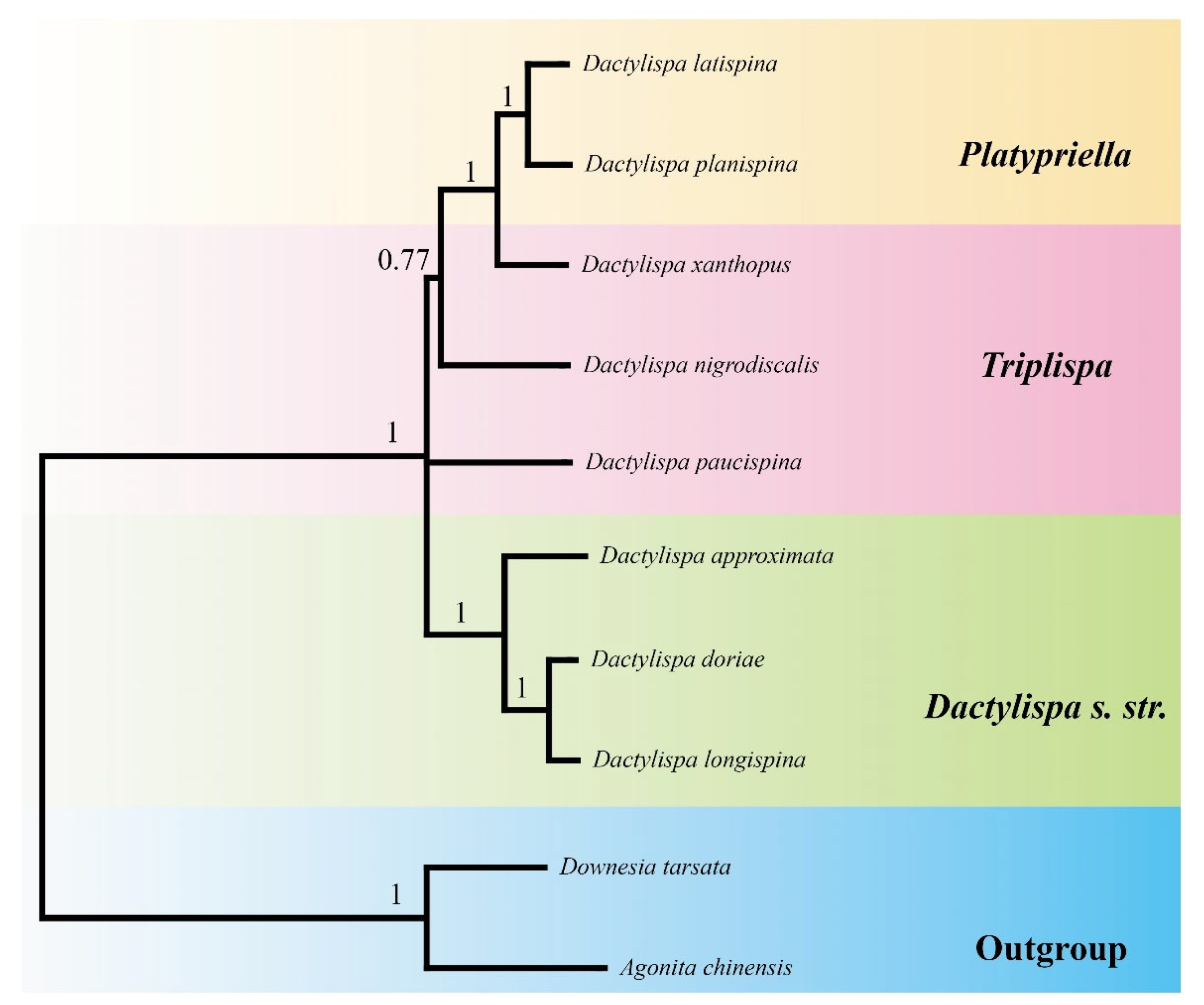
3.7. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, S.H.; Yu, P.Y.; Sun, C.H.; T’an, C.H.; Zia, Y. Fauna Sinica (Insecta: Coleoptera: Hispidae); Science Press: Beijing, China, 1986. [Google Scholar]
- Liao, C. Diversity and Host Relationship of Leaf-Mining Hispine Beetle. Master’s Thesis, Gannan Normal University, Ganzhou, China, 2015. [Google Scholar]
- Liao, C.; Xu, J.; Dai, X.; Zhao, X. Species diversity of leaf-mining hispines and of their host plants. Ecol. Sci. 2015, 34, 159–166. [Google Scholar]
- Staines, C. Catalog of the Hispines of the World (Coleoptera: Chrysomelidae: Cassidinae). Available online: https://naturalhistory.si.edu/research/entomology/collections-overview/coleoptera/catalog-hispines-world (accessed on 25 December 2016).
