

Comparison of Yersinia enterocolitica DNA Methylation at Ambient and Host Temperatures

Abstract
:1. Introduction
2. Results
2.1. DNA Sequence Analysis Confirmed Four Genes Encoding Potential Methyltransferase Enzymes
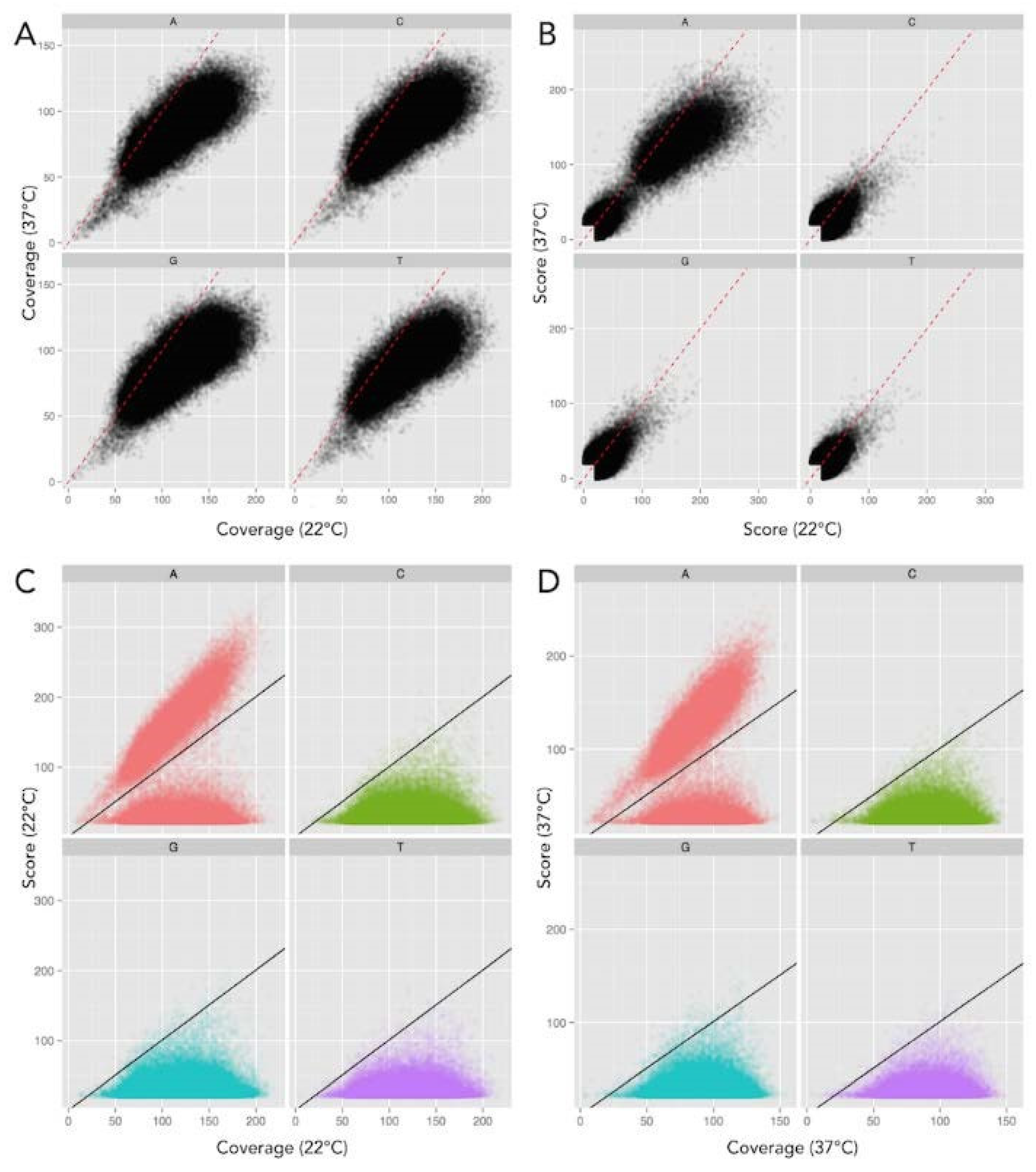
2.2. Inter-Pulse Duration Analysis Identified Methylated Nucleotides
2.3. Y. enterocolitica DNA Had No Evidence of Dcm (5mC) Methylation but Complete Methylation of All YenI Restriction Sites at Both 22 °C and 37 °C
2.4. Y. enterocolitica DNA Had Different Dam Methylation Patterns at 22 °C and 37 °C
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Culture Media, and DNA Techniques
4.2. SMRT Sequencing
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Maurelli, A.T. Temperature Regulation of Virulence Genes in Pathogenic Bacteria—A General Strategy for Human Pathogens. Microb. Pathog. 1989, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Michiels, T.; Cornelis, G.R. Secretion of Hybrid Proteins by the Yersinia Yop Export System. J. Bacteriol. 1991, 173, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Straley, S.C.; Plano, G.V.; Skrzypek, E.; Haddix, P.L.; Fields, K.A. Regulation by Ca2+ in the Yersinia low-Ca2+ response. Mol. Microbio. 1993, 8, 1005–1010. [Google Scholar] [CrossRef]
- Rebeil, R.; Ernst, R.K.; Gowen, B.B.; Miller, S.I.; Hinnebusch, B.J. Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol. 2004, 52, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Montminy, S.W.; Khan, N.; McGrath, S.; Walkowicz, M.J.; Sharp, F.; Conlon, J.E.; Fukase, K.; Kusumoto, S.; Sweet, C.; Miyake, K.; et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 2006, 7, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- de Koning-Ward, T.F.; Robins-Browne, R.M. A novel mechanism of urease regulation in Yersinia enterocolitica. FEMS Microbiol. Lett. 1997, 147, 221–226. [Google Scholar] [CrossRef]
- Minnich, S.A.; Rohde, H.N. A Rationale for Repression and/or Loss of Motility by Pathogenic Yersinia in the Mammalian Host. Adv. Exp. Med. Biol. 2007, 603, 298–311. [Google Scholar] [CrossRef]
- Rohde, J.R.; Fox, J.M.; Minnich, S.A. Thermoregulation in Yersinia enterocolitica is coincident with changes in DNA supercoiling. Mol. Microbiol. 1994, 12, 187–199. [Google Scholar] [CrossRef]
- Wilson, G.G.; Murray, N.E. Restriction and Modification Systems. Annu. Rev. Genet. 1991, 25, 585–627. [Google Scholar] [CrossRef]
- Roberts, D.; Hoopes, B.C.; McClure, W.R.; Kleckner, N. IS10 transposition is regulated by DNA adenine methylation. Cell 1985, 43, 117–130. [Google Scholar] [CrossRef]
- Collier, J.; McAdams, H.H.; Shapiro, L. A DNA methylation ratchet governs progression through a bacterial cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 17111–17116. [Google Scholar] [CrossRef]
