
Native and Non-Native Bemisia tabaci NAFME Haplotypes Can Be Implicated in Dispersal of Endemic and Introduced Begomoviruses in Oman

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Whitefly and Plant Field Collections
2.2. Total DNA Isolation from Plants and Whiteflies, PCR Amplification, DNA Sequencing, and Sequence Analysis
2.3. Begomoviral Coat Protein Gene Fragment
2.4. Whitefly Mitochondria Cytochrome Oxidase I Fragment
2.5. SNPs Classification of Haplotypes of North African–Middle East B Mitotype
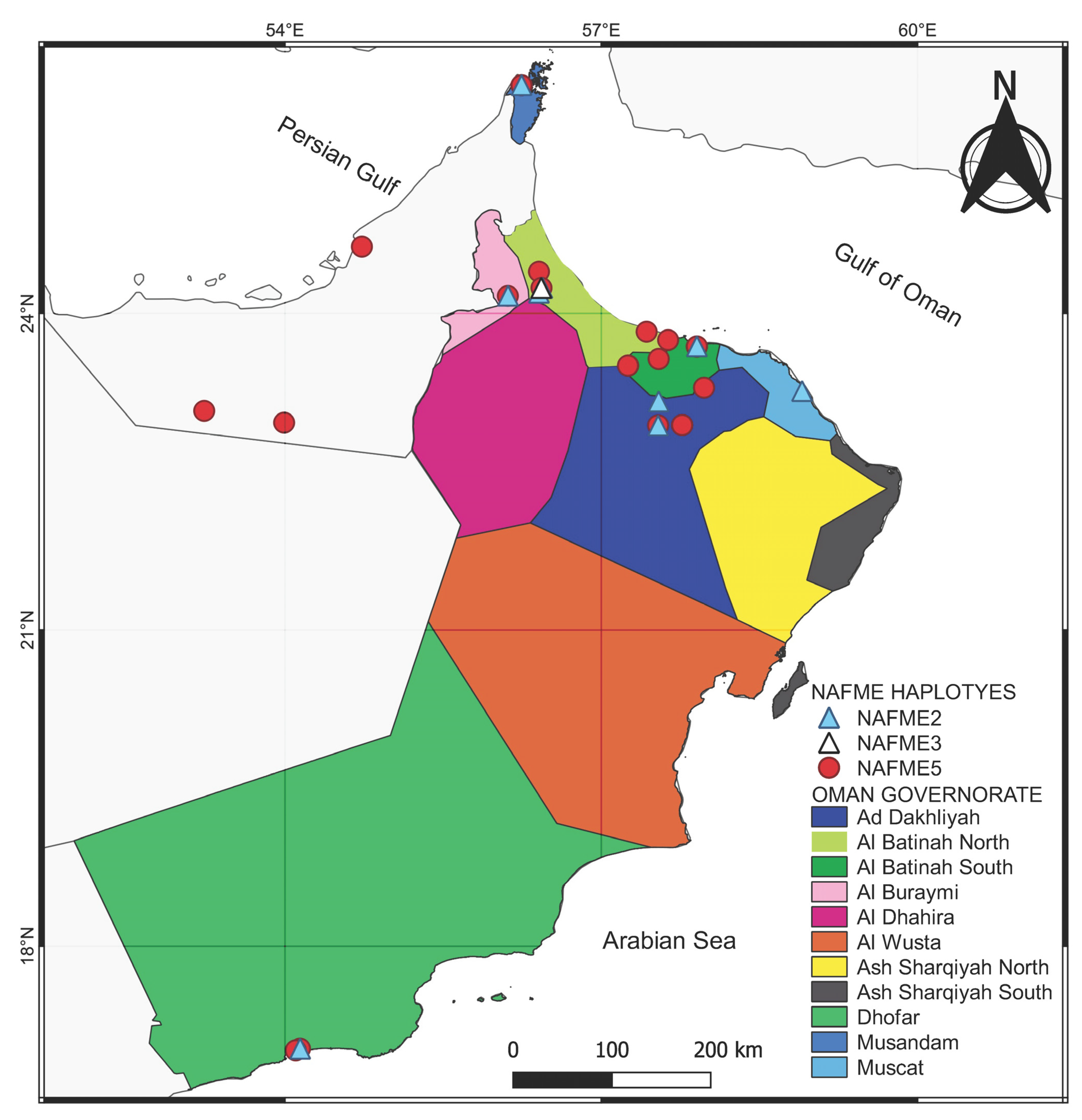
2.6. Spatial Distribution of Bemisia Tabaci NAFME Haplotypes
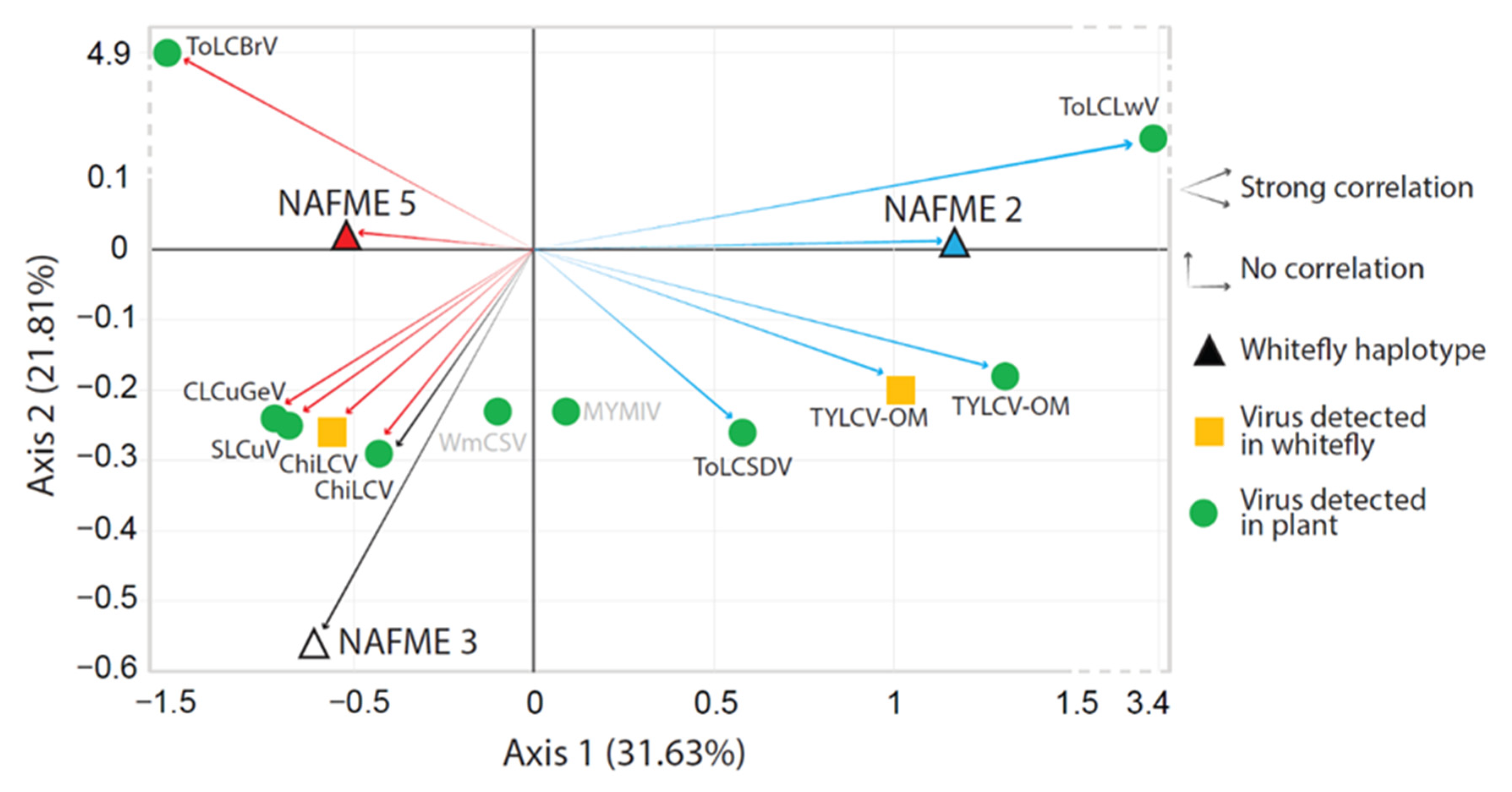
2.7. Correlation and Correspondence Analyses of Whitefly Vector–Begomovirus Associations
3. Results
3.1. Classification of Whitefly Haplotypes Based on SNPs and Phylogeographical Relationships
3.2. Provisional Begomovirus Identification and Species Associated with NAFME Haplotypes
3.3. Associations between NAFME Haplotypes and Begomoviruses
4. Discussion
4.1. Classification of Whitefly Haplotypes Based on SNPS and Phylogeographical Relationships
4.2. Provisional Begomovirus Identification and Species Associated with NAFME Haplotypes
4.3. Associations between NAFME Haplotypes and Begomoviruses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, A.J.; Mansoor, S.; Briddon, R.W. Oman: A case for a sink of begomoviruses of geographically diverse origins. Trends Plant Sci. 2014, 19, 67–70. [Google Scholar] [CrossRef]
- Melgarejo, T.A.; Kon, T.; Rojas, M.R.; Paz-Carrasco, L.; Zerbini, F.M.; Gilbertson, R.L. Characterization of a new world monopartite begomovirus causing leaf curl disease of tomato in Ecuador and Peru reveals a new direction in geminivirus evolution. J. Virol. 2013, 87, 5397–5413. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.K. Phylogenetic biology of the Bemisia tabaci sibling species group. In Bemisia: Bionomics and Management of a Global Pest; Springer: Berlin/Heidelberg, Germany, 2009; pp. 31–67. [Google Scholar]