- Santiago-Blay, J.A. Leaf-mining chrysomelids. In New Developments on the Biology of Chrysomelidae; SPB Academic Publishing: The Hague, the Netherlands, 2004; pp. 305–306. [Google Scholar]
- Banwo, O.O.; Makundi, R.H.; Abdallah, R.S.; Mbapila, J.C. First Report of Dactylispa lenta Weise (Coleoptera Chrysomelidae) as a Vector of Rice Yellow Mottle Virus. Acta Phytopathol. Entomol. Hung. 2001, 36, 189–192. [Google Scholar]
- Gupta, R.; Tara, J.S.; Chhetry, M. Bionomics of Dactylispa Dohertyi (Gestro, 1897), a new pest of apple plantations (Malus domestica Borkh.) in Jammu Region of J & K, India. Munis Entomol. Zool. 2012, 7, 754–758. [Google Scholar]
- Maulik, S. The Fauna of British India including Ceylon and Burma: Coleoptera: Chrysomelidae (Hispinae and Cassidinae); Taylor and Francis: London, UK, 1919. [Google Scholar]
- Uhmann, E. LXV.—Hispinae aus dem Britischen Museum.—VIII. Teil. 156. Beitrag zur Kenntnis der Hispinae (Coleopt., Chrysom.). Ann. Mag. Nat. Hist. 1954, 7, 497–518. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Sayadi, A.; Immonen, E.; Tellgren-Roth, C.; Arnqvist, G. The Evolution of Dark Matter in the Mitogenome of Seed Beetles. Genome Biol. Evol. 2017, 9, 2697–2706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Hewitt, G.M. Insect Mitochondrial Control Region: A Review of its Structure, Evolution and Usefulness in Evolutionary Studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- DeSalle, R.; Schierwater, B.; Hadrys, H. MtDNA: The small workhorse of evolutionary studies. Front. Biosci. Landmark 2017, 22, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Saccone, C.; Gissi, C.; Reyes, A.; Larizza, A.; Pesole, G. Mitochondrial DNA in metazoa: Degree of freedom in a frozen event. Genes 2002, 286, 3–12. [Google Scholar] [CrossRef]
- Xiao, L.; Zhang, S.; Long, C.; Guo, Q.; Xu, J.; Dai, X.; Wang, J. Complete Mitogenome of a Leaf-Mining Buprestid Beetle, Trachys auricollis, and Its Phylogenetic Implications. Genes 2019, 10, 992. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Xu, J.; Jiang, Z. Bionomics of Dactylispa approximata on Lophatherum gracile. North. Hortic. 2012, 22, 125–127. [Google Scholar]
- Lee, C. The taxonomic status of Dactylispa taiwana Takizawa, 1978 (Coleoptera: Chrysomelidae: Cassidinae). Genus. Int. J. Invertebr. Taxon. 2009, 1, 109–110. [Google Scholar]
- Wen, L.U.; Zhang, Y.; Huang, C. Study on the economic threshold of Dactylispa setifera (Chapuis). J. Agric. Biol. Sci. 2003, 22, 29–31. [Google Scholar]
- Zheng, X.L.; Zhang, Y.J.; Wang, Y.L.; Dong, Z.S.; Da-Xing, H.U.; Wen, L.U.; University, G. Observation on digestive system in Dactylispa setifera Chapuis(Coleoptera:Hispidae). J. South. Agric. 2016, 47, 223–226. [Google Scholar]
- Zaitsev, Y.M. The immature stages of the leaf-beetle genus Dactylispa (Coleoptera, Chrysomelidae) from Vietnam. Entomol. Rev. 2012, 92, 305–314. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Quant. Biol. 2014, 31, 1–3. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Nabil-Fareed, A.; Petty, N.K.; Zakour, N.L.B.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 1–10. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Res. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Guo, Q.; Xu, J.; Liao, C.; Dai, X.; Jiang, X. Complete mitochondrial genome of a leaf-mining beetle, Agonita chinensis Weise (Coleoptera: Chrysomelidae). Mitochondrial DNA B Resour. 2017, 2, 532–533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Guo, Q.; Xu, J.; Wang, X.; Dai, X. The complete mitochondrial genome of Downesia tarsata (Coleoptera: Chrysomelidae: Cassidinae). Mitochondrial DNA B Resour. 2021, 6, 1073–1074. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Zhang, L.; Guo, Z.L.; Liu, Y.J.; Shen, Y.Y.; Shao, R. High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenet. Evol. 2016, 104, 99–111. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Wang, H.L.; Yang, J.; Boykin, L.M.; Zhao, Q.Y.; Li, Q.; Wang, X.W.; Liu, S.S. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genom. 2013, 14, 401. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Complete Mitochondrial Genome Sequence of the Polychaete Annelid Platynereis dumerilii. Mol. Biol. Evol. 2001, 18, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Wang, X.Q.; Li, P.W.; Hu, X.; Wang, J.J.; Peng, P. The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae. Int. J. Mol. Sci. 2016, 17, 1843. [Google Scholar] [CrossRef] [Green Version]