- Collier, J. Epigenetic regulation of the bacterial cell cycle. Curr. Opin. Microbiol. 2009, 12, 722–729. [Google Scholar] [CrossRef]
- Low, D.A.; Weyand, N.J.; Mahan, M.J. Roles of DNA Adenine Methylation in Regulating Bacterial Gene Expression and Virulence. Infect. Immun. 2001, 69, 7197–7204. [Google Scholar] [CrossRef] [PubMed]
- Casadesús, J.; Low, D.A. Epigenetic Gene Regulation in the Bacterial World. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed]
- Srikhanta, Y.N.; Fox, K.L.; Jennings, M.P. The phasevarion: Phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 2010, 8, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.L.; Kleckner, N. The rate of Dam-mediated DNA adenine methylation in Escherichia coli. Gene 1988, 74, 189–190. [Google Scholar] [CrossRef]
- Atack, J.M.; Srikhanta, Y.N.; Fox, K.L.; Jurcisek, J.A.; Brockman, K.L.; Clark, T.A.; Boitano, M.; Power, P.M.; Jen, F.E.C.; McEwan, A.G.; et al. A biphasic epigenetic switch controls immunoevasion, virulence and niche adaptation in non-typeable Haemophilus influenzae. Nat. Commun. 2015, 6, 7828. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.J.; Han, S.-W.; Sul, W.J. Prokaryotic DNA methylation and its functional roles. J. Microbiol. 2021, 59, 242–248. [Google Scholar] [CrossRef]
- Gao, Q.; Lu, S.; Wang, Y.; He, L.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; et al. Bacterial DNA methytransferase: A key to the epigenetic world with lesons learned from proteobacteria. Front. Microbiol. 2023, 14, 1129437. [Google Scholar] [CrossRef]
- Dorman, C.J. DNA supercoiling and environmental regulation of gene expression in pathogenic bacteria. Infect. Immun. 1991, 59, 745–749. [Google Scholar] [CrossRef]
- Julio, S.M.; Heithoff, D.M.; Provenzano, D.; Klose, K.E.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA Adenine Methylase Is Essential for Viability and Plays a Role in the Pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 2001, 69, 7610–7615. [Google Scholar] [CrossRef] [PubMed]
- Fälker, S.; Schilling, J.; Schmidt, M.A.; Heusipp, G. Overproduction of DNA Adenine Methyltransferase Alters Motility, Invasion, and the Lipopolysaccharide O-Antigen Composition of Yersinia enterocolitica. Infect. Immun. 2007, 75, 4990–4997. [Google Scholar] [CrossRef]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Henikoff, S. Genome-wide analysis of DNA methylation patterns. Development 2007, 134, 3959–3965. [Google Scholar] [CrossRef]
- Reisenauer, A.; Kahng, L.S.; McCollum, S.; Shapiro, L. Bacterial DNA methylation: A cell cycle regulator? J. Bacteriol. 1999, 181, 5135–5139. [Google Scholar] [CrossRef] [PubMed]