- De Moya, R.S.; Brown, J.K.; Sweet, A.D.; Walden, K.K.; Paredes-Montero, J.R.; Waterhouse, R.M.; Johnson, K.P. Nuclear orthologs derived from whole genome sequencing indicate cryptic diversity in the Bemisia tabaci (Insecta: Aleyrodidae) complex of whiteflies. Diversity 2019, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Hadjistylli, M.; Roderick, G.K.; Brown, J.K. Global population structure of a worldwide pest and virus vector: Genetic diversity and population history of the Bemisia tabaci sibling species group. PLoS ONE 2016, 11, e0165105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Bird, J.; Frohlich, D.; Rosell, R.; Bedford, I.; Markham, P. The relevance of variability within the Bemisia tabaci species complex to epidemics caused by subgroup III geminiviruses. In Bemisia 1995: Taxonomy, Biology, Damage, Control and Management; Gerling, G., Mayer, R.T., Eds.; Intercept: Andover, MA, USA, 1995; pp. 77–89. [Google Scholar]
- Deng, D.; McGrath, P.; Robinson, D.; Harrison, B. Detection and differentiation of whitefly-transmitted geminiviruses in plants and vector insects by the polymerase chain reaction with degenerate primers. Ann. Appl. Biol. 1994, 125, 327–336. [Google Scholar] [CrossRef]
- Idris, A.M.; Shahid, M.S.; Briddon, R.W.; Khan, A.J.; Zhu, J.K.; Brown, J.K. An unusual alphasatellite associated with monopartite begomoviruses attenuates symptoms and reduces betasatellite accumulation. J. Gen. Virol. 2011, 92 Pt 3, 706–717. [Google Scholar] [CrossRef] [PubMed]
- McGrath, P.F.; Harrison, B.D. Transmission of tomato leaf curl geminiviruses by Bemisia tabaci: Effects of virus isolate and vector biotype. Ann. Appl. Biol. 1995, 126, 307–316. [Google Scholar] [CrossRef]
