
Five Mitochondrial Genomes of the Genus Eysarcoris Hahn, 1834 with Phylogenetic Implications for the Pentatominae (Hemiptera: Pentatomidae)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Mitogenome Sequencing
2.2. Genome Annotation and Sequence Analysis
2.3. Phylogenetic Analyses
3. Results
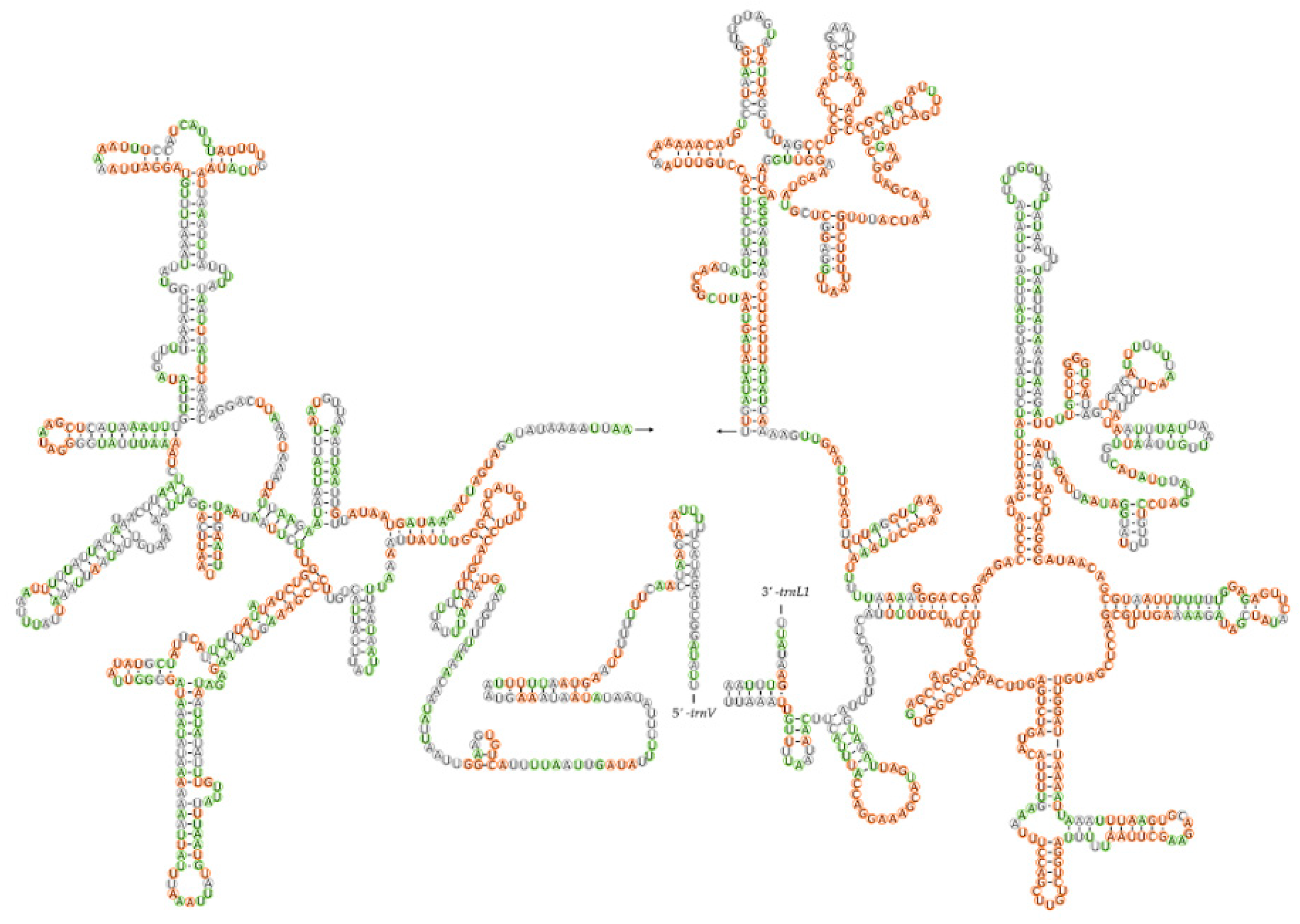
3.1. Mitochondrial Genomic Structure
3.2. Protein-Coding Genes
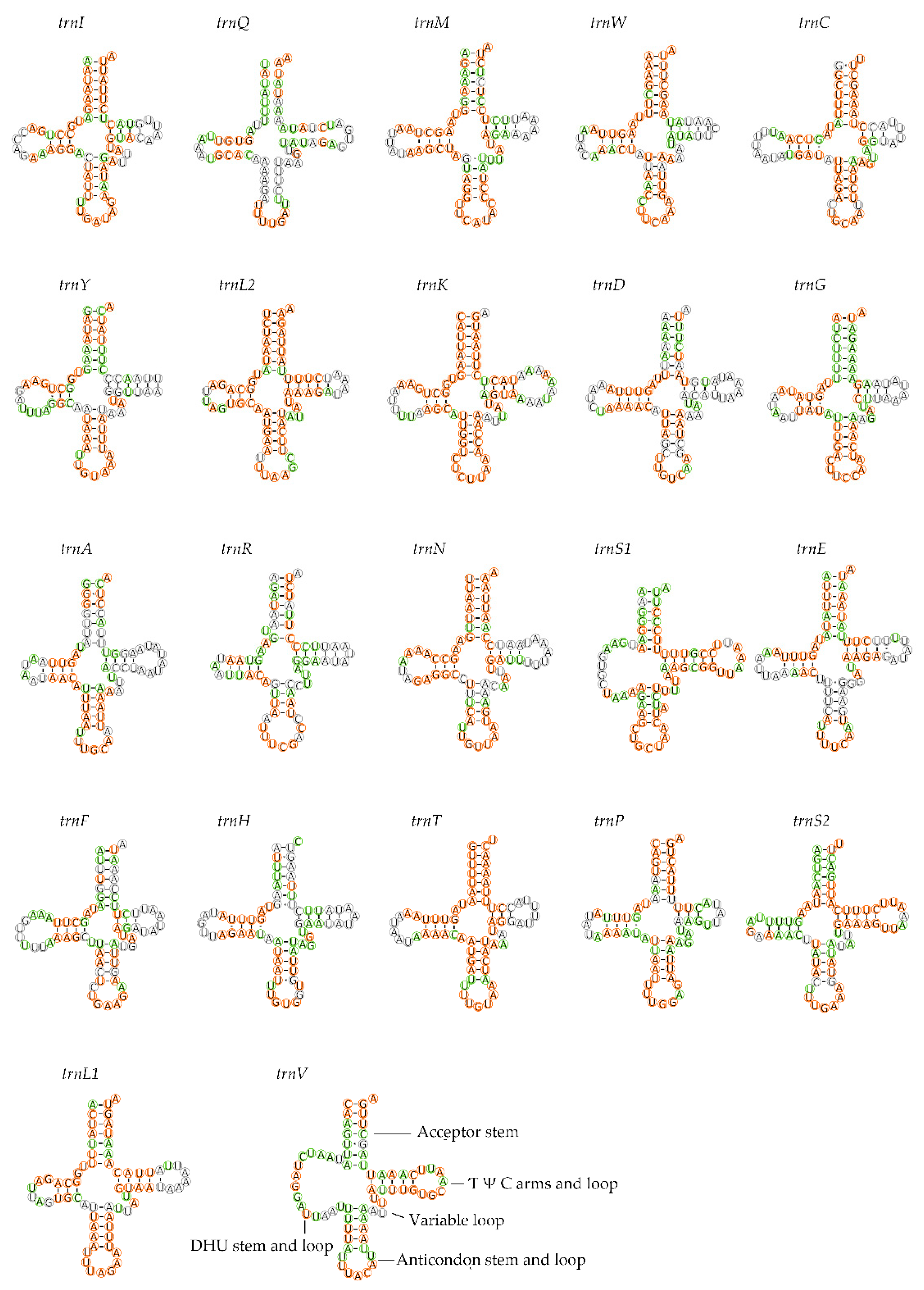
3.3. Transfer and Ribosomal RNAs
3.4. Control Region
3.5. Phylogenetic Relationships
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rider, D.A.; Schwertner, C.F.; Vilímová, J.; Rédei, D.; Kment, P.; Thomas, D.B. Higher systematics of the Pentatomoidea. In Invasive Stink Bugs and Related species (Pentatomoidea): Biology, Higher Systematics, Semiochemistry, and Management; McPherson, J.E., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 25–201. [Google Scholar]
- Distant, W.L. The Fauna of British India, including Ceylon and Burma. Rhynchota; Taylor and Francis: London, UK, 1902; Volume 1, p. 165. [Google Scholar]
- Kirkaldy, G.W. Catalogue of the Hemiptera (Heteroptera) with Biological and Anatomical References, Lists of Foodplants and Parasites, etc.; Felix, L., Ed.; Dames: Berlin, Germany, 1909; Volume 1, p. 392. [Google Scholar]
- Yang, W.Y. Economic Insect Fauna of China. Hemiptera: Pentatomidae; Science Press: Beijing, China, 1962; Fasc. 2; pp. 91–92. [Google Scholar]
- Rider, D.A. Family Pentatomidae. In Catalogue of the Heteroptera of the Palaearctic Region; Aukema, B., Rieger, C., Eds.; The Netherlands Entomological Society: Amsterdam, The Netherlands, 2006; Volume 5, pp. 298–302. [Google Scholar]
- Linnavuori, R.E. Pentatomidae and Acanthosomatidae (Heteroptera) of Nigeria and the Ivory Coast, with remarks on species of the adjacent countries in West and Central Africa. Acta Zool. Fenn. 1982, 163, 1–176. [Google Scholar]
- Wood, I.; McDonald, F.J.D. Revision of the Australian Eysarcoris group (Hemiptera: Pentatomidae). J. Aust. Entomol. Soc. 1984, 23, 253–264. [Google Scholar] [CrossRef]
