

A Comparative Analysis of Mouse Imprinted and Random X-Chromosome Inactivation


Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI 48105, USA
*
Author to whom correspondence should be addressed.
Epigenomes 2024, 8(1), 8; https://doi.org/10.3390/epigenomes8010008
Submission received: 3 January 2024
/
Revised: 1 February 2024
/
Accepted: 6 February 2024
/
Published: 10 February 2024
(This article belongs to the Special Issue X-Chromosome Inactivation)
Abstract
:The mammalian sexes are distinguished by the X and Y chromosomes. Whereas males harbor one X and one Y chromosome, females harbor two X chromosomes. To equalize X-linked gene expression between the sexes, therian mammals have evolved X-chromosome inactivation as a dosage compensation mechanism. During X-inactivation, most genes on one of the two X chromosomes in females are transcriptionally silenced, thus equalizing X-linked gene expression between the sexes. Two forms of X-inactivation characterize eutherian mammals, imprinted and random. Imprinted X-inactivation is defined by the exclusive inactivation of the paternal X chromosome in all cells, whereas random X-inactivation results in the silencing of genes on either the paternal or maternal X chromosome in individual cells. Both forms of X-inactivation have been studied intensively in the mouse model system, which undergoes both imprinted and random X-inactivation early in embryonic development. Stable imprinted and random X-inactivation requires the induction of the Xist long non-coding RNA. Following its induction, Xist RNA recruits proteins and complexes that silence genes on the inactive-X. In this review, we present a current understanding of the mechanisms of Xist RNA induction, and, separately, the establishment and maintenance of gene silencing on the inactive-X by Xist RNA during imprinted and random X-inactivation.
1. Introduction
The therian mammalian sex chromosomes differ in their gene contents; whereas the X chromosome is gene-rich, the Y chromosome is gene-poor [1,2]. The unequal gene contents on the X and Y chromosomes are hypothesized to reflect their evolution. The sex chromosomes are thought to have originated from a homologous pair of autosomes [3,4,5,6,7], and the proto-Y emerged when one autosome accumulated genes favoring male sexual differentiation [1,3,5,7]. To maintain the linked segregation of male sexual differentiation genes, the proto-Y chromosome is thought to have undergone a series of crossover-suppressing inversions to form the Y chromosome [7]. Suppressed crossing over is believed to have contributed to the degradation of genes on the Y chromosome [1,3,5,8].
Gene loss on the Y chromosome created an imbalance in X–Y gene dosage relative to the autosomes and between XX females and XY males. To compensate for gene loss on the Y chromosome, homologous X-linked genes became upregulated in males [3,9,10]. This upregulation also occurred from both X chromosomes in females, resulting in excessive X-linked gene expression in females relative to males [3,9,10]. To rectify the overexpression of X-linked genes in females, therian mammals are believed to have evolved X-chromosome inactivation as a dosage compensation mechanism [3,11].
During X-inactivation, most genes on one of the two X chromosomes in females are transcriptionally silenced, thereby equalizing X-linked gene expression between females and males [11]. Two forms of X-inactivation characterize therian mammals. Metatherian mammals (marsupials) exclusively inactivate the paternal X chromosome (Xp) in a process termed imprinted X-inactivation [12,13]. Eutherian mammals (‘placentals’) undergo imprinted as well as random X-inactivation, which results in the silencing of genes on either the Xp or the Xm (maternal-X) in individual cells [11,14]. Whereas some eutherian species, e.g., mice and voles, exhibit both imprinted and random X-inactivation, others appear to only undergo random X-inactivation, e.g., humans and rabbits [14,15,16,17,18].
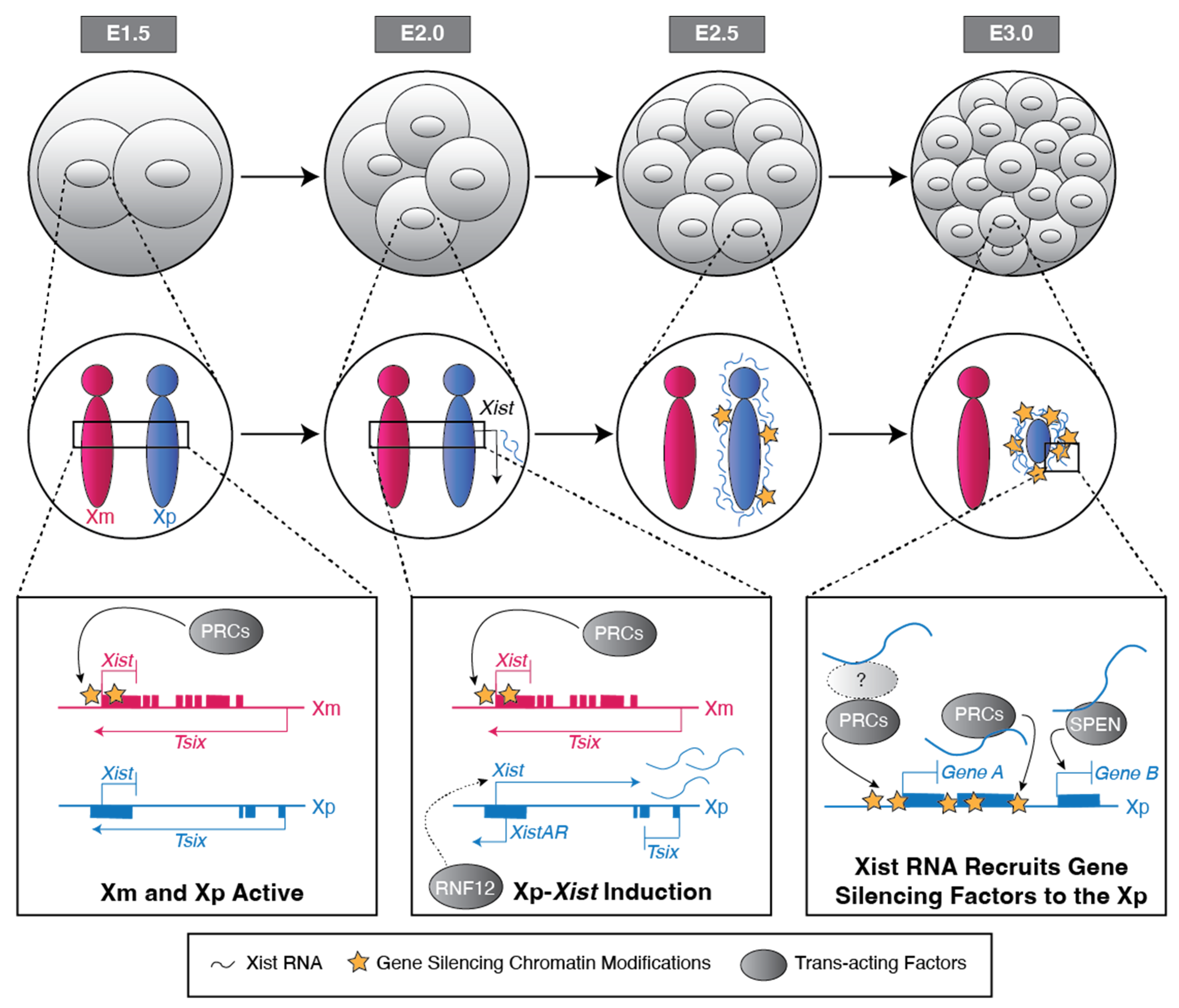
Both imprinted and random X-inactivation are established early in embryogenesis and have been studied extensively in the mouse model system. Shortly after the zygote stage, all cells in the early mouse embryo initiate imprinted X-inactivation, resulting in the silencing of genes exclusively on the Xp [18]. Imprinted X-inactivation of the Xp is subsequently maintained in the extra-embryonic trophectodermal (placental) and primitive endodermal (yolk-sac) lineages [18,19]. In the pluripotent epiblast cells of the late blastocyst (~128-cell stage), the Xp becomes reactivated [20]. As the pluripotent epiblast differentiates, each cell individually inactivates either the Xp or Xm and thus undergoes random X-inactivation [11]. Replicated copies of the randomly inactivated X chromosome are then stably maintained as inactive in most cells as the epiblast differentiates into the somatic tissues of the developing embryo. That two X chromosomes become transcriptionally divergent and that their transcriptional states are stably maintained across many cell division cycles make X-inactivation a paradigm of non-sequence-dependent, or epigenetic, transcriptional regulation.