- Díaz, F.; Endersby, N.M.; Hoffmann, A.A. Genetic structure of the whitefly Bemisia tabaci populations in Colombia following a recent invasion. Insect Sci. 2015, 22, 483–494. [Google Scholar] [CrossRef]
- Gabarra, R.; Alomar, Ò.; Castañé, C.; Goula, M.; Albajes, R. Movement of greenhouse whitefly and its predators between in-and outside of Mediterranean greenhouses. Agric. Ecosyst. Environ. 2004, 102, 341–348. [Google Scholar] [CrossRef]
- Gautam, S.; Mugerwa, H.; Buck, J.W.; Dutta, B.; Coolong, T.; Adkins, S.; Srinivasan, R. Differential Transmission of Old and New World Begomoviruses by Middle East-Asia Minor 1 (MEAM1) and Mediterranean (MED) Cryptic Species of Bemisia tabaci. Viruses 2022, 14, 1104. [Google Scholar] [CrossRef]
- Brown, J.K. The Bemisia tabaci complex: Genetic and phenotypic variation and relevance to TYLCV-vector interactions. In Tomato Yellow Leaf Curl Virus Disease; Springer Nature: Cham, Switzerland, 2007; pp. 25–56. [Google Scholar]
- Paredes-Montero, J.R.; Haq, Q.; Mohamed, A.A.; Brown, J.K. Phylogeographic and SNPs analyses of Bemisia tabaci B mitotype populations reveal only two of eight haplotypes are invasive. Biology 2021, 10, 1048. [Google Scholar] [CrossRef]
- Pan, H.; Chu, D.; Yan, W.; Su, Q.; Liu, B.; Wang, S.; Li, R. Rapid spread of tomato yellow leaf curl virus in China is aided differentially by two invasive whiteflies. PLoS ONE 2012, 7, e34817. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Chen, Q.; Guo, T.; Wang, X.; Li, P.; Liu, S. Differential efficiency of a begomovirus to cross the midgut of different species of whiteflies results in variation of virus transmission by the vectors. Sci. China Life Sci. 2018, 61, 1254–1265. [Google Scholar] [CrossRef]