- Lee, J.G.; Hong, S.S.; Kim, J.Y.; Park, K.Y.; Lim, J.W.; Lee, J.H. Occurrence of stink bugs and pecky rice damage by stink bugs in paddy fields in Gyeonggi-do, Korea. Korean J. Appl. Entomol. 2009, 48, 37–44. [Google Scholar] [CrossRef]
- Nasiruddin, M.; Roy, R.C. Rice field insect pests during the rice growing seasons in two areas of Hathazari, Chittagong. Zool. Soc. Bangl. 2012, 40, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Li, R.R.; Zhang, H.F.; Li, S.C.; Bai, M. Geometric morphometric analysis of Eysarcoris guttiger, E. annamita and E. ventralis (Hemiptera: Pentatomidae). Zool. Syst. 2017, 42, 90–101. [Google Scholar]
- Li, R.R.; Li, M.; Li, S.C.; Zhang, H.F. Further geometric morphometric analysis on the genus Eysarcoris (Hemiptera: Pentatomidae) from China. Zool. Syst. 2017, 42, 446–462. [Google Scholar]
- Li, R.R.; Li, M.; Yan, J.; Zhang, H.F. Tribe statues of Eysarcoris evaluated by scutellum geometry (Hemiptera: Pentatomidae). J. Taiyuan Norm. Univ. Nat. Sci. 2019, 18, 88–92. [Google Scholar]
- Li, R.R.; Li, M.; Zhang, H.F.; Bai, M. Intraspecific variation in Eysarcoris aeneus revealed by geometric morphometrics (Hemiptera: Pentatomidae). Acta Entomol. Sin. 2019, 62, 1081–1089. [Google Scholar]
- Zhao, Q.; Li, M.; Sun, X.; Zhang, H.F. Study on DNA taxonomy of Eysarcoris aeneus (Hemiptera: Pentatomidae: Eysarcoris). J. Shanxi Agric. Univ. Nat. Sci. 2015, 35, 241–248. [Google Scholar]
- Roca-Cusachs, M.; Jung, S. Redefining Stagonomus Gorski based on morphological and molecular data (Pentatomidae: Eysarcorini). Zootaxa 2019, 4658, 368–374. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef]
- Françoso, E.; Gomes, F.; Arias, M.C. A protocol for isolating insect mitochondrial genomes: A case study of NUMT in Melipona flavolineata (Hymenoptera: Apidae). Mitochondrial DNA 2016, 27, 2401–2404. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Liu, S.W.; Song, F.; Liu, G.F.; Yu, X.P. Comparative mitogenome analysis on species of four apple snails (Ampullariidae: Pomacea). Int. J. Biol. Macromol. 2018, 118, 525–533. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173. [Google Scholar]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Yang, P.; Jiang, F.; Chapuis, M.P.; Shali, Y.; Sword, G.A.; Kang, L. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.Y.; Xin, Z.Z.; Wang, Y.; Zhang, H.B.; Zhang, D.Z.; Wang, Z.F.; Zhou, C.L.; Tang, B.P.; Liu, Q.N. The complete mitochondrial genome of Clostera anachoreta (Lepidoptera: Notodontidae) and phylogenetic implications for Noctuoidea species. Genomics 2017, 109, 221–226. [Google Scholar] [CrossRef]
- Wu, L.W.; Chiba, H.; Lees, D.C.; Ohshima, Y.; Jeng, M.L. Unravelling relationships among the shared stripes of sailors: Mitogenomic phylogeny of Limenitidini butterflies (Lepidoptera, Nymphalidae, Limenitidinae), focusing on the genera Athyma and Limenitis. Mol. Phylogenet. Evol. 2019, 130, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Q.; Li, H.; Song, F.; Zhao, Y.S.; Wilson, J.; Cai, W.Z. Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Bolger, A.M.; Logse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucl. Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genom. 2015, 16, 460. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Wang, M.Q.; Cai, B.; Zhang, H.F.; Wei, J.F. Complete Mitochondrial Genome of Dinorhynchus dybowskyi (Hemiptera: Pentatomidae: Asopinae) and Phylogenetic Analysis of Pentatomomorpha Species. J. Insect Sci. 2018, 18, 2096–2097. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.Q.; Zhao, Q.; Li, M.; Wei, J.F.; Zhang, X.H.; Zhang, H.F. Comparative Mitogenomic Analysis of the Eurydema Genus in the Context of Representative Pentatomidae (Hemiptera: Heteroptera) Taxa. J. Insect Sci. 2019, 19, 20. [Google Scholar] [CrossRef]
- Wang, J.; Ji, Y.T.; Li, H.; Song, F.; Zhang, L.S.; Wang, M.Q. Characterization of the complete mitochondrial genome of Pentatoma semiannulata (Hemiptera: Pentatomidae). Mitochondrial DNA Part B Resour. 2021, 6, 750–752. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Zhang, Q.L.; Zhou, M.Q.; Wang, X.T.; Yang, X.Z.; Yuan, M.L. Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PerrJ 2017, 5, e3661. [Google Scholar] [CrossRef] [Green Version]