- Wion, D.; Casadesús, J. N6-methyl-adenine: An epigenetic signal for DNA–protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192. [Google Scholar] [CrossRef]
- Marinus, M.G.; Casadesús, J. Roles of DNA adenine methylation in host–pathogen interactions: Mismatch repair, transcriptional regulation, and more. FEMS Microbiol. Rev. 2009, 33, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Clark, T.A.; Morgan, R.D.; Boitano, M.; Anton, B.P.; Luong, K.; Fomenkov, A.; Turner, S.W.; Korlach, J.; Roberts, R.J. The methylomes of six bacteria. Nucleic Acids Res. 2012, 40, 11450–11462. [Google Scholar] [CrossRef]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef]
- Davis, B.M.; Chao, M.C.; Waldor, M.K. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. Curr. Opin. Microbiol. 2013, 16, 192–198. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Rev. Micro 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2009, 38, D234–D236. [Google Scholar] [CrossRef] [PubMed]
- Kinder, S.A.; Badger, J.L.; Bryant, G.O.; Pepe, J.C.; Miller, V.L. Cloning of the YenI restriction-endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R(−)M(+) mutant. Gene 1993, 136, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Fälker, S.; Schmidt, M.A.; Heusipp, G. DNA methylation in Yersinia enterocolitica: Role of the DNA adenine methyltransferase in mismatch repair and regulation of virulence factors. Microbiology 2005, 151, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Fälker, S.; Schmidt, M.A.; Heusipp, G. Altered Ca2+ Regulation of Yop Secretion in Yersinia enterocolitica after DNA Adenine Methyltransferase Overproduction Is Mediated by Clp-Dependent Degradation of LcrG. J. Bacteriol. 2006, 188, 7072–7081. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Henikoff, S.; Haughn, G.W.; Calvo, J.M.; Wallace, J.C. A Large Family of Bacterial Activator Proteins. Proc. Natl. Acad. Sci. USA 1988, 85, 6602–6606. [Google Scholar] [CrossRef]
- Maddocks, S.E.; Oyston, P.C.F. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 2008, 154, 3609–3623. [Google Scholar] [CrossRef]
- Meadow, N.D.; Fox, D.K.; Roseman, S. The bacterial phosphoenol-pyruvate: Glycose phosphotransferase system. Annu. Rev. Biochem. 1990, 59, 497–542. [Google Scholar] [CrossRef]
- Romling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef]
- Bent, Z.W.; Poorey, K.; Brazel, D.M.; LaBauve, A.E.; Sinha, A.; Curtis, D.J.; House, S.E.; Tew, K.E.; Hamblin, R.Y.; Williams, K.P.; et al. Transcriptomic Analysis of Yersinia enterocolitica Biovar 1B Infecting Murine Macrophages Reveals New Mechanisms of Extracellular and Intracellular Survival. Infect. Immun. 2015, 83, 2672–2685. [Google Scholar] [CrossRef]