- Ashfaq, M.; Hebert, P.D. DNA barcodes for bio-surveillance: Regulated and economically important arthropod plant pests. Genome 2016, 59, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Ellango, R.; Singh, S.T.; Rana, V.S.; Gayatri Priya, N.; Raina, H.; Chaubey, R.; Asokan, R. Distribution of Bemisia tabaci genetic groups in India. Environ. Entomol. 2015, 44, 1258–1264. [Google Scholar] [CrossRef]
- Islam, W.; Akutse, K.S.; Qasim, M.; Khan, K.A.; Ghramh, H.A.; Idrees, A.; Latif, S. Bemisia tabaci-mediated facilitation in diversity of begomoviruses: Evidence from recent molecular studies. Microb. Pathog. 2018, 123, 162–168. [Google Scholar] [CrossRef]
- Götz, M.; Winter, S. Diversity of Bemisia tabaci in Thailand and Vietnam and indications of species replacement. J. Asia-Pac. Entomol. 2016, 19, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Al-Shihi, A.A.; Hanson, P.; Al-Sadi, A.M.; Al-Yahyai, R.A.; Briddon, R.W.; Deadman, M.; Shahid, M.S. Evaluation of tomato inbred lines for resistance to the tomato yellow leaf curl disease complex in Oman. Crop Prot. 2018, 110, 91–98. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Shahid, M.S.; Raza, A.; Al-Sadi, A.M.; Briddon, R.W. Identification of Chilli leaf curl virus associated with tomato leaf curl betasatellite infecting Mentha in Oman. Can. J. Plant Pathol. 2019, 41, 291–295. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Brown, J.K.; Idris, A.M.; Torres-Jerez, I.; Banks, G.K.; Wyatt, S.D. The core region of the coat protein gene is highly useful for establishing the provisional identification and classification of begomoviruses. Arch. Virol. 2001, 146, 1581–1598. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Di Rienzo, J.; Casanoves, F.; Balzarini, M.; Gonzalez, L.; Tablada, M.; Robledo, C.W. InfoStat Versión 2013, Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Argentina. 2013. Available online: http://www.Infostat.com.ar (accessed on 16 March 2021).
