
Understanding the Role of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi Based on Metagenomic DNA Analysis

Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Taxonomic Assignment
2.3. Analysis of Diversity of Carbohydrate-Active Enzymes Producing Freely Living Prokaryotes in the C. gestroi gut
2.4. Analysis of the Role of the Termite Gut Prokaryotes in Important Metabolic Pathways
3. Results
3.1. Overall Gut Microbial Diversity
3.2. Diversity of Free-Living Archaea in the Termite Gut
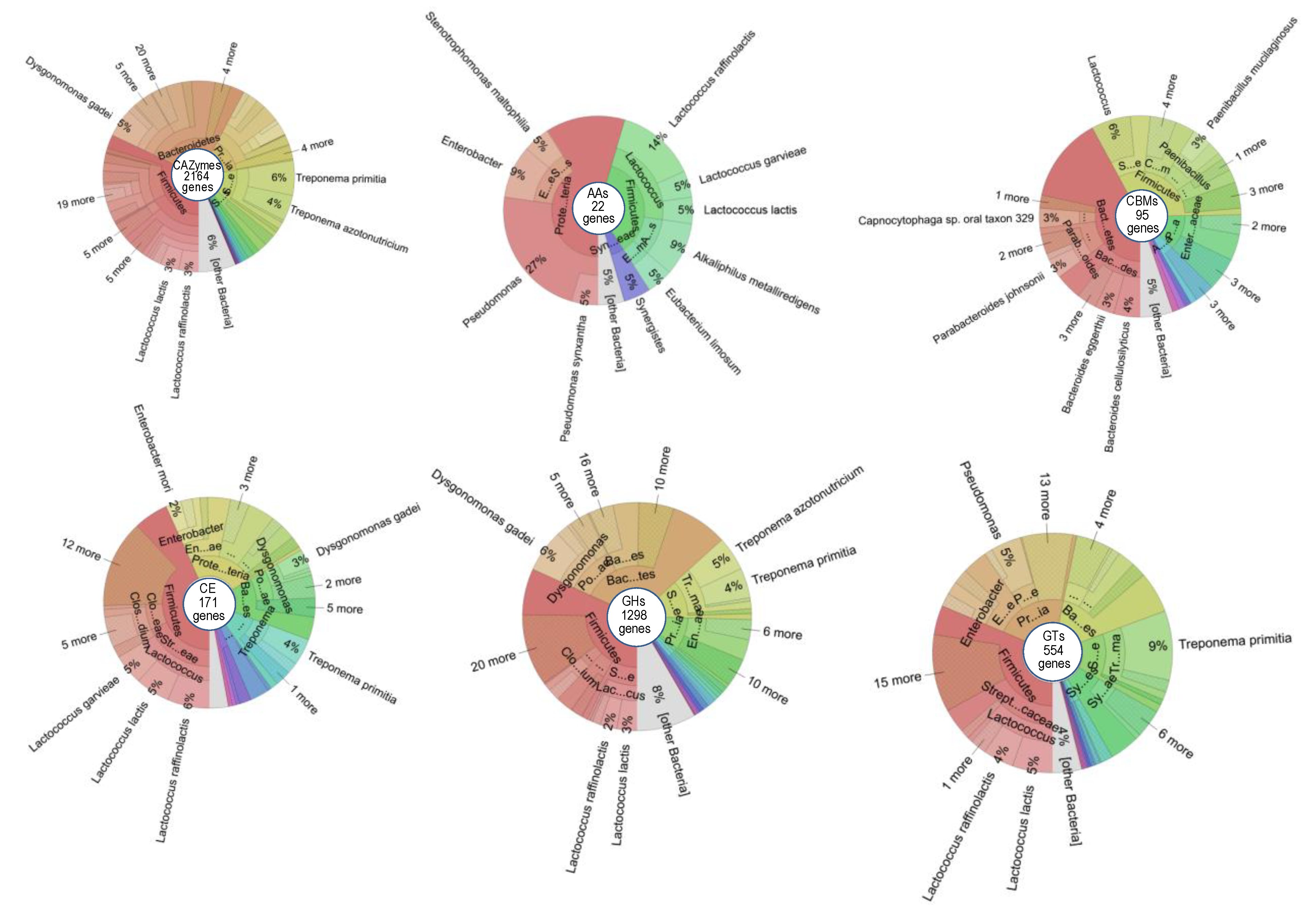
3.3. The Carbohydrate-Active Enzymes of Prokaryotes Free-Living in the C. gestroi Gut
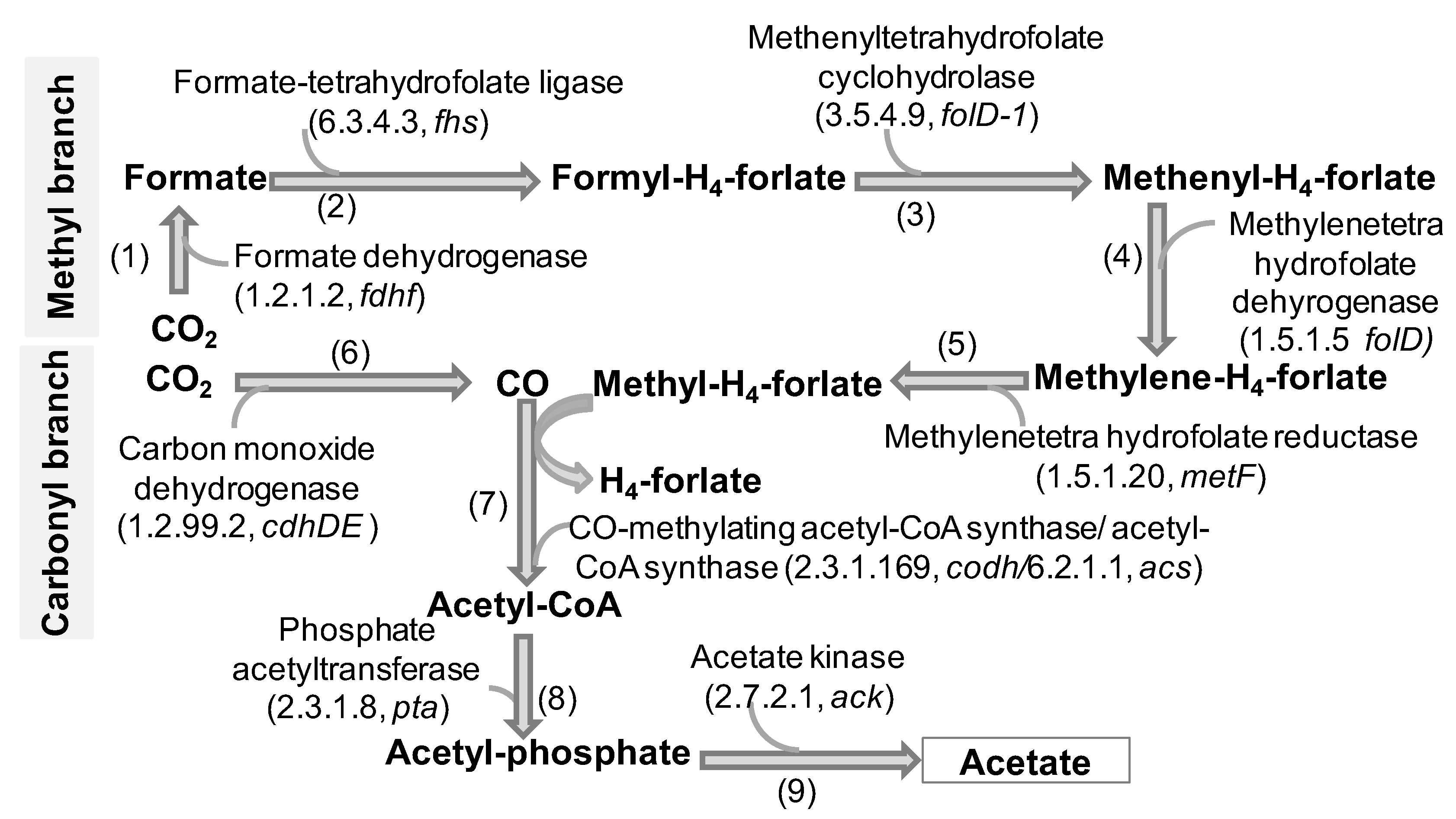
3.4. Diversity and The Role of Freely Living Gut Prokaryotes in Reductive Acetogenesis
3.5. The Role of Freely Living Gut Prokaryotes in Methanogenesis
3.6. The Role of Freely Living Gut Prokaryotes in Methane Metabolism
3.7. The Role of Freely Living Gut Prokaryotes in Sulfur Metabolism
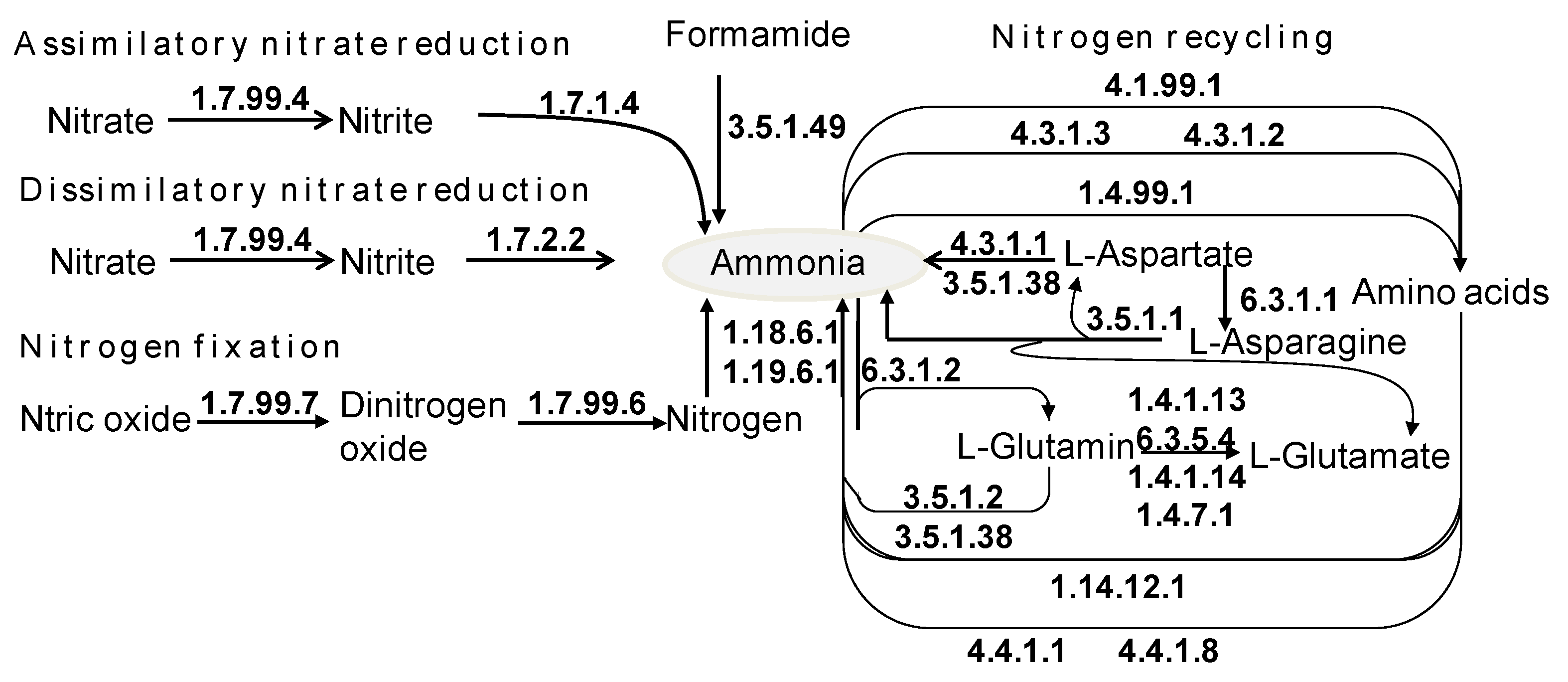
3.8. The Role of Freely Living Gut Prokaryotes in Nitrogen Recycling
3.9. The Role of Freely Living Gut Prokaryotes in Antibiotic Synthesis
3.10. The Role of Freely Living Gut Prokaryotes in Chemical Degradation
4. Discussion
4.1. Diversity of Prokaryotic Community Living Freely in C. gestroi Gut Reveals Unique Attributes of the Dominant Types
4.2. Contribution of CAZymes of Free-Living Prokaryotes of C. gestroi Gut in Lignocellulose Digestion
4.3. Free-Living Prokaryotes of C. gestroi Gut Interact with Each Other to Exhibit Diverse Nutritional Functions and Provide Health Benefits to Their Hosts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ali, H.R.K.; Ropy, A.; Batt, M.A. The Differences in the Digestive System and Enzymes between Soldiers and Workers of the Subterranean Termite, Psammotermes hypostoma Desneux (Rhinotermitidae: Isoptera). Egypt. Acad. J. Biol. Sciences. A Entomol. 2019, 12, 49–61. [Google Scholar] [CrossRef]
- Noda, S.; Iida, T.; Kitade, O.; Nakajima, H.; Kudo, T.; Ohkuma, M. Endosymbiotic Bacteroidales Bacteria of the Flagellated Protist Pseudotrichonympha grassii in the Gut of the Termite Coptotermes formosanus. Appl. Environ. Microbiol. 2005, 71, 8811–8817. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Hongoh, Y.; Usami, R.; Kudo, T.; Ohkuma, M. Spatial Distribution of Bacterial Phylotypes in the Gut of the Termite Reticulitermes speratus and the Bacterial Community Colonizing the Gut Epithelium. FEMS Microbiol. Ecol. 2005, 54, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Hongoh, Y.; Noda, S.; Yoshida, Y.; Usami, R.; Kudo, T.; Ohkuma, M. Phylogenetic and Morphological Diversity of Bacteroidales Members Associated with the Gut Wall of Termites. Biosci. Biotechnol. Biochem. 2006, 70, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.S. Termite Evolution: A Primal Knock on Wood or a Hearty Mouthful of Dirt. Curr. Biol. 2019, 29, R1126–R1129. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Yang, G.Y.; Liang, S.Y.; Zhou, Q.H.; Gaal, A.H.; Mo, G.C. Multipartite Symbioses in Fungus-Growing Termites (Blattodea: Termitidae, Macrotermitinae) for the Degradation of Lignocellulose. Insect 2021, 28, 1512–1529. [Google Scholar] [CrossRef]
- Arora, J.; Kinjo, Y.; Šobotník, J.; Buček, A.; Clitheroe, C.; Stiblik, P.; Roisin, Y.; Žifčáková, L.; Park, Y.C.; Kim, K.Y.; et al. The Functional Evolution of Termite Gut Microbiota. Microbiome 2022, 10, 78. [Google Scholar] [CrossRef]
- Do, T.H.; Nguyen, T.T.; Nguyen, T.N.; Le, Q.G.; Nguyen, C.; Kimura, K.; Truong, N.H. Mining Biomass-Degrading Genes through Illumina-Based de Novo Sequencing and Metagenomic Analysis of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi Harvested in Vietnam. J. Biosci. Bioeng. 2014, 118, 665–671. [Google Scholar] [CrossRef]
- Austin, J.W.; Szalanski, A.L.; Cabrera, B.J. Phylogenetic Analysis of the Subterranean Termite Family Rhinotermitidae (Isoptera) by Using the Mitochondrial Cytochrome Oxidase II Gene. Entomol. Soc. Am. 2004, 97, 548–555. [Google Scholar] [CrossRef]
- Trinh, V.H.; Tran, T.H.; Nguyen, T.H. Diversity of termite species in Vietnam. In Proceedings of the 7th conference of the Pacific Rim Termite Research Group, Singapore, 1–2 March 2010. [Google Scholar]
- Nguyen, M.D.; Bui, T.L.; Do, T.N.A.; Nguyen, T.M.; Nguyen, V.Q.; Trinh, V.H. Data on Species Composition of Termites (Insecta: Isoptera) in Bac Huong Hoa Nature Reserve, Quang Tri Province. VNU J. Sci. Nat. Sci. Technol. 2016, 32, 18–25. [Google Scholar]
- Nguyen, T.M.; Nguyen, Q.H.; Nguyen, M.D.; Nguyen, V.Q.; To, T.M.D. Species Composition and Distribution of Termites in Ngoc Linh Nature Reserve, Quang Nam Province, Vietnam. IOP Conf. Ser. Earth Environ. Sci. 2020, 425, 012004. [Google Scholar] [CrossRef]