Stable imprinted and random X-inactivation in eutherian mammals requires the Xist long non-coding RNA (lncRNA). Xist RNA is upregulated from the future inactive-X [21] and functions in cis to silence genes [22,23,24]. X-inactivation can be separated into at least three phases: initiation, establishment, and maintenance. Initiation is defined as the induction of Xist RNA from the future inactive-X. Once induced, Xist RNA ‘coats’ the inactive-X and directly or indirectly recruits proteins and complexes to the inactive-X. In the establishment phase, the proteins and complexes recruited by Xist RNA silence gene expression on the future inactive-X. During the maintenance phase, the silenced state of the inactive-X is stably propagated across cell divisions in descendant cells. Discussed below is our current understanding of the mechanisms regulating Xist RNA induction and, separately, establishment and maintenance of gene silencing on the inactive X chromosome during imprinted as well as random X-inactivation.
2. Regulation of Xist in Imprinted X-Inactivation
In imprinted X-inactivation, Xist RNA is expressed exclusively from the Xp [18]. In principle, Xist RNA expression from the Xp but not the Xm could be due to a chromatin-based mark on the Xm or the Xp. The transmission of an Xm-specific epigenetic imprint from the oocyte may prevent Xist expression from and inactivation of the Xm in the embryo. Conversely, the inheritance of an Xp-specific imprint from the sperm might facilitate Xist RNA expression from and inactivation of the Xp in the early female embryo. One model of the Xp imprint posits that the silencing of the sex chromosomes during meiosis in males, termed meiotic sex chromosome inactivation (MSCI), is the basis for the selective inactivation of the Xp in the early embryo [25,26]. MSCI silences genes on the X and Y sex chromosomes, which together form a heterochromatic ‘sex body’ during the pachytene stage of meiotic prophase I during spermatogenesis [27,28]. However, much work has shown that the Xp is active prior to the initiation of imprinted X-inactivation in the early embryo [29,30,31], suggesting a lack of inherited silencing of the Xp in zygotes and preimplantation embryos. Any impact of MSCI on imprinted X-inactivation must thus manifest only after the Xp becomes transcriptionally active in the zygote.
Much work has also explored whether and how the Xm carries the chromatin-based imprint in imprinted X-inactivation. Early evidence for an Xm epigenetic imprint arose from studies examining mouse embryos with supernumerary X chromosomes (e.g., XmXmY, XmXmXp). Preimplantation-stage embryos with two Xm’s resist Xist induction and maintain both Xm’s in a transcriptionally active state [32,33,34,35]. In contrast, individual cells in early embryos harboring two Xp’s initially express Xist RNA from both X chromosomes but later display Xist expression from only one of the two Xp’s [36]. These studies indicated that whereas the Xm effectively repels Xist induction in the early embryo, the Xp is more epigenetically labile and can both induce and repress Xist expression. The Xm is thus believed to carry a stringent germline imprint that prevents its inactivation during imprinted X-inactivation in the early embryo.
The Xist antisense lncRNA, Tsix, has been of particular interest to understand how inactivation of the Xm is forestalled in the early mouse embryo. Whereas Xist is expressed from the inactive-X, Tsix expression marks the active-X [37,38,39]. Xist is thus induced from the paternal-X, and Tsix is expressed from the maternal-X in imprinted X-inactivation [37,39,40]. Embryos that inherit a Tsix mutation on the Xm ectopically express Xist from both the Xm (Xm-Xist) and the Xp (Xp-Xist) in cells that would otherwise exclusively express Xp-Xist [39,40], suggesting that Tsix is a cis repressor of Xm-Xist. The maternal germline imprint may thus prevent Xm-Xist expression in the early embryo by inducing Tsix expression [40]. However, tests to determine the temporal requirement of Tsix in repressing Xist during embryonic development revealed that Tsix is required to repress Xist on the Xm beginning only at the late blastocyst stage [41]. Tsix is thus dispensable to repress Xm-Xist at the onset of imprinted X-inactivation during the post-zygotic early mouse embryonic stages [41]. A Tsix-independent regulatory mechanism thus represses Xm-Xist in the zygote and preimplantation embryonic stages.
The H3 Lysine 27 tri-methylation (H3K27me3) chromatin mark emerged as a candidate Xm germline epigenetic imprint upon the discovery that H3K27me3 is enriched in the Xm-Xist locus in mouse oocytes and early embryos [42,43]. Xp-Xist, by contrast, is devoid of H3K27me3 enrichment in the sperm and early embryos (Figure 1) [42,43]. H3K27me3 is catalyzed by the Polycomb Repressive Complex 2 (PRC2) [44,45,46,47]. Upon depletion of PRC2 activity in oocytes, Xm-Xist is de-repressed in the early mouse embryo [43,48]. Early embryos generated from PRC2-mutant oocytes express Xist RNA from both the Xm and Xp [43,48]. In later-stage preimplantation embryos, this biallelic Xist RNA induction is stochastically resolved into one or the other X chromosome expressing Xist in most cells, thus resulting in randomization of X-inactivation in cells that would normally undergo imprinted X-inactivation [43].
In addition to PRC2, the related PRC1 complex may also ensure the repression of Xm-Xist. PRC1 catalyzes the gene silencing-associated histone modification H2A lysine 119 mono-ubiquitination (H2AK119ub1) [49]. H2AK119ub1, like H3K27me3, is enriched at the Xm-Xist locus in mouse oocytes and zygotes, but not at the Xp-Xist locus in sperm or early embryos [50,51]. Like the absence of PRC2 activity, the absence of the PRC1 subunits PCGF1 and PCGF6 in the oocyte results in ectopic Xm-Xist induction in early embryos [51]. The enrichment dynamics of H3K27me3 and H2AK119ub1 at the Xm-Xist locus differ during early embryogenesis. Whereas H3K27me3 is enriched at Xm-Xist across preimplantation development of wild-type (WT) embryos, H2AK119ub1 becomes depleted at Xm-Xist from the two-cell to blastocyst embryonic stages before being enriched again [50]. An interpretation of these findings is that both oocyte-derived marks are required to repress Xm-Xist in the early embryo and that H3K27me3 may serve to recruit PRC1 and establish H2AK119ub1 at Xm-Xist during preimplantation mouse embryogenesis [50].
In contrast to mice, human zygotes do not express the core PRC2 components until zygotic genome activation at the ~eight-cell stage of embryogenesis [43,52]. Imprinted X-inactivation may thus arise in a species-specific manner due to the presence of oocyte-derived silencing complexes that target Xm-Xist for silencing, e.g., the PRC proteins. The reasons why the oocyte-derived PRC proteins do not also target the Xp-Xist locus in the early mouse embryo are unclear. The resistance of Xp-Xist to repression may in principle arise from either the absence of factor(s) in the sperm and early embryo that function to recruit the PRCs to Xm-Xist in the oocyte, or, formally, the presence of a paternally inherited chromatin mark or chromosome conformation state, e.g., due to MSCI, that forestalls PRC2 occupancy at Xp-Xist.