- Greenacre, M.J.; Groenen, P.J. Weighted euclidean biplots. J. Classif. 2016, 33, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, D.; Torres-Jerez, I.; Bedford, I.; Markham, P.G.; Brown, J. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef]
- Saleh, D.; Laarif, A.; Clouet, C.; Gauthier, N. Spatial and host-plant partitioning between coexisting Bemisia tabaci cryptic species in Tunisia. Popul. Ecol. 2012, 54, 261–274. [Google Scholar] [CrossRef]
- Blackmer, J.; Byrne, D. Environmental and physiological factors influencing phototactic flight of Bemisia tabaci. Physiol. Entomol. 1993, 18, 336–342. [Google Scholar] [CrossRef]
- Blackmer, J.; Byrne, D.; Tu, Z. Behavioral, morphological, and physiological traits associated with migratory Bemisia tabaci (Homoptera: Aleyrodidae). J. Insect Behav. 1994, 8, 251–267. [Google Scholar] [CrossRef]
- Byrne, D.N. Migration and dispersal by the sweet potato whitefly, Bemisia tabaci. Agric. For. Meteorol. 1999, 97, 309–316. [Google Scholar] [CrossRef]
- Naranjo, S.E.; Castle, S.J.; Barro, P.J.D.; Liu, S.-S. Population dynamics, demography, dispersal and spread of Bemisia tabaci. In Bemisia: Bionomics and Management of a Global Pest; Springer: Berlin/Heidelberg, Germany, 2009; pp. 185–226. [Google Scholar]
- Paredes-Montero, J.R.; Ibarra, M.A.; Arias-Zambrano, M.; Peralta, E.L.; Brown, J.K. Phylo-biogeographical distribution of whitefly Bemisia tabaci (Insecta: Aleyrodidae) mitotypes in Ecuador. Ecosphere 2020, 11, e03154. [Google Scholar] [CrossRef]
- Cock, M. Bemisia tabaci, an Update 1986–1992 on the Cotton Whitefly with an Annotated Bibliography; CAB IIBC Silwood Park: Berks, UK, 1993. [Google Scholar]
- Akhtar, S.; Khan, A.J.; Singh, A.S.; Briddon, R.W. Identification of a disease complex involving a novel monopartite begomovirus with beta- and alphasatellites associated with okra leaf curl disease in Oman. Arch. Virol. 2014, 159, 1199–1205. [Google Scholar] [CrossRef]
- Shafiq, M.; Sattar, M.N.; Shahid, M.S.; Al-Sadi, A.M.; Briddon, R.W. Interaction of Watermelon chlorotic stunt virus with satellites. Australas. Plant Pathol. 2021, 50, 117–128. [Google Scholar] [CrossRef]
- Shahid, M.S.; Al-Sadi, A.M. First identification of Tomato leaf curl Palampur virus in Oman: Detection and characterization. J. Plant Prot. Res. 2022, 62, 295–301. [Google Scholar]
- Shahid, M.S. Characterization of Tomato leaf curl Palampur virus naturally infecting wild melon in Oman. Indian Phytopathol. 2022, 76, 215–221. [Google Scholar] [CrossRef]
- Hosseinzadeh, M.R.; Shams-Bakhsh, M.; Osaloo, S.K.; Brown, J.K. Phylogenetic relationships, recombination analysis, and genetic variability among diverse variants of Tomato yellow leaf curl virus in Iran and the Arabian Peninsula: Further support for a TYLCV center of diversity. Arch. Virol. 2014, 159, 485–497. [Google Scholar] [CrossRef]