- Bölker, M.; Kahnmann, R. The Escherichia coli Regulatory Protein OxyR Discriminates between Methylated and Unmethylated States of the Phage Mu-Mom Promoter. EMBO J. 1989, 8, 2403–2410. [Google Scholar] [CrossRef]
- Casadesús, J.; Low, D.A. Programmed Heterogeneity: Epigenetic Mechanisms in Bacteria. J. Biol. Chem. 2013, 288, 13929–13935. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Wholey, W.Y.; Parker, B.W.; Kim, M.; Jakob, U. NemR Is a Bleach-sensing Transcription Factor. J. Biol. Chem. 2013, 288, 13789–13798. [Google Scholar] [CrossRef] [PubMed]
- Hassett, D.J.; Cohen, M.S. Bacterial adaptation to oxidative stress: Implications for pathogenesis and interaction with phagocytic cells. FASEB J. 1989, 3, 2574–2582. [Google Scholar] [CrossRef]
- Muskhelishvili, G.; Travers, A.A. Integration of syntactic and semantic properties of the DNA code reveals chromosomes as thermodynamic machines converting energy into information. Cell. Mol. Life Sci. 2013, 70, 4555–4567. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A.; Muskhelishvili, G. DNA structure and function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef]
- Wang, L.; Ling, Y.; Jiang, H.; Qiu, Y.; Qiu, J.; Chen, H.; Yang, R.; Zhou, D. AphA is required for biofilm formation, motility, and virulence in pandemic Vibrio parahaemolyticus. Intern. J. Food Microbiol. 2013, 160, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Kovacikova, G.; Lin, W.; Skorupski, K. Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol. Microbiol. 2004, 53, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Kovacikova, G.; Lin, W.; Skorupski, K. Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol. Microbiol. 2005, 57, 420–433. [Google Scholar] [CrossRef]
- Chester, B.; Stotzky, G. Temperature-dependent cultural and biochemical characteristics of rhamnose-positive Yersinia enterocolitica. J. Clin. Microbiol. 1976, 3, 119–127. [Google Scholar] [CrossRef]
- van der Woude, M.; Hale, W.B.; Low, D.A. Formation of DNA methylation patterns: Nonmethylated GATC sequences in gut and pap operons. J. Bacteriol. 1998, 180, 5913–5920. [Google Scholar] [CrossRef]
- Bobrov, A.G.; Kirillina, O.; Ryjenkov, D.A.; Waters, C.M.; Price, P.A.; Fetherston, J.D.; Mack, D.; Goldman, W.E.; Gomelsky, M.; Perry, R.D. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 2011, 79, 533–551. [Google Scholar] [CrossRef]
- Buchanan, S.K. Bacterial metal detectors. Mol. Microbiol. 2005, 58, 1205–1209. [Google Scholar] [CrossRef]