SMCHD1 (structural maintenance of chromosomes hinge domain containing protein 1) also contributes to Xm-Xist repression in pre-implantation embryos [53]. SMCHD1 is thought to function primarily as a chromatin remodeler and compactor [54,55]. The absence of oocyte-derived SMCHD1 results in the incompletely penetrant de-repression of Xm-Xist in ~75% of female and ~50% of male early embryos [53]. However, Xm-Xist repression is restored upon zygotic SMCHD1 expression [53]. Evidence further suggests that SMCHD1 functions downstream of PRC2 and PRC1, suggesting that SMCHD1 enforces rather than initiates Xm-Xist silencing [53,56].
The induction of Xist RNA from the Xp requires an antisense transcript at the 5′ end of Xist called XistAR (Figure 1) [57]. Like Xist RNA, XistAR is expressed exclusively from the Xp in early mouse embryos and in cultured extraembryonic endoderm (XEN) and trophoblast stem cells (TSCs) that stably maintain imprinted X-inactivation ex vivo [57]. Xp-XistAR mutant embryos fail to properly upregulate Xist RNA and perish early in embryogenesis [57,58].
The X-linked loci Ftx and Xert have also been tested as cis-regulators of Xp-Xist in imprinted X-inactivation. Ftx resides ~150 kb upstream of Xist and encodes a lncRNA [59,60,61]. Xert is located ~200 kb upstream of Xist, contains an Xist enhancer cluster, and transcribes a lncRNA [62]. The Ftx locus is dispensable for imprinted X-inactivation in pre-implantation embryos [63]. However, recent findings demonstrate that deleting two enhancer elements within the Xert and Ftx loci, respectively, abrogates Xist RNA induction in eight-cell embryos [64]. This observation suggests that coordination between these cis-acting loci in the X-inactivation center may be necessary to upregulate Xist expression during imprinted X-inactivation.
The induction of Xp-Xist RNA also requires the X chromosome-encoded protein RNF12 (also known as RLIM; Figure 1). RNF12 is an E3 ubiquitin ligase that functions in part to ubiquitinate and mark proteins for degradation [65,66]. A female-specific function of RNF12 is supported by mouse breeding data. Whereas Rnf12–Y hemizygous-mutant males are viable and fertile, heterozygous Rnf12−/+ female mice that inherit an Rnf12-mutant allele on the Xm but not the Xp are inviable [67]. Rnf12−/+ female embryos exhibit defective Xp-Xist upregulation and X-linked gene silencing, which may contribute to their lethality [67].
RNF12 is expressed more highly in female pre-implantation embryos relative to males [67]. This elevated expression of RNF12 might underpin the induction of Xist selectively in females. Rnf12−/− female embryos, though, initiate but do not maintain Xp-Xist expression in the absence of both oocyte-derived and zygotically expressed RNF12 [68]. Furthermore, although RNF12 is required to maintain the expression of Xp-Xist in the early mouse embryo, it is insufficient to induce Xist from the Xm. These observations suggest that (1) an epigenetic imprint, i.e., H3K27me3 or H2AK119ub1, prevents RNF12-mediatied induction of Xm-Xist; and/or, (2) RNF12 expression is below the threshold required to induce Xm-Xist. In support of the latter, overexpression of RNF12 in early mouse embryos results in Xist induction from both the Xm and Xp [69].
3. Mechanisms of Gene Silencing on the Inactive-X during Imprinted X-Inactivation
PRC2 and PRC1 not only deposit the H3K27me3 and H2AK119ub1 marks, respectively, to repress Xm-Xist, but may also contribute to gene silencing on the inactive-Xp during imprinted X-inactivation. Xist RNA either directly or indirectly recruits both PRC complexes to the inactive-X [70,71]. Soon after Xp-Xist RNA induction, the Xp becomes enriched for the core components of both PRC2 and PRC1 as well as for the H3K27me3 and H2AK119ub1 marks that the two complexes catalyze [70,71,72,73,74,75].
Tests to determine the contributions of oocyte-derived and zygotic PRC2 revealed that PRC2 may contribute to the establishment of X-linked gene silencing during imprinted X-inactivation. Mutant female embryos lacking both the maternal (oocyte-derived) and zygotic (mz−/−) PRC2 core component EED as well as embryos lacking only maternal EED (m−/−) equally induce Xist randomly from either the Xm or Xp [43]. However, Eed mz−/− embryos exhibit a greater defect in Xp gene silencing relative to Eed m−/− embryos [43]. These observations suggest that oocyte-derived and zygotic PRC2 together may be required to establish gene silencing on the inactive-Xp during the establishment of imprinted X-inactivation [43].
In addition to enforcing the silencing of Xm-Xist, SMCHD1 may contribute to the establishment of gene silencing on the inactive-X during imprinted X-inactivation. Smchd1−/+ mutant female embryos that lack oocyte-derived SMCHD1 ectopically express a subset of paternal X-linked genes that are normally silenced in the placenta at embryonic day (E) 14.5 [53]. Formally, either the initial absence of oocyte-derived SMCHD1 or the haploinsufficiency of SMCHD1 during embryogenesis might explain this gene silencing defect. However, relative to these Smchd1−/+ mutant embryos, female heterozygotes mutant for paternal Smchd1 and WT for maternal Smchd1 (Smchd1 +/−) show a milder paternal X-linked gene silencing defect in the placenta [53]. Therefore, oocyte-derived SMCHD1 appears to be required to establish or maintain silencing of a subset of Xp-linked genes. Zygotically-expressed SMCHD1 may additionally contribute to the maintenance of gene silencing of a set of X-linked genes during imprinted X-inactivation [76,77]. SMCHD1 homozygous mutant (Smchd1−/−) female post-implantation embryos exhibit de-repression of a handful of X-linked genes tested thus far in the extraembryonic ectoderm and trophoblast lineages [76,77]. However, X chromosome-wide gene expression changes in the extraembryonic tissues of Smchd1−/− female embryos await further testing. Smchd1−/− female embryos display defective trophoblast giant cell development and do not survive past mid-gestation, whereas Smchd1−/− males are viable [76]. The female-specific lethality of Smchd−/− later-stage embryos is consistent with a role for SMCHD1 in maintaining imprinted X-inactivation.
The autosomally encoded SPEN protein (also known as MINT or SHARP) is another key regulator that establishes gene silencing on the inactive-Xp (Figure 1). SPEN interacts with histone deacetylases (HDACs) [78], which help silence genes by removing acetyl marks on histones [79]. Mouse blastocyst-stage embryos lacking both oocyte-derived and zygotic SPEN (Spen mz−/−) exhibit a pronounced defect in the establishment of gene silencing on the Xist-expressing Xp [80]. However, early embryos lacking oocyte-derived SPEN but expressing zygotic SPEN (Spen−/+) are not defective in Xp gene silencing [80]. This suggests that expression of zygotic SPEN is sufficient to establish gene silencing during imprinted X-inactivation. Both zygotically-mutant Spen−/− female and male embryos perish during mid-gestation between E12.5 to E14.5 [81]. The lethality of both Spen−/− female and male embryos at a similar and relatively late embryonic stage argues against a broad and female-specific role for SPEN in X-inactivation.