- Zaidi, S.; Shakir, S.; Farooq, M.; Amin, I.; Mansoor, S. First report of a novel strain of Tomato yellow leaf curl virus causing yellow leaf curl disease on cluster bean in Pakistan. Plant Dis. 2017, 101, 1071. [Google Scholar]
- Ahmed, N.; Zaidi, S.S.-E.-A.; Amin, I.; Scheffler, B.E.; Mansoor, S. Tomato leaf curl Oman virus and associated Betasatellite causing leaf curl disease in tomato in Pakistan. Eur. J. Plant Pathol. 2021, 160, 249–257. [Google Scholar] [CrossRef]
- Al Shihi, A.A.; Al Sadi, A.M.; Deadman, M.; Briddon, R.W.; Shahid, M.S. Identification of a distinct strain of Cotton leaf curl Gezira virus infecting tomato in Oman. J. Phytopathol. 2018, 166, 199–205. [Google Scholar] [CrossRef]
- Shahid, M.S. Molecular and biological characterization of Chilli leaf curl virus and associated betasatellite infecting Cucurbita maxima in Oman. Virus Dis. 2020, 31, 378–382. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field sample Identification Number | Collection Year | Location/ GPS Coord. | Plant Host and Family | Agroecosystem | Plant-Infecting Begomo- Virus * | Begomovirus Predicted Center of Origin | Whitefly-Associated Begomovirus | Begomoviral Core Coat Protein Accession Number | Cytochrome Oxidase I Gene Accession Number | Bemisia tabaci Haplotype Identification by SNPs |
|---|---|---|---|---|---|---|---|---|---|---|
| 004-12 | 2015 | Al Barka/ 23.6837° N 57.9049° E | Tomato Solanaceae | Crop | CLCuGeV/ChiLCV | North Africa/Indian subcontinent | ChiLCV | OM326816 | OM368461 | NAFME5 |
| WSB-1CO13FR | 2015 | Al Qurayyat/ 23.2652° N 58.9034° E | Tomato Solanaceae | Crop | ChiLCV | Indian subcontinent | No begomovirus detected | NA ** | OM368471 | NAFME5 |
| 010-5 | 2015 | Al-Batinah/ 23.5684° N 57.5432° E | Mungbean Fabaceae | Crop | MYMIV | Indian subcontinent | No begomovirus detected | NA | OM368460 | NAFME5 |
| 002-5 | 2015 | Al Barka/ 23.6857° N 57.9048° E | Watermelon Cucurbitaceae | Crop | ChiLCV | Indian subcontinent | ChiLCV | OM326817 | OM368454 | NAFME5 |
| 005-1HamFR | 2015 | Al Qurayyat/ 23.2652° N 58.9034° E | Cucumber Cucurbitaceae | Crop | TYLCV-OM | Mediterranean and/or Middle East | No begomovirus detected | NA | OM368451 | NAFME5 |
| 008-1_28FR | 2016 | Al Barka/ 23.6837° N 57.9049° E | Tomato Solanaceae | Crop | ToLCLwV | Middle East | ToLCLwV | OM326823 | OM368473 | NAFME2 |
| WSE-2CO13FR | 2016 | Salalah/ 17.0302° N 54.1435° E | Mint Lamiaceae | Home garden | ChiLCV | Indian subcontinent | ChiLCV | OM326818 | OM368468 | NAFME2 |