- Nguyen, D.K.; Nguyen, T.V.; Trinh, V.H.; Nguyen, V.Q.; Le, V.T.; Nguyen, T.H.; Vu, V.N.; Ngo, T.S.; Vo, T.H. Fauna of Vietnam. Termite: Isoptera.; The Science and Technology of Ha Noi: Ha Noi, Vietnam, 2007. [Google Scholar]
- Li, H.F.; Fujisaki, I.; Su, N.-Y. Predicting Habitat Suitability of Coptotermes gestroi (Isoptera: Rhinotermitidae) with Species Distribution Models. J. Econ. Entomol. 2013, 106, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Franco Cairo, J.P.L.; Leonardo, F.C.; Alvarez, T.M.; Ribeiro, D.A.; Buchli, F.; Costa-Leonardo, A.M.; Carazzolle, M.F.; Costa, F.F.; Paes Leme, A.F.; Pereira, G.A.; et al. Functional Characterization and Target Discovery of Glycoside Hydrolases from the Digestome of the Lower Termite Coptotermes gestroi. Biotechnol. Biofuels 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.G.; Do, T.H.; Truong, N.H. Using Bioinformatic Tools in Exploited Gene Encoding Enzyme to Decompose Lignocellulose from Metagenome of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi. Vietnam. J. Biotechnol. 2016, 14, 39–47. [Google Scholar] [CrossRef]
- Lin, H.; Chen, W.; Ding, H. AcalPred: A Sequence-Based Tool for Discriminating between Acidic and Alkaline Enzymes. PLoS ONE 2013, 8, e75726. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.G.; Do, T.H.; Truong, N.H. In Silico Mining for Alkaline Enzymes from Metagenomic Dna Data of Gut Microbes of the Lower Termite Coptotermes gestroi in Vietnam. Acad. J. Biol. 2016, 38, 374–383. [Google Scholar] [CrossRef]
- Lilburn, T.G.; Kim, K.S.; Ostrom, N.E.; Byzek, K.R.; Leadbetter, J.R.; Breznak, J.A. Nitrogen Fixation by Symbiotic and Free-Living Spirochetes. Science 2001, 292, 2495–2498. [Google Scholar] [CrossRef]
- Hongoh, Y.; Sharma, V.K.; Prakash, T.; Noda, S.; Toh, H.; Taylor, T.D.; Kudo, T.; Sakaki, Y.; Toyoda, A.; Hattori, M.; et al. Genome of an Endosymbiont Coupling N2 Fixation to Cellulolysis within Protist Cells in Termite Gut. Science 2008, 322, 1108–1109. [Google Scholar] [CrossRef]
- Hirschler, B. Termite Guts May Hold Key to Better Biofuels. Environment, The Thomson Reuters Trust Principles. London. 2007. Available online: https://www.reuters.com/article/environment-termites-biofuel-dc-idUSL2187737820071121 (accessed on 28 September 2023).