PRC2 and PRC1 are also required to maintain gene silencing on the inactive-Xp (Figure 1). When zygotic PRC2 or PRC1 are absent, defects in Xp-linked gene silencing emerge in the post-implantation embryonic stages in differentiating extraembryonic cells, which normally maintain imprinted inactivation of the Xp [82,83,84]. Female embryos lacking the PRC2 subunit EED (Eed−/−) exhibit prominent Xp-linked gene silencing defects in the extraembryonic ectoderm and to some extent in the extraembryonic endoderm of post-implantation gastrulation-stage embryos [83]. Furthermore, relative to Eed−/− male embryos, Eed−/− female embryos exhibit reduced development of trophoblast giant cells, possibly due to defective imprinted X-inactivation in the trophectoderm lineage [85]. Similarly, female embryos lacking the PRC1 subunit RNF2 (also known as RING1B; Rnf2−/−) exhibit de-repression of X-linked genes in the extraembryonic ectoderm, albeit to a lesser extent than Eed−/− female embryos [83]. The de-repression of X-linked genes in the extraembryonic cells of later-stage embryos in the absence of either EED or RNF2 supports the requirements of both PRC2 and PRC1 in maintaining imprinted X-inactivation of the Xp. The presence of oocyte-derived PRC2 and PRC1 components, though, may mask a broader Xp-linked gene silencing defect in the Eed−/− and Rnf2−/− zygotically mutant early embryos.
The absence of PRC2 or PRC1 results in defective silencing of both overlapping and unique sets of Xp-linked genes in the extra-embryonic tissues [82,86], suggesting both redundant and independent targeting of genes on the inactive-X by PRC2 and PRC1 during imprinted X-inactivation. Loss of both PRC2 and PRC1 together in the early embryo may reveal a synergistic role for the two complexes in silencing Xp-linked genes.
4. Regulation of Xist in Random X-Inactivation
Random X-inactivation initiates when the pluripotent embryonic epiblast begins to differentiate, just after implantation of the mouse embryo [87,88,89]. The onset of random X-inactivation has been studied to some extent in developing mouse embryos and more extensively in differentiating mouse embryonic stem cells (ESCs). Like the pluripotent epiblast cells in the developing embryo, pluripotent female ESCs harbor two active X chromosomes [90,91,92,93]. And, like the differentiating pluripotent epiblast cells in the female embryo, differentiating female ESCs randomly inactivate one of their two X chromosomes in individual cells [93].
In random X-inactivation, Xist RNA is induced from either the Xm or Xp in individual cells [11]. Early models of random X-inactivation posited that cellular mechanisms ‘counted’ the number of X chromosomes and then ‘chose’ one of the two X chromosomes for silencing [92,94,95]. If a cell counts two or more X chromosomes per diploid set of autosomes, it will trigger inactivation of all X chromosomes except for one. The choice step would subsequently decide which of the X chromosome(s) to inactivate.
The counting, choice, and inactivation steps underlying random X-inactivation have long been thought to be controlled by a several-hundred kilobase region of the X chromosome termed the X-inactivation center (XIC) [93,96,97]. Xist maps to the XIC and the XIC is thought to encode all sequences that regulate Xist (Table 1) [93,96,97]. The XIC is thus thought to be both necessary and sufficient to induce X-inactivation [93,96,97].
The XIC-derived Xist antisense Tsix lncRNA was put forth as a regulator of both counting and choice by controlling Xist expression and thus X-inactivation [37,98]. The requirement of Tsix in X chromosome counting was initially tested in XY male and XO ESCs. In XY and XO cells, the counting process would prevent Xist induction from and X-inactivation of the single X chromosome. If Tsix controls counting, loss of Tsix is expected to trigger ectopic Xist expression and inactivation of the single X chromosome in the XY and XO cells. Initial studies provided conflicting results for the role of Tsix in counting. One study suggested that deleting a 65 kb sequence containing a ~37 kb proximal portion of the Tsix locus might interfere with counting, as mutant XO cells ectopically expressed Xist [94]. Other studies with Tsix-mutant (Tsix −Y) male ESCs reported transient ectopic Xist expression in a subset of the cells, which became extinguished upon extended culture [37,99], suggesting that the counting step was largely intact in the absence of Tsix. To test the function of Tsix in choice, Tsix heterozygous mutant (Tsix +/−) female ESCs were differentiated to induce Xist. These Tsix +/− ESCs exhibited biased inactivation of the Tsix-mutant X chromosome in all cells, consistent with a defect in choice [37,40]. That only a single X is inactivated, though, suggested that X chromosome counting remained intact in Tsix +/− females [37,40].
To reconcile the disparate roles attributed to Tsix in X chromosome counting and choice, a set of studies examined the temporal impact of Tsix loss on Xist induction in developing embryos and differentiating pluripotent cells [41,100,101]. If Tsix is required for counting, Xist should be ectopically expressed from the single X chromosome in Tsix −Y males during the early stages of differentiation corresponding to when random X-inactivation initiates in female cells. However, Tsix −Y males failed to ectopically induce Xist early in the differentiation of pluripotent cells [100,101]. Instead, Tsix −Y males ectopically expressed Xist RNA from their sole X chromosome upon further differentiation, corresponding to the stage when X-inactivation is in the maintenance phase in females [100,101]. This result suggests that Tsix is not required for the counting step in X-inactivation but is required to prevent inactivation of the single active-X only upon further differentiation of the cells [100,101].
If Tsix is required for the choice step of X-inactivation, Tsix +/− females should only exhibit Xist induction from the Tsix-mutant X chromosome and not from the WT X chromosome during the initiation stage of random X-inactivation. However, Tsix +/− female early embryonic epiblast cells and cultured epiblast stem cells (EpiSCs), which capture the epiblast lineage just after the establishment of X-inactivation, were found to induce Xist randomly from either the WT or the Tsix-mutant X chromosome [100,101]. After the induction of random X-inactivation, however, differentiating Tsix +/− cells that initially inactivated the WT X chromosome ectopically induced Xist from and silenced genes on the Tsix-mutant X chromosome, thus inactivating both X chromosomes [100,101]. Due to deficient X-linked gene expression, these cells became subject to rapid counter-selection, resulting in a surviving population of cells that had originally inactivated the Tsix-mutant X chromosome [100,101]. Tsix is thus required to prevent inactivation of the active-X only after random X-inactivation has commenced appropriately in females [100]. These findings suggest that Tsix is not required for the choice step in X-inactivation.
The Jpx locus in the XIC has also been proposed to regulate X chromosome counting and choice. Jpx resides ~10 kb upstream of Xist on the X chromosome [102]. The Jpx gene lacks an open reading frame and encodes a lncRNA [102,103]. Initial studies proposed that the Jpx lncRNA functions as a trans-acting dosage-sensitive factor to regulate X chromosome counting and choice [102,103]. Heterozygous loss of Jpx (Jpx +/−) in differentiating female ESCs led to defective Xist induction from either of the two X chromosomes and cellular lethality [102]. Furthermore, overexpression of an autosomally integrated Jpx transgene caused a modest level of ectopic Xist induction in differentiating male ESCs [103].
Follow-up investigations, however, have not recapitulated the initial observations of Jpx function. An independent study found that Jpx +/− heterozygous differentiating female ESCs were viable and could induce Xist [104]. These Jpx +/− female ESCs were also able to robustly contribute to chimeras [104]. Furthermore, independent analysis of differentiating male ESCs harboring autosomal integrations of multi-copy Jpx transgenes did not recapitulate the ectopic Xist induction by Jpx overexpression [105].
In addition to these findings, a recent study revealed that Jpx −/− female mice are viable and are born at near-expected Mendelian ratios [106]. Moreover, embryonic fibroblasts derived from these Jpx −/− female embryos exhibit Xist expression equivalent to that of WT female fibroblasts [106]. Collectively, the above observations indicate that Jpx is dispensable in regulating Xist and random X-inactivation.