| 008-2_28FR | 2016 | Al Buraymi/ 24.1679° N 56.1149° E | Tomato Solanaceae | Crop | TYLCV-OM | Mediterranean and/or Middle East | TYLCV-OM | OM326813 | OM368463 | NAFME2 |
| WS-1CO13FR | 2016 | Al Barka/ 23.6878° N 57.9059° E | Tomato Solanaceae | Crop | MYMIV/TYLCV-OM | Indian subcontinent/Middle East | TYLCV-OM | OM326812 | OM368475 | NAFME2 |
| 010-4 | 2016 | Khasab/ 26.1644° N 56.2426° E | Senna spp. Fabaceae | Weed | ChiLCV | Indian subcontinent | No begomovirus detected | NA | OM368450 | NAFME5 |
| 008-1 | 2016 | Salalah/ 17.0302° N 54.1415° E | Eggplant Solanaceae | Home garden | No begomovirus detected | NA | No begomovirus detected | NA | OM368465 | NAFME5 |
| 002-3 | 2016 | Khasab/ 26.1654° N 56.2426° E | Tomato Solanaceae | Crop | CLCuGeV | North Africa | No begomovirus detected | NA | OM368452 | NAFME5 |
| WSB-2CO13FR | 2016 | Al Buraymi/ 24.1679° N 56.1149° E | Tomato Solanaceae | Crop | TYLCV—OM | Mediterranean and/or Middle East | TYLCV-OM | OM326811 | OM368473 | NAFME2 |
| WSL-1CO13FR | 2016 | Khasab/ 26.1664° N 56.2432° E | Muskmelon Cucurbitaceae | Crop | No begomovirus detected | NA | No begomovirus detected | NA | OM368472 | NAFME2 |
| WSO-2CO13FR | 2016 | Sohar/ 24.396° N 56.408° E | Tobacco Solanaceae | Crop | ChiLCV | Indian subcontinent | ChiLCV | OM326819 | OM368463 | NAFME5 |
| WK-1CO13FR | 2016 | Khasab/ 26.1654° N 56.2432° E | Squash Cucurbitaceae | Crop | SLCuV | Central and North America | No begomovirus detected | NA | OM368467 | NAFME5 |
| WKM-1CO13FR | 2016 | Salalah/ 17.014° N 54.1013° E | Urtica Urticaceae | Weed | ChiLCV | Indian subcontinent | ChiLCV | OM326820 | OM368457 | NAFME5 |
| 008-4 | 2016 | Al Suwayq/ 23.8262° N 57.4288° E | Okra Malvaceae | Crop | CLCuGeV | Sudan | No begomovirus detected | NA | OM368449 | NAFME5 |
| 005-2HamFR | 2016 | Khasab/ 26.1654° N 56.2432° E | Red pumpkin Cucurbitaceae | Crop | No begomovirus detected | NA | No begomovirus detected | NA | OM368476 | NAFME5 |
| WSM-2CO13FR | 2017 | Al-Batinah/ 24.2421° N 56.4320° E | Tomato Solanaceae | Crop | ToLCBrV | Middle East | ToLCBrV | OM326815 | OM368464 | NAFME5 |
| 010-3 | 2017 | Rustaq/ 23.5058° N 57.2539° E | Eggplant Solanaceae | Home garden | No begomovirus detected | NA | No begomovirus detected | NA | 844/ OM368455 | NAFME5 |
| 008-3 | 2017 | Al Buraymi/ 24.1660° N 56.1130° E | Pumpkin Cucurbitaceae | Crop | WmCSV | Northern Middle East | No begomovirus detected | NA | OM368453 | NAFME5 |
| WSO-1CO13FR | 2017 | Sohar/ 24.196° N 56.408° E | Muskmelon Cucurbitaceae | Crop | ChiLCV | Indian subcontinent | No begomovirus detected | NA | OM368474 | NAFME2 |