- White-Ziegler, C.A.; Davis, T.R. Genome-Wide Identification of H-NS-Controlled, Temperature-Regulated Genes in Escherichia coli K-12. J. Bacteriol. 2009, 191, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- White-Ziegler, C.A.; Angus Hill, M.L.; Braaten, B.A.; van der Woude, M.; Low, D.A. Thermoregulation of Escherichia coli pap transcription: H-NS is a temperature-dependent DNA methylation blocking factor. Mol. Microbiol. 1998, 28, 1121–1137. [Google Scholar] [CrossRef]
- Gerganova, V.; Berger, M.; Zaldastanishvili, E.; Sobetzko, P.; Lafon, C.; Mourez, M.; Travers, A.A.; Muskhelishvili, G. Chromosomal position shift of a regulatory gene alters the bacterial phenotype. Nucleic Acids Res. 2015, 43, 8215–8226. [Google Scholar] [CrossRef] [PubMed]
- Kozdon, J.B.; Melfi, M.D.; Luong, K.; Clark, T.A.; Boitano, M.; Wang, S.; Zhou, B.; Gonzalez, D.; Collier, J.; Turner, S.W.; et al. Global methylation state at base-pair resolution of the Caulobacter genome throughout the cell cycle. Proc. Natl. Acad. Sci. USA 2013, 110, E4658–E4667. [Google Scholar] [CrossRef] [PubMed]
- Blyn, L.B.; Braaten, B.A.; Low, D.A. Regulation of pap pilin phase variation by a mechanism involving differential dam methylation states. EMBO J. 1990, 9, 4045–4054. [Google Scholar] [CrossRef]
- Schadt, E.E.; Banerjee, O.; Fang, G.; Feng, Z.; Wong, W.H.; Zhang, X.; Kislyuk, A.; Clark, T.A.; Luong, K.; Keren-Paz, A.; et al. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 2013, 23, 129–141. [Google Scholar] [CrossRef]
- Travers, K.J.; Chin, C.S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.R.; Howard, S.; Wren, B.W.; Holden, M.T.G.; Crossman, L.; Challis, G.L.; Churcher, C.; Mungall, K.; Brooks, K.; Chillingworth, T.; et al. The Complete Genome Sequence and Comparative Genome Analysis of the High Pathogenicity Yersinia enterocolitica Strain 8081. PLoS Genet. 2006, 2, e206. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Total # of Motifs | 22 °C | 37 °C | |||
|---|---|---|---|---|---|
| # of Atypical Methylation Detected | # of Fully Methylation Detected | # of Atypical Methylation Detected | # of Fully Methylation Detected | ||
| 5′-GATC-3′ (Dam) | |||||
| Chromosome | 26,664 | 28 | 26,636 | 50 | 26,614 |
| pYV Plasmid | 454 | 7 | 447 | 7 | 447 |
| 5′-CTGCAG-3′ (YenI) | |||||
| Chromosome | 554 | 0 | 554 | 0 | 554 |
| pYV Plasmid | 11 | 0 | 11 | 0 | 11 |
| 5′-CCWGG-3′ (Dcm) | |||||
| Chromosome | 7237 | 0 | 0 | 0 | 0 |
| pYV Plasmid | 88 | 0 | 0 | 0 | 0 |
| Position | 22 °C a | 37°C a | Name | Location b | Cat. c | Description |
|---|---|---|---|---|---|---|
| 20357 | ◼◼ | ◻◻ | YEr002 | ORF | rR | 23S rRNA |
| 315457 | ◼◼ | ◼◻ | YEr007 | ORF, −160 from 3′ end | rR | 16S rRNA |
| 315631 | ◼◻ | ◼◻ | +16 from 3′ end of 16S rRNA | |||
| 356036 | ◼◼ | ◼◻ | YEr010 | ORF, −158 from 3′ end | rR | 16S rRNA |
| 356210 | ◼◼ | ◼◻ | +16 from 3′ end of 16S rRNA | |||