The XIC-derived Ftx lncRNA is also a proposed positive regulator of Xist [59,60,61]. Differentiating Ftx +/− female ESCs display biased Xist expression from the WT X chromosome [60]. Transcription of the Ftx locus, not the lncRNA that it produces, is thought to be required for Xist regulation since inhibition of Ftx transcription diminishes Xist upregulation [60]. However, Xist can still be upregulated in cis in the absence of Ftx and other nearby putative X-linked Xist regulators [104]. Furthermore, Ftx −/− females are born and survive to adulthood [61]. Xist is downregulated in Ftx −/− female mice, though, and a subset of X-linked genes are de-repressed, suggesting that Ftx is required for robust Xist induction and X-inactivation [61]. The mild phenotype of Ftx −/− female mice suggests that other factors/sequences induce Xist in cis.
The Xert locus and enhancer cluster within the XIC is another proposed positive cis-regulator of Xist [62]. In female ESCs, heterozygous loss of Xert biases Xist induction, with 65–80% of cells inducing Xist RNA from the WT X chromosome [62]. Furthermore, overexpression of Xert in male ESCs in cis by CRISPR activation causes ectopic Xist induction [62]. Xert may work additively with Ftx to induce Xist in cis, as heterozygous deletion of Xert and Ftx in ESCs together leads to expression of Xist RNA exclusively from the WT X [62]. The additive functions of Xert and Ftx in vitro suggest that the combined loss of Xert and Ftx may result in a more pronounced developmental phenotype relative to Ftx −/− female mice. A definitive test of the requirement of Xert alone or together with Ftx in inducing Xist and random X-inactivation awaits the generation of mutant mice.
In contrast to Ftx and Xert, the X-linked Linx locus is thought to negatively regulate Xist in cis [107,108]. Linx maps within the XIC ~150 kb downstream of the Xist promoter and is co-expressed with Tsix in the embryonic lineage before random X-inactivation [107,108]. Linx could, therefore, function independently or through the regulation of Tsix to negatively regulate Xist. Heterozygous deletion of the Linx promoter, though, does not alter Tsix expression [107]. In female post-implantation embryos, heterozygous deletion of the Linx promoter results in a modest bias (54%) in X-inactivation, favoring the mutant X chromosome [107]. More recently, the Lppnx lncRNA has been described to arise from the region corresponding to the Linx locus [109,110]. Deletion of the Lppnx promoter, which appears to be nearly identical to the Linx promoter, similarly results in a modest bias in X-inactivation of the mutant X in mid-gestational female embryos [109,110]. Due to the repetitive sequences in the Linx/Lppnx region, whether these two loci are distinct remains unclear [111]. The mild effects observed upon deletion of the Linx/Lppnx promoters, however, suggest that other factors contribute to Xist repression.
A competing model posits that physical pairing of the two X chromosomes at the onset of random X-inactivation in females underlies X chromosome counting and choice. Early studies using DNA fluorescent in situ hybridization in differentiating female ESCs demonstrated that loci near Xist on the two X chromosomes transiently co-localize at the onset of random X-inactivation [112,113,114]. These studies posited that this transient co-localization facilitates counting and choice of only one of the two X chromosomes for the induction of Xist and X-inactivation [112,113,114]. Several XIC loci have been nominated to modulate X-pairing, including Tsix, Xite, and Xpr [112,113,115]. Xite is a lncRNA-expressing sequence thought to enhance Tsix expression [116,117]. Xpr (X-pairing region) contains the Xpct (Slc16a2) gene, which encodes for a thyroid hormone transporter [113]. Initial work showed that the Xpr sequence could trigger ectopic Xist expression in a subset of cells when autosomally integrated [112,113].
Several pieces of evidence collected more recently suggest that X-pairing may not be essential for counting and choice in females. Large heterozygous deletions encompassing the putative X-pairing loci Tsix, Xite, and the Xpr do not abrogate Xist induction [104,118]. Furthermore, XX-XY heterokaryons that contain separate XX and XY nuclei in a shared cytoplasm nevertheless exhibit Xist RNA coating of the sole X chromosome in the XY nuclei at a frequency consistent with independent initiation of X-inactivation of one of the three X chromosomes in these cells [104]. This finding suggests that in the absence of pairing, the X chromosome in the XY nucleus of an XX-XY heterokaryon is capable of inducing Xist [104]. Separately, another set of experiments attempted to prevent pairing in female ESCs by tethering one or both X chromosomes to the nuclear lamina and found that Xist RNA was still induced from one of the two X chromosomes upon the tethering [119]. Taken together, these experiments suggest that X-pairing is not required for either the counting or choice steps of X-inactivation.
Recent time-course observations of Xist RNA expression challenge the counting and choice model of X-inactivation. These analyses have revealed transient biallelic expression of Xist from the two parental X chromosomes when Xist is first induced in the epiblast lineage of developing WT female mouse embryos (Figure 2) [87,88,120,121]. The counting and choice model predicts monoallelic induction of Xist from one of the two X chromosomes at the onset of random X-inactivation in female cells [121,122]. Thus, biallelic induction of Xist from both X chromosomes at the onset of random X-inactivation argues against the counting and choice model of random X-inactivation (Figure 2). Rather, the induction of Xist from both X chromosomes suggests that the activity and expression dose of one or more X-linked trans-acting factors suffices to induce Xist during the initiation of random X-inactivation [121,122,123]. Upon the stochastic and non-synchronous induction of Xist from both alleles, the expression of X-linked inducer(s) of Xist is expected to be reduced, thereby preventing robust Xist induction from both X chromosomes (Figure 2) [121,122].
Rnf12, which may fall within the XIC, was the first X chromosome encoded protein proposed to be a stochastic inducer of Xist [121]. Rnf12 +/− female ESCs exhibit reduced Xist expression levels [104]. Conversely, transgenic overexpression of RNF12 in male ESCs results in ectopic Xist induction [105]. Rnf12 is amongst the earliest genes to be silenced when X-inactivation is being established [29,105,121], thereby potentially preventing robust Xist induction from both X chromosomes. RNF12, therefore, matches the stipulations of an X-linked factor that stochastically induces Xist in female cells. However, the epiblast-specific loss of RNF12 in female mouse embryos does not ablate Xist expression or cause lethality [124]. These Rnf12−/− epiblast cells, though, do show reduced Xist RNA expression relative to WT female epiblast cells [124]. RNF12 may thus be required for robust Xist induction during random X-inactivation [124]. Nevertheless, these results suggest that additional X-linked factors act in trans to stochastically induce Xist, either independently of or together with RNF12.
The X-linked KDM5C demethylase of histone H3 lysine 4 di- and trimethylation (H3K4me2/3) [125,126,127] has also been recently shown to induce Xist in a dose-dependent manner [128]. Kdm5c −/− females are inviable, whereas Kdm5c −Y males are viable and fertile [128,129]. Kdm5c −/− female embryos and differentiating female ESCs exhibit a >80% reduction in Xist induction relative to WT female embryos [128]. KDM5C over-expression ectopically induces Xist in male ESCs, suggesting that KDM5C functions in a dose-dependent manner to induce Xist [128]. Kdm5c escapes X-inactivation and is expressed from both the active and the inactive-X chromosomes in females [130]. However, upon the establishment of X-inactivation, Kdm5c expression is reduced relative to when both Xs are active prior to X-inactivation [130,131]. Thus, although Kdm5c escapes X- inactivation, the downregulation of KDM5C expression upon the commencement of X-inactivation may forestall robust Xist induction and inactivation of the second X chromosome. KDM5C function and expression dynamics, therefore, are consistent with KDM5C inducing Xist in a dose-dependent and stochastic manner.