| WS-2CO13FR | 2017 | Nizwa/ 22.9371° N 57.5383° E | Pumpkin Cucurbitaceae | Crop | TYLCV-OM | Mediterranean and/or Middle East | TYLCV-OM | OM326810 | OM368476 | NAFME2 |
| WS-3CO13FR | 2017 | Al-Batinah/ 23.1664° N 57.5432° E | Cucumber Cucurbitaceae | Green house | WmCSV/TYLCV-OM | Mediterranean and/or Middle East | TYLCV-OM | OM326809 | OM368470 | NAFME2 |
| Ham-2FR | 2018 | Al-Batinah/ 23.1664° N 57.5432° E | Muskmelon Cucurbitaceae | Small-scale | WmCSV | Northern Middle East | No begomovirus detected | NA | OM368477 | NAFME5 |
| 002-2 | 2018 | Khasab/ 26.1644° N 56.2426° E | Cucumber Cucurbitaceae | Green house | MYMIV | Indian subcontinent | No begomovirus detected | NA | OM368459 | NAFME5 |
| 004-2 | 2018 | Al Barka/ 23.6867° N 57.9058° E | Tomato Solanaceae | Crop | ChiLCV | Indian subcontinent | ChiLCV | OM326821 | OM368466 | NAFME5 |
| WK-2CO13FR | 2018 | Rustaq/ 23.5058° N 57.2519° E | Papaya Caricaceae | Garden | TYLCV-OM | Mediterranean and/or Middle East | TYLCV-OM | OM326808 | OM368456 | NAFME5 |
| WSM-1CO13FR | 2018 | Samail/ 23.2969° N 57.9731° E | Papaya Caricaceae | Garden | ChiLCV | Indian subcontinent | ChiLCV | OM326822 | OM368462 | NAFME5 |
| WSE-1CO13FR | 2018 | Al-Batinah/ 24.2420° N 56.4399° E | Tomato Solanaceae | Crop | ToLCSDV | North Africa | No begomovirus detected | NA | OM368477 | NAFME2 |
| WSL-2CO13FR | 2018 | Al-Batinah/ 24.2420° N 56.4299° E | Pumpkin Cucurbitaceae | Small-scale | ChiLCV | Indian subcontinent | No begomovirus detected | NA | OM368472 | NAFME3 |
| WKM-2CO13FR | 2018 | Al Masnaah/ 23.7474° N 57.6326° E | Watermelon Cucurbitaceae | Crop | WmCSV | Northern Middle East | No begomovirus detected | NA | OM368458 | NAFME5 |
| WSG-1CO13FR | 2018 | Nizwa/ 22.9392° N 57.5378° E | Pepper Solanaceae | Crop | ChiLCV | Indian subcontinent | ChiLCV | OM326814 | OM368451 | NAFME5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahid, M.S.; Paredes-Montero, J.R.; Ashfaq, M.; Al-Sadi, A.M.; Brown, J.K. Native and Non-Native Bemisia tabaci NAFME Haplotypes Can Be Implicated in Dispersal of Endemic and Introduced Begomoviruses in Oman. Insects 2023, 14, 268. https://doi.org/10.3390/insects14030268
Shahid MS, Paredes-Montero JR, Ashfaq M, Al-Sadi AM, Brown JK. Native and Non-Native Bemisia tabaci NAFME Haplotypes Can Be Implicated in Dispersal of Endemic and Introduced Begomoviruses in Oman. Insects. 2023; 14(3):268. https://doi.org/10.3390/insects14030268
Chicago/Turabian StyleShahid, Muhammad Shafiq, Jorge R. Paredes-Montero, Muhammad Ashfaq, Abdullah M. Al-Sadi, and Judith K. Brown. 2023. "Native and Non-Native Bemisia tabaci NAFME Haplotypes Can Be Implicated in Dispersal of Endemic and Introduced Begomoviruses in Oman" Insects 14, no. 3: 268. https://doi.org/10.3390/insects14030268