| 373662 | ◻◻ | ◼◻ | YE0315 | −172 from TSS | M | putative membrane transport proteins, similar to Salmonella TonB |
| YE0316 | −206 from TSS | MT | putative DNA-binding protein | |||
| 399373 | ◼◼ | ◻◻ | YE0335 | −75 from TSS | MT | HemN, coproporphyrinogen III oxidase |
| 623091 | ◻◻ | ◼◻ | YE0546 | ORF | M | putative glycosyltransferase |
| 888993 | ◻◻ | ◼◻ | YE0762 | ORF | MT | CysD, sulfate adenylyltransferase subunit 2 |
| 1042835 | ◻◻ | ◼◼ | YE0914 | −36 from TSS | R | putative LysR-type transcriptional regulator |
| 1104202 | ◻◻ | ◼◻ | YE0981 | −74 from TSS | H | hypothetical protein |
| 1111932 | ◼◼ | ◼◻ | YE0990 | ORF | MT | AYP/GTP-binding protein |
| 1113753 | ◼◼ | ◼◻ | YE0992 | ORF | H | hypothetical protein |
| 1228189 | ◻◻ | ◼◻ | YE1098 | −182 from TSS | MT | GutA, pts system, glucitol/sorbitol-specific iic2 component |
| 1403906 | ◻◻ | ◼◻ | YE1259 | −51 from TSS | R | putative PadR-like family transcriptional regulator |
| 1468949 | ◼◼ | ◼◻ | YE1322 | ORF | V | putative RTX-family protein |
| 1469615 | ◼◼ | ◼◻ | ||||
| 1471616 | ◼◼ | ◻◻ | ||||
| 1471736 | ◼◼ | ◼◻ | ||||
| 1471902 | ◼◼ | ◼◻ | ||||
| 1879399 | ◼◼ | ◼◻ | YE1683 | ORF | R | putative prophage encoded two-component system response regulator |
| 1881251 | ◼◼ | ◼◻ | YE1684 | ORF | R | putative prophage encoded two-component system histidine kinase |
| 2354334 | ◼◼ | ◼◻ | YE2154 | ORF. near 5′ | MT | Rnt, ribonuclease T |
| 2436018 | ◼◼ | ◼◻ | YE2225 | −288 from TSS | R | putative cyclic-di-GMP phosphodiesterase |
| 2598581 | ◼◼ | ◼◻ | YE2407 | ORF | V | putative hemolysin |
| 2602469 | ◼◼ | ◼◻ | YE2409 | +30 from 3′ end of mviN | MT | MviN, putative membrane-associated protein |
| 2852693 | ◼◼ | ◼◻ | YE2635 | ORF | putative metallo-beta-lactamase superfamily protein | |
| 3352496 | ◼◼ | ◼◻ | YE3082 | ORF | MT | RfbX, putative O-antigen transporter |
| 3548081 | ◼◼ | ◼◻ | YEr017 | ORF | rR | 23S rRNA |
| 3549372 | ◼◻ | ◻◻ | ||||
| 3550740 | ◼◼ | ◼◻ | YEr018 | +17 from 3′ end of 16S rRNA | rR | 16S rRNA |
| 3655022 | ◼◼ | ◼◻ | YE3343 | ORF. near 3′ | MT | OutL, general secretion pathway protein L |
| 3739389 | ◻◻ | ◼◻ | YE3423 | −85 from TSS | R | ArsR-family transcriptional regulator |
| YE3424 | −66 from TSS | M | putative zinc metallopeptidases | |||
| 4243325 | ◼◼ | ◻◻ | YEr022 | +17 from 3′ end of 16S rRNA | rR | 16S rRNA |
| 4441739 | ◻◻ | ◼◼ | YE4070 | −100 from TSS | MT | putative oligogalacturonate-specific porin protein |
| 2443 | ◼◼ | ◼◻ | YEP0004 | ORF | V | YopQ, virulence plasmid protein |
| 2654 | ◼◻ | ◼◻ | ORF | |||
| 15967 | ◼◻ | ◼◼ | YEP0018 | ORF | MT | LcrD, low calcium response locus membrane protein d |
| 21305 | ◼◻ | ◼◻ | YEP0026 | ORF | V | YscP, putative type III secretion protein |