Like KDM5C, KDM6A is an X-inactivation escapee that has also been suggested to induce Xist RNA [130,132]. KDM6A functions as a demethylase of H3K27me3 [133,134,135]. Loss of KDM6A results in defective Xist induction in differentiating female ESCs [132]. Kdm6a homozygous mutant female (Kdm6a−/−) mice perish midway through gestation between E10.5 to E12.5, whereas Kdm6a−Y males are viable and fertile [136]. Whether the lethality of Kdm6a−/− female mice is related to defective random X-inactivation awaits the characterization of Kdm6a−/− embryos. The absence of Rnf12, Kdm5c, or Kdm6a individually does not appear to completely abrogate Xist RNA induction [124,128,132]. It is thus likely that multiple X-linked factors function cooperatively in a dose-dependent manner to robustly induce Xist in females.
The Kdm5c and Kdm6a loci both reside far outside the XIC. That these non-XIC-encoded factors are required for Xist induction during random X-inactivation challenges the notion that all Xist regulators reside in the XIC. These observations prompt the exploration of other non-XIC X-linked loci in regulating Xist and random X-inactivation.
Emerging evidence also suggests that the autosomally encoded SPEN protein induces Xist during random X-inactivation [137]. Spen−/− female ESCs were initially reported to exhibit defective Xist induction upon differentiation [137]. However, a separate study of Spen−/− female ESCs did not replicate this observation [138]. Since SPEN is autosomally encoded, its expression is expected to be equal between females and males. To induce Xist selectively in females, SPEN may need to be recruited to the Xist locus either directly or indirectly by X chromosome-encoded factors that are differentially expressed between females and males.
5. Mechanisms of Gene Silencing on the Inactive-X during Random X-Inactivation
SPEN has a significant role in establishing X-linked gene silencing during random X-inactivation. By profiling the proteins that bind Xist RNA, several independent groups found that SPEN directly interacts with Xist RNA [78,139,140]. SPEN interacts with Xist RNA’s repeat A through an RNA-recognition motif (RRM) [78,139,141]. Xist repeat A is required for robust Xist RNA-mediated X-linked gene silencing, and the loss of SPEN results in a similar X-linked gene silencing defect relative to the loss of repeat A [142,143].
SPEN is recruited to the inactive X concomitantly with Xist RNA induction, suggesting that SPEN contributes to the establishment of X-linked gene silencing during random X-inactivation [80]. SPEN harbors a SPOC domain (Spen paralog and ortholog C-terminal domain) that is necessary for SPEN-mediated X-linked gene silencing [78,80,144]. The SPEN SPOC domain interacts with the SMRT/NCoR co-repressor complex, which recruits histone deacetylases (HDACs) to target loci [78,80,144,145]. By deacetylating histone tails, HDACs may contribute to X-linked gene silencing [144]. However, ablation of the SPOC domain does not completely abrogate X-linked gene silencing [146], suggesting that other domains of SPEN and/or other factors contribute to the establishment of X-linked gene silencing during random X-inactivation. The absence of any reported female-specific defects and the relatively late lethality of Spen-/- female and male embryos (E12.5–E14.5), though, argue against an essential role of SPEN in the establishment of random X-inactivation [81].
PRC2 and PRC1 also contribute to X-linked gene silencing establishment during random X-inactivation. Loss of the core PRC2 subunit SUZ12 in female ESCs leads to defective establishment of silencing of a subset of X-linked genes upon differentiation [143]. Similarly, ablation of the PRC1 subunits PCGF3 and PCGF5 in differentiating female ESCs results in defective silencing of a subset of X-linked genes [146]. The loss of PCGF3 and PCGF5 also abrogates Xist RNA-mediated recruitment and accumulation of PRC1-catalyzed H2AK119ub1 and to some extent PRC2-catalyzed H3K27me3 on X-linked genes [143,147,148]. The impact of PCGF3 and PCGF5 loss on H3K27me3 accumulation has led to the proposal that PRC1 recruits PRC2 to loci on the inactive-X [143,147,148]. The order of recruitment of the two PRC complexes to the inactive-X has been the subject of much recent debate.
Some studies have suggested that PRC2 and PRC1 directly interact with Xist RNA [149], whereas others propose that PRC2 and PRC1 are recruited through indirect interactions with Xist RNA [147,148,150]. Various approaches using crosslinking followed by mass spectrometry (MS) have not consistently shown direct interactions between the subunits of PRC2 or PRC1 with Xist RNA. One study that employed UV-crosslinking identified the PRC2 subunit EZH2 as one of more than 100 hits inferred to be direct Xist RNA interactors [140]. A different study using formaldehyde crosslinking found several subunits of PRC1, but not PRC2, to directly interact with Xist RNA [139]. However, profiling of direct protein–Xist RNA interactions using UV-crosslinking after stable isotope labelling of amino acids in culture (SILAC) for quantitative MS did not yield any subunits of PRC2 or PRC1 [78]. Xist RNA may thus recruit PRC2 and PRC1 to the inactive-X indirectly. PRC2 may be indirectly recruited to the inactive-X through a direct interaction of the PRC2 accessory subunit JARID2 with Xist RNA [151]. Similarly, the direct Xist RNA-binding protein hnRNPK may be an indirect recruiter of PRC1 to the inactive-X [139,148].
SMCHD1 is also thought to maintain X-linked gene silencing during random X-inactivation downstream of the PRCs and SPEN [146]. SMCHD1 has been proposed to interact directly with Xist RNA [140]. However, a study using PAR-CLIP (photoactivatable ribonucleoside-enhanced cross linking and immunoprecipitation) found that SMCHD1 does not directly interact with any RNAs, including Xist RNA [152]. Instead, evidence suggests that SMCHD1 is indirectly recruited to the inactive-X through its interaction with hnRNPK, similar to the proposed mechanism of hnRNPK recruiting PRC1 to the inactive-X [148,152]. In support of this mechanism, loss of Xist RNA repeats B and C, to which hnRNPK binds, ablates SMCHD1 accumulation on the inactive-X [152]. The recruitment of SMCHD1 downstream of PRC1 is consistent with a primary role of SMCHD1 in maintaining X-linked gene silencing [146].
6. Open Questions
Our understanding of the necessity of cis-acting loci within the XIC in regulating Xist expression during imprinted X-inactivation is limited relative to random X-inactivation (Table 1). The XistAR and Tsix lncRNAs have established roles in Xp-Xist induction and in the maintenance of Xm-Xist repression, respectively, during imprinted X-inactivation [41,57]. In contrast, other cis-acting loci either do not appear to have a role or await further testing as regulators of Xist in imprinted X-inactivation.

{kind=link}
{kind=link}
Table 1.
Summary of factors regulating Xist RNA induction during imprinted vs. random X-inactivation. All studies were performed in mouse embryos or cells.
Table 1.