- Nauer, P.A.; Hutley, L.B.; Arndt, S.K. Termite Mounds Mitigate Half of Termite Methane Emissions. Proc. Natl. Acad. Sci. USA 2018, 115, 13306–13311. [Google Scholar] [CrossRef]
- Myer, A.; Myer, M.H.; Trettin, C.C.; Forschler, B.T. The Fate of Carbon Utilized by the Subterranean Termite Reticulitermes flavipes. Ecosphere 2021, 12, e03872. [Google Scholar] [CrossRef]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood-Ljungdahl Pathway of CO2 Fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar] [CrossRef] [PubMed]
- Pester, M.; Brune, A. Hydrogen Is the Central Free Intermediate during Lignocellulose Degradation by Termite Gut Symbionts. ISME J. 2007, 1, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Heise, P.; Liu, Y.; Degenkolb, T.; Vogel, H.; Schäberle, T.F.; Vilcinskas, A. Antibiotic-Producing Beneficial Bacteria in the Gut of the Burying Beetle Nicrophorus vespilloides. Front. Microbiol. 2019, 10, 1178. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Tanaka, J.; Namihira, T.; Shinzato, N. Antibiotics Production by an Actinomycete Isolated from the Termite Gut. J. Basic Microbiol. 2012, 52, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, Y.; Huang, F.; Liu, S.; Gao, T.; Zhang, Y. Diversity and Antimicrobial Activities of Culturable Actinomycetes from Odontotermes formosanus (Blattaria: Termitidae). BMC Microbiol. 2022, 22, 80. [Google Scholar] [CrossRef]
- Krishanti, N.P.R.A.; Zulfina, D.; Wikantyoso, B.; Zulfitri, A.; Yusuf, S. Antimicrobial Production by an Actinomycetes Isolated from the Termite Nest. J. Trop. Life Sci. 2018, 8, 279–288. [Google Scholar]
- Benndorf, R.; Guo, H.; Sommerwerk, E.; Weigel, C.; Garcia-Altares, M.; Martin, K.; Hu, H.; Küfner, M.; De Beer, Z.W.; Poulsen, M.; et al. Natural Products from Actinobacteria Associated with Fungus-Growing Termites. Antibiotics 2018, 7, 83. [Google Scholar] [CrossRef]
- Mokoena, N.; Mathiba, K.; Tsekoa, T.; Steenkamp, P.; Rashamuse, K. Application of Termite Hindgut Metagenome Derived Carboxyl Ester Hydrolases in the Modification of Cephalosporin Substrates. Biochem. Biophys. Rep. 2015, 4, 44–51. [Google Scholar] [CrossRef]
- Scharf, M. Termites as Targets and Models for Biotechnology. Annu. Rev. Entomol. 2015, 60, 77–102. [Google Scholar] [CrossRef]
- Michaud, C.; Hervé, V.; Dupont, S.; Dubreuil, G.; Bézier, A.M.; Meunier, J.; Brune, A.; Dedeine, F. Efficient but Occasionally Imperfect Vertical Transmission of Gut Mutualistic Protists in a Wood-Feeding Termite. Mol. Ecol. 2020, 29, 308–324. [Google Scholar] [CrossRef]
- Hervé, V.; Liu, P.; Dietrich, C.; Sillam-Dussès, D.; Stiblik, P.; Šobotník, J.; Brune, A. Phylogenomic Analysis of 589 Metagenome-Assembled Genomes Encompassing All Major Prokaryotic Lineages from the Gut of Higher Termites. PeerJ 2020, 8, e8614. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, T.; Lo, N.; Dietrich, C.; Šobotník, J.; Sidek, S.; Roisin, Y.; Brune, A.; Evans, T.A. Rampant Host Switching Shaped the Termite Gut Microbiome. Curr. Biol. 2018, 28, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Do, T.H.; Duong, T.H.; Le, Q.G.; Dao, T.K.; Nguyen, T.T.; Nguyen, T.Q.; Nguyen, T.T.H.; Kimura, K.; Truong, N.H. Identification of Vietnamese Coptotermes Pest Species Based on the Sequencing of Two Regions of 16S rRNA Gene. Bull. Insectology 2014, 67, 131–136. [Google Scholar]
- Hongoh, Y. Diversity and Genomes of Uncultured Microbial Symbionts in the Termite Gut. Biosci. Biotechnol. Biochem. 2010, 74, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed]
- Schuchmann, K.; Müller, V. Autotrophy at the Thermodynamic Limit of Life: A Model for Energy Conservation in Acetogenic Bacteria. Nat. Rev. Microbiol. 2014, 12, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Oshima, K.; Suda, W.; Sakamoto, M.; Iino, T.; Noda, S.; Hongoh, Y.; Hattori, M.; Ohkuma, M. Distribution and Evolution of Nitrogen Fixation Genes in the Phylum Bacteroidetes. Microbes Environ. 2015, 30, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Enagbonma, B.J.; Babalola, O.O. Metagenomics Reveals the Microbiome Multifunctionalities of Environmental Importance from Termite Mound Soils. Bioinform. Biol. Insights 2023, 17, 11779322231184025. [Google Scholar] [CrossRef]
- Butera, G.; Ferraro, C.; Alonzo, G.; Colazza, S.; Quatrini, P. The Gut Microbiota of the Wood-Feeding Termite Reticulitermes lucifugus (Isoptera; Rhinotermitidae). Ann. Microbiol. 2016, 66, 253–260. [Google Scholar] [CrossRef]
- Su, L.; Yang, L.; Huang, S.; Su, X.; Li, Y.; Wang, F.; Wang, E.; Kang, N.; Xu, J.; Song, A. Comparative Gut Microbiomes of Four Species Representing the Higher and the Lower Termites. J. Insect Sci. 2016, 16, 97. [Google Scholar] [CrossRef]