| 23409 | ◼◻ | ◼◼ | YEP0029 | ORF | V | YscS, putative type III secretion protein |
| 24586 | ◼◼ | ◼◻ | YEP0031 | ORF | V | YscU, putative type III secretion protein |
| 25258 | ◼◻ | ◼◼ | ORF | |||
| 45942 | ◼◼ | ◼◻ | YEP0064 | −86 from TSS | H | putative pseudogene |
| 48481 | ◼◻ | ◼◼ | YEP0066 | −158 from TSS | M | putative YadA invasin |
| 66135 | ◼◼ | ◼◻ | YEP0096 | ORF | MT | putative plasmid copy control protein |
| Position | 22°C a | 37°C a | Name | Location b | Cat. c | Description |
|---|---|---|---|---|---|---|
| 316977 | ◻◻ | ◻◻ | YEr008 | ORF | rR | 23s rRNA |
| 357556 | ◻◻ | ◻◻ | YEr011 | ORF | rR | 23s rRNA |
| 1105938 | ◻◻ | ◻◻ | YE0983 | −131 from TSS | H | hypothetical protein |
| 1105951 | ◻◻ | ◻◻ | −144 from TSS | |||
| 1998525 | ◻◻ | ◻◻ | YE1808 | ORF | MT | YenI, methyltransferase-endonuclease |
| 1998670 | ◻◻ | ◻◻ | ||||
| 2057696 | ◻◻ | ◻◻ | YE1876 | −106 from TSS | MT | Ogl, oligogalacturonate lyase |
| 2351648 | ◻◻ | ◻◻ | YE2151 | −23 from TSS | R | transcriptional repressor NemR |
| 2435768 | ◻◻ | ◻◻ | YE2225 | −38 from TSS | R | putative cyclic-di-GMP phosphodiesterase. |
| 2435785 | ◻◻ | ◻◻ | −55 from TSS | |||
| 2435803 | ◻◻ | ◻◻ | −73 from TSS | |||
| 2656973 | ◻◻ | ◻◻ | YE2469 | −369 from TSS | MT | ArgM, bifunctional succinylornithine transaminase/acetylornithine transaminase |
| 2857854 | ◻◻ | ◻◻ | YE2639 | −318 from TSS | M | putative transporter protein |
| 3351041 | ◻◻ | ◻◻ | YE3081 | ORF | MT | WbcD, putative 6-deoxy-D-Gul transferase |
| YE3080 | −191 from TSS | MT | WbcE, putative glycosyl transferases | |||
| 4241957 | ◻◻ | ◻◻ | YEr021 | ORF | rR | 23s rRNA |
| 4243004 | ◻◻ | ◻◻ | YEt079 | ORF | tRNA-Ala | |
| 65753 | ◻◻ | ◻◻ | YEP0094 | ORF | MT | IS541 transposase |
| YEP0095 | −240 from TSS | MT | putative plasmid copy number |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Hofwegen, D.J.; Hovde, C.J.; Minnich, S.A. Comparison of Yersinia enterocolitica DNA Methylation at Ambient and Host Temperatures. Epigenomes 2023, 7, 30. https://doi.org/10.3390/epigenomes7040030
Van Hofwegen DJ, Hovde CJ, Minnich SA. Comparison of Yersinia enterocolitica DNA Methylation at Ambient and Host Temperatures. Epigenomes. 2023; 7(4):30. https://doi.org/10.3390/epigenomes7040030
Chicago/Turabian StyleVan Hofwegen, Dustin J., Carolyn J. Hovde, and Scott A. Minnich. 2023. "Comparison of Yersinia enterocolitica DNA Methylation at Ambient and Host Temperatures" Epigenomes 7, no. 4: 30. https://doi.org/10.3390/epigenomes7040030
APA StyleVan Hofwegen, D. J., Hovde, C. J., & Minnich, S. A. (2023). Comparison of Yersinia enterocolitica DNA Methylation at Ambient and Host Temperatures. Epigenomes, 7(4), 30. https://doi.org/10.3390/epigenomes7040030