Summary of factors regulating Xist RNA induction during imprinted vs. random X-inactivation. All studies were performed in mouse embryos or cells.
| Region | Regulatory Factor | Imprinted X-Inactivation | Random X-Inactivation |
|---|---|---|---|
| XIC | Xist Expression in Mutants | Xist Expression in Mutants | |
| Tsix lncRNA | Xm-Xist ↑↑↑ Tsix −/+ pre-implantation embryos [41] | Xist ↑↑↑ from WT active-X Tsix +/− post-implantation embryonic epiblasts [100] | |
| XistAR lncRNA | Xp-Xist ↓↓↓ XistAR +/− pre-implantation embryos [57] | Biased Xist expression from WT X XistAR +/– EpiSCs [57] | |
| Jpx lncRNA | Awaits further testing | No reported change Jpx −/− mouse embryonic fibroblasts [106] | |
| Ftx locus and/or lncRNA | No change Ftx +/− pre-implantation embryos [63] | Biased Xist expression from WT X Differentiating Ftx +/− ESCs [60] | |
| Xert locus and/or lncRNA | Awaits further testing | Biased Xist expression from WT X Xert +/− promoter deletion; differentiating ESCs [62] | |
| Linx/Lppnx locus and/or lncRNA | Awaits further testing | Biased Xist expression from mutant X Linx/Lppnx +/− promoter deletion; post-implantation embryonic epiblasts [107,109] | |
| RNF12 | Xp-Xist ↓↓↓ Rnf12 −/+ and Rnf12 −/− pre-implantation embryos [67,68] | Xist ↓ Rnf12 −/− post-implantation embryonic epiblasts [124] | |
| Non-XIC | KDM5C | No reported change Kdm5c −/− pre-implantation embryos [128] | Xist ↓↓↓ Kdm5c −/− post-implantation embryonic epiblasts [128] |
| KDM6A | Awaits further testing | Xist ↓↓ Differentiating Kdm6a–/– ESCs [132] | |
| Auto- somal | SPEN | No reported change Spen −/− pre-implantation embryos [80] | Variable reports [137,138] |
| PRC2/1 | Randomized Xist expression Pre-implantation embryos lacking oocyte-derived PRC2 or PRC1 [43,48,51] | No reported change PRC2 or PRC1 null post-implantation embryonic epiblasts [82,153] | |
| SMCHD1 | Incompletely penetrant Xm-Xist ↑ Pre-implantation embryos lacking oocyte-derived SMCHD1 [53] | No reported change Smchd1 −/− post-implantation embryos [76] |
Distinct trans-acting factors regulate Xist during imprinted vs. random X-inactivation [121,122] (Table 1). For example, PRC2 and PRC1 are required to prevent Xist expression from the active X (the Xm) during imprinted X-inactivation but not during random X-inactivation [67,68]. Conversely, KDM5C and SPEN appear to be required for Xist induction during random but not imprinted X-inactivation [80,128].
Testing the requirement of factors for Xist induction in both imprinted and random X-inactivation can be made more conclusive through the examination of mutant mouse embryos. Many factors implicated in the induction of random X-inactivation have been tested in stem cells but not in embryos (Table 1). Some factors, e.g., SPEN, exhibit varied results in stem cell models of random X-inactivation [103,104,137,138]; therefore, conducting tests in embryos may provide deeper insights.
7. Conclusions
Our improved understanding of Xist RNA induction as well as the establishment and maintenance of X-linked gene silencing during X-inactivation derives from technological advances, which have propelled a re-examination of previous conclusions. Although the mechanisms of Xist RNA induction during imprinted and random X-inactivation are distinct, both forms of X-inactivation appear to rely largely on the same set of factors to establish and maintain X-linked gene silencing. The use of mouse embryos has provided valuable insights into the mechanisms of both imprinted and random X-inactivation, as the embryos undergo both types of inactivation. The extent to which the models derived from the mouse apply to the onset of X-inactivation in other therian mammals, including humans, remains largely unknown and will be important to elucidate in the future.
Author Contributions
R.M.M.: original draft preparation and editing; S.K.: supervision, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
R.M. is supported by the US National Institutes of Health (NIH)-funded Michigan Predoctoral Training Program in Genetics (T32GM007544) and the US NIH-funded Career Training in Reproductive Biology Program (T32HD079342). Work in the Kalantry lab is funded by the US NIH (R01GM124571 and R01HD095463).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Clair Harris for critically reading the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef]
- Charlesworth, B.; Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1563–1572. [Google Scholar] [CrossRef]
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Springer: Berlin/Heidelberg, Germany, 1967; x; 192p. [Google Scholar]
- Charlesworth, B. Model for evolution of Y chromosomes and dosage compensation. Proc. Natl. Acad. Sci. USA 1978, 75, 5618–5622. [Google Scholar] [CrossRef]
- Cortez, D.; Marin, R.; Toledo-Flores, D.; Froidevaux, L.; Liechti, A.; Waters, P.D.; Grutzner, F.; Kaessmann, H. Origins and functional evolution of Y chromosomes across mammals. Nature 2014, 508, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.; Schmidt, M.M. Mammalian sex chromosomes: Design or accident? Curr. Opin. Genet. Dev. 1992, 2, 890–901. [Google Scholar]
- Lahn, B.T.; Page, D.C. Four evolutionary strata on the human X chromosome. Science 1999, 286, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Lamb, M.J. The evolution of heteromorphic sex chromosomes. Biol. Rev. 1990, 65, 249–276. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Hiatt, J.B.; Nguyen, D.K.; Ercan, S.; Sturgill, D.; Hillier, L.W.; Schlesinger, F.; Davis, C.A.; Reinke, V.J.; Gingeras, T.R.; et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet. 2011, 43, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Halsall, J.A.; Antczak, P.; O’Neill, L.P.; Falciani, F.; Turner, B.M. Relative overexpression of X-linked genes in mouse embryonic stem cells is consistent with Ohno’s hypothesis. Nat. Genet. 2011, 43, 1169–1170. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Richardson, B.J.; Czuppon, A.B.; Sharman, G.B. Inheritance of glucose-6-phosphate dehydrogenase variation in kangaroos. Nat. New Biol. 1971, 230, 154–155. [Google Scholar] [CrossRef]
- Sharman, G.B. Late DNA replication in the paternally derived X chromosome of female kangaroos. Nature 1971, 230, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Yeh, M.; Fairbanks, V.F. The normal human female as a mosaic of X-chromosome activity: Studies using the gene for C-6-PD-deficiency as a marker. Proc. Natl. Acad. Sci. USA 1962, 48, 9–16. [Google Scholar] [CrossRef]
- Reik, W.; Lewis, A. Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat. Rev. Genet. 2005, 6, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.A.; Dementyeva, E.V.; Shevchenko, A.I.; Pavlova, S.V.; Grigor’eva, E.V.; Zhelezova, A.I.; Vandeberg, J.L.; Zakian, S.M. Dynamics of the two heterochromatin types during imprinted X chromosome inactivation in vole Microtus levis. PLoS ONE 2014, 9, e88256. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.L.; Lyon, M.F. X chromosome inactivation studied by injection of a single cell into the mouse blastocyst. Nature 1971, 231, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Takagi, N.; Sasaki, M. Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 1975, 256, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Obata, Y.; Tada, M.; Goto, Y.; Nakatsuji, N.; Tan, S.; Kono, T.; Takagi, N. Imprint switching for non-random X-chromosome inactivation during mouse oocyte growth. Development 2000, 127, 3101–3105. [Google Scholar] [CrossRef] [PubMed]
- Mak, W.; Nesterova, T.B.; de Napoles, M.; Appanah, R.; Yamanaka, S.; Otte, A.P.; Brockdorff, N. Reactivation of the paternal X chromosome in early mouse embryos. Science 2004, 303, 666–669. [Google Scholar] [CrossRef]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A Gene from the Region of the Human X-Inactivation Center Is Expressed Exclusively from the Inactive X-Chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef]