- Do, T.H.; Dao, T.K.; Nguyen, K.H.V.; Le, N.G.; Nguyen, T.M.P.; Le, T.L.; Phung, T.N.; van Straalen, N.M.; Roelofs, D.; Truong, N.H. Metagenomic Analysis of Bacterial Community Structure and Diversity of Lignocellulolytic Bacteria in Vietnamese Native Goat Rumen. Asian-Australas J. Anim. Sci. 2018, 31, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Do, T.H.; Le, N.G.; Dao, T.K.; Nguyen, T.M.P.; Le, T.L.; Luu, H.L.; Nguyen, K.H.V.; Nguyen, V.L.; Le, L.A.; Phung, T.N.; et al. Metagenomic Insights into Lignocellulose-Degrading Genes through Illumina-Based de Novo Sequencing of the Microbiome in Vietnamese Native Goats’ Rumen. J. Gen. Appl. Microbiol. 2018, 64, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.K.; Do, T.H.; Le, N.G.; Nguyen, H.D.; Nguyen, T.Q.; Le, T.T.H.; Truong, N.H. Understanding the Role of Prevotella Genus in the Digestion of Lignocellulose and Other Substrates in Vietnamese Native Goats’ Rumen by Metagenomic Deep Sequencing. Animals 2021, 11, 3257. [Google Scholar] [CrossRef] [PubMed]
- Güllert, S.; Fischer, M.A.; Turaev, D.; Noebauer, B.; Ilmberger, N.; Wemheuer, B.; Alawi, M.; Rattei, T.; Daniel, R.; Schmitz, R.A.; et al. Deep Metagenome and Metatranscriptome Analyses of Microbial Communities Affiliated with an Industrial Biogas Fermenter, a Cow Rumen, and Elephant Feces Reveal Major Differences in Carbohydrate Hydrolysis Strategies. Biotechnol. Biofuels 2016, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Calusinska, M.; Marynowska, M.; Bertucci, M.; Untereiner, B.; Klimek, D.; Goux, X.; Sillam-Dussès, D.; Gawron, P.; Halder, R.; Wilmes, P.; et al. Integrative Omics Analysis of the Termite Gut System Adaptation to Miscanthus Diet Identifies Lignocellulose Degradation Enzymes. Commun. Biol. 2020, 3, 275. [Google Scholar] [CrossRef]
- Marynowska, M.; Goux, X.; Sillam-Dussès, D.; Rouland-Lefèvre, C.; Halder, R.; Wilmes, P.; Gawron, P.; Roisin, Y.; Delfosse, P.; Calusinska, M. Compositional and Functional Characterisation of Biomass-Degrading Microbial Communities in Guts of Plant Fibre- and Soil-Feeding Higher Termites. Microbiome 2020, 8, 96. [Google Scholar] [CrossRef]
- Tokuda, G.; Mikaelyan, A.; Fukui, C.; Matsuura, Y.; Watanabe, H.; Fujishima, M.; Brune, A. Fiber-Associated Spirochetes Are Major Agents of Hemicellulose Degradation in the Hindgut of Wood-Feeding Higher Termites. Proc. Natl. Acad. Sci. USA 2018, 115, E11996–E12004. [Google Scholar] [CrossRef]
- King, J.H.P.; Mahadi, N.M.; Bong, C.F.J.; Ong, K.H.; Hassan, O. Bacterial Microbiome of Coptotermes curvignathus (Isoptera: Rhinotermitidae) Reflects the Coevolution of Species and Dietary Pattern. Insect Sci. 2014, 21, 584–596. [Google Scholar] [CrossRef]
- Husseneder, C.; Ho, H.-Y.; Blackwell, M. Comparison of the Bacterial Symbiont Composition of the Formosan Subterranean Termite from Its Native and Introduced Range. Open Microbiol J 2010, 4, 53–66. [Google Scholar] [CrossRef]
- Telmadarrehei, T.; Tang, J.D.; Raji, O.; Rezazadeh, A.; Narayanan, L.; Shmulsky, R.; Jeremic, D. A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment. Insects 2020, 11, 681. [Google Scholar] [CrossRef]
- Vikram, S.; Arneodo, J.D.; Calcagno, J.; Ortiz, M.; Mon, M.L.; Etcheverry, C.; Cowan, D.A.; Talia, P. Diversity Structure of the Microbial Communities in the Guts of Four Neotropical Termite Species. PeerJ 2021, 9, e10959. [Google Scholar] [CrossRef]
- Rossmassler, K.; Dietrich, C.; Thompson, C.; Mikaelyan, A.; Nonoh, J.O.; Scheffrahn, R.H.; Sillam-Dussès, D.; Brune, A. Metagenomic Analysis of the Microbiota in the Highly Compartmented Hindguts of Six Wood- or Soil-Feeding Higher Termites. Microbiome 2015, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Brune, A. Microbial Symbioses in the Digestive Tract of Lower Termites. In Beneficial Microorganisms in Multicellular Life Forms; Rosenberg, E., Gophna, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–25. ISBN 978-3-642-21679-4. [Google Scholar]
- Ufnar, J.A.; Wang, S.Y.; Christiansen, J.M.; Yampara-Iquise, H.; Carson, C.A.; Ellender, R.D. Detection of the nifH Gene of Methanobrevibacter smithii: A Potential Tool to Identify Sewage Pollution in Recreational Waters. J. Appl. Microbiol. 2006, 101, 44–52. [Google Scholar] [CrossRef] [PubMed]
- McGovern, E.; McCabe, M.S.; Cormican, P.; Popova, M.; Keogh, K.; Kelly, A.K.; Kenny, D.A.; Waters, S.M. Plane of Nutrition Affects the Phylogenetic Diversity and Relative Abundance of Transcriptionally Active Methanogens in the Bovine Rumen. Sci. Rep. 2017, 7, 13047. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.J.F.; Martz, E.; Holmes, D.E.; Zhou, Z.; Nonnenmann, S.S.; Lovley, D.R. The Archaellum of Methanospirillum hungatei Is Electrically Conductive. mBio 2019, 10, e00579-19. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.; Deere, T.M.; Chanderban, M.; McIntosh, G.J.; Lessner, D.J. Methanosarcina Acetivorans. Trends Microbiol. 2023, 31, 320–321. [Google Scholar] [CrossRef]