- Su, T.; Liang, A. Comparative analysis of seven mitochondrial genomes of Phymatostetha (Hemiptera: Cercopidae) and phylogenetic implications. Int. J. Biol. Macromol. 2019, 125, 1112–1117. [Google Scholar] [CrossRef]
- Ren, L.; Shang, Y.; Yang, L.; Shen, X.; Chen, W.; Wang, Y.; Cai, J.; Guo, Y. Comparative analysis of mitochondrial genomes among four species of muscid flies (Diptera: Muscidae) and its phylogenetic implications. Int. J. Biol. Macromol. 2019, 127, 357–364. [Google Scholar] [CrossRef]
- Hong, M.Y.; Jeong, H.C.; Kim, M.J.; Jeong, H.U.; Lee, S.H.; Kim, I. Complete mitogenome sequence of the jewel beetle, Chrysochroa fulgidissima (Coleoptera: Buprestidae). Mitochondrial DNA 2009, 20, 46–60. [Google Scholar] [CrossRef]
- Amorim, I.C.; Melo, A.S.; Cruz, G.A.d.S.; Wallau, G.d.L.; Moura, R.d.C.d. Dichotomius (Luederwaldtinia) schiffleri (Coleoptera: Scarabaeidae) mitochondrial genome and phylogenetic relationships within the superfamily Scarabaeoidea. Mitochondrial DNA Part B 2017, 2, 887–888. [Google Scholar] [CrossRef] [Green Version]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.D. The Ka/Ks ratio:diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Tomiczek, B.; Sojo, V.; dos Reis, M. A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol. Biol. 2015, 1201, 65–90. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Nie, R.-E.; Yang, X.-K. Research progress in mitochondrial genomes of Coleoptera. Acta Biochim. Biophys. Sin. 2014, 57, 860–868. [Google Scholar]
- Hebert, P.D.; Ratnasingham, S.; de Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 2003, 270 (Suppl. 1), S96–S99. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, D.C.; Raychoudhury, R.; Lavrov, D.V.; Werren, J.H. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp nasonia (hymenoptera: Pteromalidae). Mol. Biol. Evol. 2008, 25, 2167–2180. [Google Scholar] [CrossRef] [Green Version]
- Smietanka, B.; Burzynski, A.; Wenne, R. Comparative genomics of marine mussels (Mytilus spp.) gender associated mtDNA: Rapidly evolving atp8. J. Mol. Evol. 2010, 71, 385–400. [Google Scholar] [CrossRef]
- Shen, X.; Li, X.; Sha, Z.; Yan, B.; Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China Life Sci. 2012, 55, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Chaboo, C.S. Biology and phylogeny of the Cassidinae Gyllenhal sensu lato (tortoise and leaf-mining beetles) Coleoptera Chrysomelid. Bull. Am. Mus. Nat. Hist. 2007, 305, 1–250. [Google Scholar] [CrossRef] [Green Version]
- Wilf, P.; Labandeira, C.C.; Kress, W.J.; Staines, C.L.; Windsor, D.M.; Allen, A.L.; Johnson, K.R. Timing the radiations of leaf beetles: Hispines on gingers from latest cretaceous to recent. Science 2000, 289, 291–294. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Sekerka, L.; Liao, C.; Long, C.; Xu, J.; Dai, X.; Guo, Q. The First Eight Mitogenomes of Leaf-Mining Dactylispa Beetles (Coleoptera: Chrysomelidae: Cassidinae) Shed New Light on Subgenus Relationships. Insects 2021, 12, 1005. https://doi.org/10.3390/insects12111005
Zhang S, Sekerka L, Liao C, Long C, Xu J, Dai X, Guo Q. The First Eight Mitogenomes of Leaf-Mining Dactylispa Beetles (Coleoptera: Chrysomelidae: Cassidinae) Shed New Light on Subgenus Relationships. Insects. 2021; 12(11):1005. https://doi.org/10.3390/insects12111005
Chicago/Turabian StyleZhang, Shengdi, Lukáš Sekerka, Chengqing Liao, Chengpeng Long, Jiasheng Xu, Xiaohua Dai, and Qingyun Guo. 2021. "The First Eight Mitogenomes of Leaf-Mining Dactylispa Beetles (Coleoptera: Chrysomelidae: Cassidinae) Shed New Light on Subgenus Relationships" Insects 12, no. 11: 1005. https://doi.org/10.3390/insects12111005