- Pita, S.; Panzera, F.; Vela, J.; Mora, P.; Palomeque, T.; Lorite, P. Complete mitochondrial genome of Triatoma infestans (Hemiptera, Reduviidae, Triatominae), main vector of Chagas disease. Infect. Genet. Evol. 2017, 54, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gao, C.Q.; Cui, Y.; Xie, Q.; Bu, W.J. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS ONE 2013, 8, e55381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navajas, M.; Conte, Y.L.; Solignac, M.; Cros-Arteil, S.; Cornuet, J.M. The complete sequence of the mitochondrial genome of the honeybee ectoparasite mite Varroa destructor (Acari: Mesostigmata). Mol. Biol. Evol. 2002, 19, 2313–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.F.; Zheng, L.Y. Studies on the cytotaxonomy of five species of the family Pentatomidae (Hemiptera: Heteroptera). Entomotaxonomia 2001, 23, 265–276. [Google Scholar]
- Puton, A. Catalogue des Hémiptères Hétéroptères d’Europe; Deyrolle: Paris, France, 1869; p. 40. [Google Scholar]
- Gross, G.F. Handbook of the Flora and Fauna of South Australia. Plant-Feeding and Other Bugs (Hemiptera) of South Australia. Heteroptera—Part 2; Handbooks Committee, South Australian Government: Adelaide, Australia, 1976; pp. 251–501.






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Li, M.; Yan, J.; Bai, M.; Zhang, H. Five Mitochondrial Genomes of the Genus Eysarcoris Hahn, 1834 with Phylogenetic Implications for the Pentatominae (Hemiptera: Pentatomidae). Insects 2021, 12, 597. https://doi.org/10.3390/insects12070597
Li R, Li M, Yan J, Bai M, Zhang H. Five Mitochondrial Genomes of the Genus Eysarcoris Hahn, 1834 with Phylogenetic Implications for the Pentatominae (Hemiptera: Pentatomidae). Insects. 2021; 12(7):597. https://doi.org/10.3390/insects12070597
Chicago/Turabian StyleLi, Rongrong, Min Li, Jiang Yan, Ming Bai, and Hufang Zhang. 2021. "Five Mitochondrial Genomes of the Genus Eysarcoris Hahn, 1834 with Phylogenetic Implications for the Pentatominae (Hemiptera: Pentatomidae)" Insects 12, no. 7: 597. https://doi.org/10.3390/insects12070597