- Graber, J.R.; Breznak, J.A. Physiology and Nutrition of Treponema primitia, an H2/CO2-Acetogenic Spirochete from Termite Hindguts. Appl. Environ. Microbiol. 2004, 70, 1307–1314. [Google Scholar] [CrossRef]
- Ohkuma, M.; Noda, S.; Hattori, S.; Iida, T.; Yuki, M.; Starns, D.; Inoue, J.; Darby, A.C.; Hongoh, Y. Acetogenesis from H2 plus CO2 and Nitrogen Fixation by an Endosymbiotic Spirochete of a Termite-Gut Cellulolytic Protist. Proc. Natl. Acad. Sci. USA 2015, 112, 10224–10230. [Google Scholar] [CrossRef]
- Ikeda-Ohtsubo, W.; Strassert, J.F.H.; Köhler, T.; Mikaelyan, A.; Gregor, I.; McHardy, A.C.; Tringe, S.G.; Hugenholtz, P.; Radek, R.; Brune, A. “Candidatus Adiutrix intracellularis”, an Endosymbiont of Termite Gut Flagellates, Is the First Representative of a Deep-Branching Clade of Deltaproteobacteria and a Putative Homoacetogen. Environ. Microbiol. 2016, 18, 2548–2564. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Alberdi, A.; Deng, J.; Zhong, Z.; Si, H.; Zheng, C.; Zhou, H.; Wang, J.; Yang, Y.; et al. Comparative Microbiome Analysis Reveals the Ecological Relationships between Rumen Methanogens, Acetogens, and Their Hosts. Front. Microbiol. 2020, 11, 1311. [Google Scholar] [CrossRef]
- Ohashi, Y.; Igarashi, T.; Kumazawa, F.; Fujisawa, T. Analysis of Acetogenic Bacteria in Human Feces with Formyltetrahydrofolate Synthetase Sequences. Biosci. Microflora 2007, 26, 37–40. [Google Scholar] [CrossRef]
- Dröge, S.; Limper, U.; Emtiazi, F.; Schönig, I.; Pavlus, N.; Drzyzga, O.; Fischer, U.; König, H. In vitro and in vivo Sulfate Reduction in the Gut Contents of the Termite Mastotermes Darwiniensis and the Rose-Chafer Pachnoda Marginata. J. Gen. Appl. Microbiol. 2005, 51, 57–64. [Google Scholar] [CrossRef]
- Sayavedra, L.; Li, T.; Bueno Batista, M.; Seah, B.K.B.; Booth, C.; Zhai, Q.; Chen, W.; Narbad, A. Desulfovibrio diazotrophicus Sp. Nov., a Sulfate-Reducing Bacterium from the Human Gut Capable of Nitrogen Fixation. Environ. Microbiol. 2021, 23, 3164–3181. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Singh, D.; Chen, S. Aromatic Compound Degradation by the Wood-Feeding Termite Coptotermes formosanus (Shiraki). Int. Biodeterior. Biodegrad. 2011, 65, 744–756. [Google Scholar] [CrossRef]
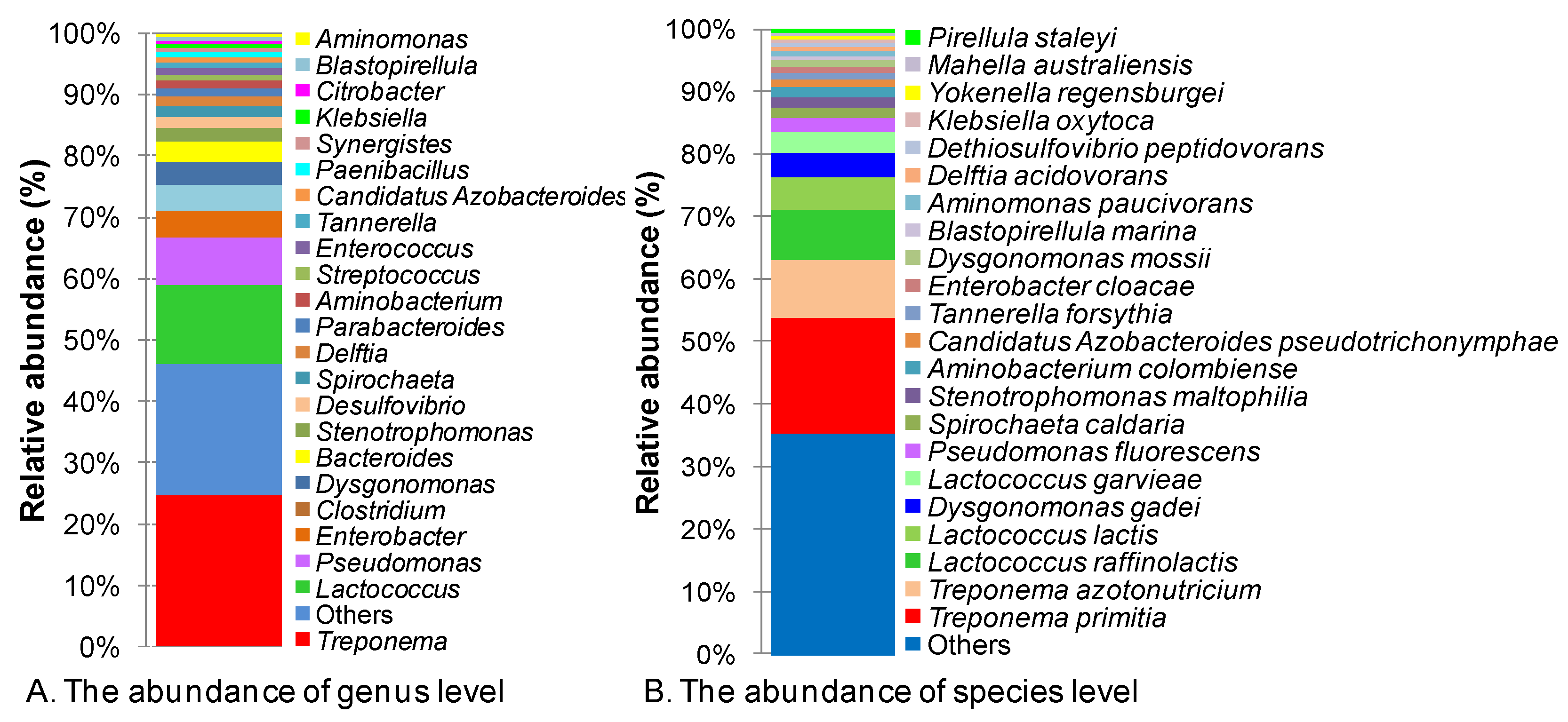





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAZy Family | Domain Number | % | CAZy Family | Gene Number | % | CAZy Family | Gene Number | % | CAZy Family | Gene Number | % |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA: | 22/22 genes | 1.011 | GH3 | 90 | 4.14 | GH115 | 5 | 0.23 | GT: 554/554 genes | 25.471 | |
| AA4 | 7 | 0.32 | GH1 | 77 | 3.54 | GH137 | 5 | 0.23 | GT2 | 170 | 7.82 |
| AA10 | 6 | 0.28 | GH29 | 67 | 3.08 | GH141 | 5 | 0.23 | GT4 | 92 | 4.23 |
| AA3 | 5 | 0.23 | GH109 | 52 | 2.39 | GH142 | 5 | 0.23 | GT51 | 73 | 3.36 |
| AA6 | 2 | 0.09 | GH23 | 51 | 2.34 | GH144 | 5 | 0.23 | GT5 | 35 | 1.61 |
| AA2 | 1 | 0.05 | GH78 | 48 | 2.21 | GH37 | 5 | 0.23 | GT35 | 27 | 1.24 |
| AA7 | 1 | 0.05 | GH92 | 35 | 1.61 | GH76 | 5 | 0.23 | GT28 | 20 | 0.92 |
| CBM: | 101/95 genes | 4.644 | GH73 | 30 | 1.38 | GH102 | 4 | 0.18 | GT83 | 20 | 0.92 |
| CBM67 | 25 | 1.15 | GH95 | 30 | 1.38 | GH108 | 4 | 0.18 | GT19 | 16 | 0.74 |
| CBM32 | 16 | 0.74 | GH77 | 29 | 1.33 | GH113 | 4 | 0.18 | GT9 | 16 | 0.74 |
| CBM48 | 11 | 0.51 | GH106 | 25 | 1.15 | GH133 | 4 | 0.18 | GT26 | 12 | 0.55 |
| CBM62 | 8 | 0.37 | GH2 | 24 | 1.10 | GH136 | 4 | 0.18 | GT1 | 10 | 0.46 |
| CBM13 | 5 | 0.23 | GH18 | 22 | 1.01 | GH103 | 3 | 0.14 | GT8 | 10 | 0.46 |
| CBM34 | 6 | 0.28 | GH20 | 21 | 0.97 | GH120 | 3 | 0.14 | GT30 | 8 | 0.37 |
| CBM6 | 6 | 0.28 | GH4 | 21 | 0.97 | GH140 | 3 | 0.14 | GT27 | 5 | 0.23 |
| CBM51 | 5 | 0.23 | GH5 | 20 | 0.92 | GH19 | 3 | 0.14 | GT81 | 5 | 0.23 |
| CBM9 | 5 | 0.23 | GH127 | 19 | 0.87 | GH27 | 3 | 0.14 | GT101 | 4 | 0.18 |
| CBM20 | 3 | 0.14 | GH51 | 19 | 0.87 | GH63 | 3 | 0.14 | GT21 | 4 | 0.18 |
| CBM35 | 2 | 0.09 | GH31 | 17 | 0.78 | GH11 | 2 | 0.09 | GT3 | 4 | 0.18 |
| CBM41 | 2 | 0.09 | GH28 | 16 | 0.74 | GH128 | 2 | 0.09 | GT14 | 3 | 0.14 |
| CBM73 | 2 | 0.09 | GH38 | 16 | 0.74 | GH129 | 2 | 0.09 | GT20 | 3 | 0.14 |
| CBM22 | 1 | 0.05 | GH42 | 16 | 0.74 | GH139 | 2 | 0.09 | GT32 | 3 | 0.14 |
| CBM5 | 1 | 0.05 | GH65 | 15 | 0.69 | GH145 | 2 | 0.09 | GT56 | 3 | 0.14 |
| CBM50 | 1 | 0.05 | GH10 | 14 | 0.64 | GH24 | 2 | 0.09 | GT11 | 2 | 0.09 |
| CBM66 | 1 | 0.05 | GH35 | 14 | 0.64 | GH50 | 2 | 0.09 | GT84 | 2 | 0.09 |
| CBM77 | 1 | 0.05 | GH130 | 13 | 0.60 | GH55 | 2 | 0.09 | GT10 | 1 | 0.05 |
| CE: | 172/171 genes | 7.908 | GH30 | 13 | 0.60 | GH94 | 2 | 0.09 | GT104 | 1 | 0.05 |
| CE10 | 45 | 2.07 | GH36 | 13 | 0.60 | GH110 | 1 | 0.05 | GT25 | 1 | 0.05 |
| CE4 | 37 | 1.70 | GH57 | 13 | 0.60 | GH123 | 1 | 0.05 | GT39 | 1 | 0.05 |
| CE1 | 32 | 1.47 | GH88 | 13 | 0.60 | GH143 | 1 | 0.05 | GT70 | 1 | 0.05 |
| CE9 | 31 | 1.43 | GH105 | 12 | 0.55 | GH15 | 1 | 0.05 | GT73 | 1 | 0.05 |
| CE11 | 10 | 0.46 | GH25 | 12 | 0.55 | GH17 | 1 | 0.05 | GT94 | 1 | 0.05 |
| CE7 | 5 | 0.23 | GH97 | 12 | 0.55 | GH53 | 1 | 0.05 | PL: 26/24 genes | 1.195 | |
| CE15 | 3 | 0.14 | GH125 | 10 | 0.46 | GH66 | 1 | 0.05 | PL1 | 6 | 0.28 |
| CE3 | 3 | 0.14 | GH32 | 10 | 0.46 | GH68 | 1 | 0.05 | PL22 | 5 | 0.23 |
| CE14 | 2 | 0.09 | GH9 | 10 | 0.46 | GH74 | 1 | 0.05 | PL8 | 4 | 0.18 |
| CE8 | 2 | 0.09 | GH39 | 8 | 0.37 | GH85 | 1 | 0.05 | PL9 | 3 | 0.14 |
| CE2 | 1 | 0.05 | GH116 | 7 | 0.32 | GH89 | 1 | 0.05 | PL11 | 2 | 0.09 |
| CE6 | 1 | 0.05 | GH8 | 7 | 0.32 | GH91 | 1 | 0.05 | PL17 | 2 | 0.09 |
| GH: | 1300/1298 genes | 59.770 | GH16 | 6 | 0.28 | GH93 | 1 | 0.05 | PL5 | 2 | 0.09 |
| GH13 | 145 | 6.67 | GH26 | 6 | 0.28 | GH99 | 1 | 0.05 | PL6 | 1 | 0.05 |
| GH43 | 91 | 4.18 | GH67 | 6 | 0.28 | PL7 | 1 | 0.05 | |||
| Phylum | Genus | Species | Pathway Module | Gene Number |
|---|---|---|---|---|
| Spirochaetes | 85 | |||
| Treponema | 80 | |||
| Treponema azotonutricium | Assimilatory sulfate reduction | 1 | ||
| Treponema azotonutricium | Cysteine/acetate biosynthesis | 20 | ||
| Treponema phagedenis | Assimilatory sulfate reduction | 2 | ||
| Treponema phagedenis | Cysteine/acetate biosynthesis | 1 | ||
| Treponema primitia | Assimilatory sulfate reduction | 7 | ||
| Treponema primitia | Cysteine/acetate biosynthesis | 37 | ||
| Proteobacteria | 80 | |||
| Pseudomonas | Assimilatory sulfate reduction | 5 | ||
| Pseudomonas | Cysteine/acetate biosynthesis | 9 | ||
| Pseudomonas | Dissimilatory sulfate reduction | 7 | ||
| Pseudomonas fluorescens | Assimilatory sulfate reduction | 1 | ||
| Pseudomonas fluorescens | Cysteine/acetate biosynthesis | 2 | ||
| Enterobacter | Assimilatory sulfate reduction | 3 | ||
| Enterobacter | Cysteine/acetate biosynthesis | 8 | ||
| Enterobacter | Dissimilatory sulfate reduction | 1 | ||
| Desulfovibrio | Assimilatory sulfate reduction | 4 | ||
| Desulfovibrio | Cysteine/acetate biosynthesis | 2 | ||
| Desulfovibrio | Dissimilatory sulfate reduction | 1 | ||
| Stenotrophomonas | Stenotrophomonas maltophilia | Cysteine/acetate biosynthesis | 3 | |
| Stenotrophomonas | Stenotrophomonas maltophilia | Dissimilatory sulfate reduction | 1 | |
| Delftia | Delftia acidovorans | Assimilatory sulfate reduction | 1 | |
| Delftia | Delftia acidovorans | Cysteine/acetate biosynthesis | 2 | |
| Salmonella | Salmonella enterica | Cysteine/acetate biosynthesis | 1 | |
| Salmonella | Salmonella enterica | Dissimilatory sulfate reduction | 1 | |
| Firmicutes | 72 | |||
| Lactococcus | Cysteine/acetate biosynthesis | 27 | ||
| Clostridium | Assimilatory sulfate reduction | 6 | ||
| Clostridium | Cysteine/acetate biosynthesis | 9 | ||
| Bacteroidetes | Assimilatory sulfate reduction | 8 | ||
| Bacteroidetes | Cysteine/acetate biosynthesis | 22 | ||
| Bacteroidetes | Dissimilatory sulfate reduction | 7 | ||
| Dysgonomonas | Dysgonomonas mossii | Cysteine/acetate biosynthesis | 1 | |
| Dysgonomonas | Dysgonomonas mossii | Dissimilatory sulfate reduction | 1 | |
| Candidatus Azobacteroides | Candidatus Azobacteroides pseudotrichonymphae | Assimilatory sulfate reduction | 4 | |
| Candidatus Azobacteroides | Candidatus Azobacteroides pseudotrichonymphae | Cysteine/acetate biosynthesis | 1 |
| No. | Map | Degradation Pathway | Gene Number | Key Role of Important Enzymes | |
|---|---|---|---|---|---|
| EC | |||||
| 1 | map00632 | Benzoate degradation via CoA ligation | 555 | 4.1.3.17 | The last step to produce pyruvate, oxaloacetate, succinyl-CoA |
| 2.3.1.16, 2.3.1.174 | The last step to produce pyruvate, oxaloacetate, succinyl-CoA | ||||
| 2.3.1.9 | The last step to generate succinyl-CoA | ||||
| 2 | map00624 | Methylnaphthalene degradation | 502 | 1.14.13.1 | The last step to generate acetyl-CoA |
| 3 | map00626 | Naphthalene and anthracene degradation | 323 | 1.14.13.1 | The last step to generate catechol to enter the pathway for benzoate degradation or generate dihydroxynapthoate entering napthalene degradation pathway |
| 4 | map00625 | Tetrachloroethene degradation | 287 | 6.2.1-, 3.7.1-, 6.2.1-, 3.1.2.-, 1.1.-.- | The last step to generate catechol that can be further processed in benzoate degradation or generate methylcatechol to enter the pathway of xylene degradation |
| 5 | map00362 | Benzoate degradation via hydroxylation | 170 | 2.3.1.16, 2.3.1.174, 1.3.1.25 | The last step to generate benzoyl-CoA |
| 6 | map00641 | 3-Chloroacrylic acid degradation | 150 | 1.2.1.3 | The last step to generate catechol |
| 7 | map00361 | Hexachlorocyclohexane degradation | 149 | 3.1.1.45, 3.8.1.2 | The most important step to generate 3-chloroacrylic acid |
| 8 | map00281 | Geraniol degradation | 125 | 2.3.1.16 | The last step to generate CO2, maleylacetate, glycolate |
| 9 | map00633 | Trinitrotoluene degradation | 110 | 1.2.7.1, 1.12.99.6 | The last step to generate 3-methylcrolonyl-CoA |
| 10 | map00791 | Atrazine degradation | 80 | 3.5.1.5, 3.5.1.54 | A step to generate 2,4-diamino 6 hydroxylaminotoluene |
| 11 | map00930 | Caprolactam degradation | 65 | 1.1.1.35 | The last step to generate CO2 |
| 12 | map00643 | Styrene degradation | 50 | 2.8.3.1 | The last step to generate3-oxoadipyl-CoA to enter the pathway of benzoate degradation |
| 3.7.1.2 | The last step to generate L-lactate | ||||
| 13 | map00623 | 2,4-Dichlorobenzoate degradation | 49 | 4.1.3.39 | The last step to convert xylen into acetoacetate and fumarate |
| 14 | map00622 | Toluene and xylene degradation | 49 | 1.2.1.28, 4.1.3.39 | The last step to generate pyruvate acetaldehyde |
| 15 | map00627 | 1,4-Dichlorobenzene degradation | 35 | 3.1.1.45 | An important step to convert toluenze into hydroxybenzoate and benzoate before entering the pathway of benzoate degradation |
| 16 | map00631 | 1,2-Dichloroethane degradation | 23 | 3.8.1.2, 3.8.1.3 | The last step to generate pyruvate or acetaldehyde |
| 17 | map00364 | Fluorobenzoate degradation | 22 | 3.1.1.45 | An important step to generate maleylacetate before entering the pathway of benzoate degradation |
| 18 | map00629 | Carbazole degradation | 8 | 4.1.3.39 | The last step to generate glycolate |
| Termite | Firmicutes/Bacteroi- detes | % Relative Abundance of Bacteroidetes, Spirochaetes, Firmicutes, and Fibrobacteres | Strategy Used to Study Bacterial Community | Ref. |
|---|---|---|---|---|
| Wood-feeding lower termite Reticulitermes flaviceps | 1.3 | 78.9 | Pyrosequencing of the 16S rRNA gene amplicons from gut | [43] |
| Wood-feeding lower termite Tsaitermes ampliceps | 0.8 | 84.4 | ||
| Wood-feeding higher termite Mironasutitermes shangchengensis | 1.0 | 75.9 | ||
| Fungus-feeding higher termite Odontotermes formosanus | 0.6 | 73.7 | ||
| Subterranean lower termite Reticulitermes virginicus | 0.6 | 81.8 | V3 and V4 hyper-variable regions | [53] |
| Hardwood-feeding higher termite Microcerotermes strunckii | 1 | 76.0 | Deep sequencing of amplified 16S rRNA and ITS genes | [54] |
| Softwood-feeding higher termite Nasutitermes corniger | 1 | 90.0 | ||
| Grass-feeding higher termite Cornitermes cumulans | 1.8 | |||
| Oil/grass-feeding higher termite Termes riograndensis | 2.5 | |||
| Coptotermes gestroi | 1.9 | 67.8 | Free-living bacteria in the gut, whole metagenome sequencing, diversity analysis based on alignment against NR database | In this study |
| Lower termite | 0.2 | 70.9 | Gut metagenomes of 74 termite samples belonging to 4 groups, analyzed based on 40 markers | [7] |
| Non-Macrotermitinae wood-feeding Termitidae | 6.5 | 95.0 | ||
| Soil-feeding termites (SF) | 1.4 | 65.4 | ||
| Fungal-cultivating termites (FC) | 0.3 | 68.0 | ||
| Cockroach (CR) | 0.0 | 83.2 | ||
| Lower termite | 1.0 | 35.0 | Gut metagenomes of 74 termite samples belonging to 4 groups, analyzed based on 16S rDNA amplicon | |
| Non-Macrotermitinae wood-feeding Termitidae | 1.3 | 36.3 | ||
| Soil-feeding termites (SF) | 1.3 | 35.1 | ||
| Fungal-cultivating termites (FC) | 1.0 | 35.3 | ||
| Wood-feeding lower termite Coptotermes curvignathus | 0.2 | 81.5 | 16S rDNA cloning and sequencing by Sanger | [51] |
| Wood-feeding lower termite Coptotermes formosanus | 0.7 | 86.9 | 16S rDNA cloning and sequencing by Sanger | [52] |
| Mound-building higher termite Cornitermes sp. (Co191) | 1.6 | 79.2 | Three-compartment metagenomes of 6 termite gut samples analyzed by 16S rRNA V4 region | [55] |
| Soil-feeding higher termite Cubitermes ugandensis (Cu122) | 2.0 | 75.2 | ||
| Higher termiteMicrocerotermes parvus (Mp193) | 0.9 | 86.5 | ||
| Higher termiteNasutitermes corniger (Nc150) | 3.1 | 78.5 | ||
| Higher termite Neocapritermes taracua (Nt197) | 3.8 | 72.5 | ||
| Higher termite Termes hospes (Th196) | 3.2 | 80.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, T.H.; Dao, T.K.; Nguyen, H.D.; Truong, N.H. Understanding the Role of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi Based on Metagenomic DNA Analysis. Insects 2023, 14, 832. https://doi.org/10.3390/insects14110832
Do TH, Dao TK, Nguyen HD, Truong NH. Understanding the Role of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi Based on Metagenomic DNA Analysis. Insects. 2023; 14(11):832. https://doi.org/10.3390/insects14110832
Chicago/Turabian StyleDo, Thi Huyen, Trong Khoa Dao, Hong Duong Nguyen, and Nam Hai Truong. 2023. "Understanding the Role of Free-Living Bacteria in the Gut of the Lower Termite Coptotermes gestroi Based on Metagenomic DNA Analysis" Insects 14, no. 11: 832. https://doi.org/10.3390/insects14110832