- Marahrens, Y.; Loring, J.; Jaenisch, R. Role of the Xist gene in X chromosome choosing. Cell 1998, 92, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Marahrens, Y.; Panning, B.; Dausman, J.; Strauss, W.; Jaenisch, R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997, 11, 156–166. [Google Scholar] [CrossRef] [PubMed]
- McKee, B.D.; Handel, M.A. Sex chromosomes, recombination, and chromatin conformation. Chromosoma 1993, 102, 71–80. [Google Scholar] [CrossRef]
- Huynh, K.D.; Lee, J.T. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 2003, 426, 857–862. [Google Scholar] [CrossRef]
- Ohno, S.; Kaplan, W.D.; Kinosita, R. Conjugation of the heteropycnotic X and Y chromosomes of the rat spermatocyte. Exp. Cell Res. 1957, 12, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.M. Meiotic Silencing in Mammals. Annu. Rev. Genet. 2015, 49, 395–412. [Google Scholar] [CrossRef]
- Kalantry, S.; Purushothaman, S.; Bowen, R.B.; Starmer, J.; Magnuson, T. Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature 2009, 460, 647–651. [Google Scholar] [CrossRef]
- Namekawa, S.H.; Payer, B.; Huynh, K.D.; Jaenisch, R.; Lee, J.T. Two-step imprinted X inactivation: Repeat versus genic silencing in the mouse. Mol. Cell Biol. 2010, 30, 3187–3205. [Google Scholar] [CrossRef]
- Patrat, C.; Okamoto, I.; Diabangouaya, P.; Vialon, V.; Le Baccon, P.; Chow, J.; Heard, E. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 5198–5203. [Google Scholar] [CrossRef]
- Matsui, J.; Goto, Y.; Takagi, N. Control of Xist expression for imprinted and random X chromosome inactivation in mice. Hum. Mol. Genet. 2001, 10, 1393–1401. [Google Scholar] [CrossRef]
- Shao, C.S.; Takagi, N. An Extra Maternally Derived X-Chromosome Is Deleterious to Early Mouse Development. Development 1990, 110, 969–975. [Google Scholar] [CrossRef]
- Goto, Y.; Takagi, N. Tetraploid embryos rescue embryonic lethality caused by an additional maternally inherited X chromosome in the mouse. Development 1998, 125, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Takagi, N.; Adler, I.D. Parental imprinting on the mouse X chromosome: Effects on the early development of X0, XXY and XXX embryos. Genet. Res. 1993, 62, 139–148. [Google Scholar] [CrossRef]
- Okamoto, I.; Tan, S.; Takagi, N. X-chromosome inactivation in XX androgenetic mouse embryos surviving implantation. Development 2000, 127, 4137–4145. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999, 21, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Mise, N.; Goto, Y.; Nakajima, N.; Takagi, N. Molecular cloning of antisense transcripts of the mouse Xist gene. Biochem. Biophys. Res. Commun. 1999, 258, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Sado, T.; Wang, Z.; Sasaki, H.; Li, E. Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 2001, 128, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell 2000, 103, 17–27. [Google Scholar] [CrossRef]
- Maclary, E.; Buttigieg, E.; Hinten, M.; Gayen, S.; Harris, C.; Sarkar, M.K.; Purushothaman, S.; Kalantry, S. Differentiation-dependent requirement of Tsix long non-coding RNA in imprinted X-chromosome inactivation. Nat. Commun. 2014, 5, 4209. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 2016, 63, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Cloutier, M.; Trotter, M.; Hinten, M.; Gayen, S.; Du, Z.H.; Xie, W.; Kalantry, S. Conversion of random X-inactivation to imprinted X-inactivation by maternal PRC2. eLife 2019, 8, e44258. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Chen, Z.Y.; Yin, Q.Z.; Zhang, Y. Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev. 2018, 32, 1525–1536. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, L.J.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Djekidel, M.N.; Zhang, Y. Distinct dynamics and functions of H2AK119ub1 and H3K27me3 in mouse preimplantation embryos. Nat. Genet. 2021, 53, 551–563. [Google Scholar] [CrossRef]
- Mei, H.L.; Kozuka, C.; Hayashi, R.; Kumon, M.; Koseki, H.; Inoue, A. H2AK119ub1 guides maternal inheritance and zygotic deposition of H3K27me3 in mouse embryos. Nat. Genet. 2021, 53, 539–550. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, Y.; Wang, L.; Wang, L.; Wang, H.; Xu, Q.; Xiang, Y.; Chen, C.; Kong, F.; Xia, W.; et al. Evolutionary epigenomic analyses in mammalian early embryos reveal species-specific innovations and conserved principles of imprinting. Sci. Adv. 2021, 7, eabi6178. [Google Scholar] [CrossRef]
- Wanigasuriya, I.; Kinkel, S.A.; Beck, T.; Roper, E.A.; Breslin, K.; Lee, H.J.; Keniry, A.; Ritchie, M.E.; Blewitt, M.E. Maternal SMCHD1 controls both imprinted Xist expression and imprinted X chromosome inactivation. Epigenetics Chromatin 2022, 15, 26. [Google Scholar] [CrossRef]
- Chen, K.; Hu, J.; Moore, D.L.; Liu, R.; Kessans, S.A.; Breslin, K.; Lucet, I.S.; Keniry, A.; Leong, H.S.; Parish, C.L.; et al. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl. Acad. Sci. USA 2015, 112, E3535–E3544. [Google Scholar] [CrossRef]
- Gurzau, A.D.; Blewitt, M.E.; Czabotar, P.E.; Murphy, J.M.; Birkinshaw, R.W. Relating SMCHD1 structure to its function in epigenetic silencing. Biochem. Soc. Trans. 2020, 48, 1751–1763. [Google Scholar] [CrossRef]
- Wanigasuriya, I.; Gouil, Q.; Kinkel, S.A.; del Fierro, A.T.; Beck, T.; Roper, E.A.; Breslin, K.; Stringer, J.; Hutt, K.; Lee, H.J.; et al. Smchd1 is a maternal effect gene required for genomic imprinting. Elife 2020, 9, e55529. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.K.; Gayen, S.; Kumar, S.; Maclary, E.; Buttigieg, E.; Hinten, M.; Kumari, A.; Harris, C.; Sado, T.; Kalantry, S. An Xist-activating antisense RNA required for X-chromosome inactivation. Nat. Commun. 2015, 6, 8564. [Google Scholar] [CrossRef] [PubMed]
- Ohhata, T.; Hoki, Y.; Sasaki, H.; Sado, T. Crucial role of antisense transcription across the Xist promoter in Tsix-mediated Xist chromatin modification. Development 2008, 135, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Chureau, C.; Chantalat, S.; Romito, A.; Galvani, A.; Duret, L.; Avner, P.; Rougelle, C. Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum. Mol. Genet. 2011, 20, 705–718. [Google Scholar] [CrossRef]
- Furlan, G.; Hernandez, N.G.; Huret, C.; Galupa, R.; van Bemmel, J.G.; Romito, A.; Heard, E.; Morey, C.; Rougeulle, C. The Ftx Noncoding Locus Controls X Chromosome Inactivation Independently of Its RNA Products. Mol. Cell 2018, 70, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, Y.; Soma, M.; Shiura, H.; Sado, T.; Hasuwa, H.; Abe, K.; Kohda, T.; Ishino, F.; Kobayashi, S. Female mice lacking Ftx lncRNA exhibit impaired X-chromosome inactivation and a microphthalmia-like phenotype. Nat. Commun. 2018, 9, 3829. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.F.; Schwammle, T.; Kautz, P.; Robson, M.; Schopflin, R.; Lustig, L.R.; Brandenburg, L.; Dunkel, I.; Vechiatto, C.; Ntini, E.; et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Mol. Cell 2022, 82, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Soma, M.; Fujihara, Y.; Okabe, M.; Ishino, F.; Kobayashi, S. Ftx is dispensable for imprinted X-chromosome inactivation in preimplantation mouse embryos. Sci. Rep. 2014, 4, 5181. [Google Scholar] [CrossRef] [PubMed]
- Ravid Lustig, L.; Sampath Kumar, A.; Schwammle, T.; Dunkel, I.; Noviello, G.; Limberg, E.; Weigert, R.; Pacini, G.; Buschow, R.; Ghauri, A.; et al. GATA transcription factors drive initial Xist upregulation after fertilization through direct activation of long-range enhancers. Nat. Cell Biol. 2023, 25, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Ostendorff, H.P.; Bossenz, M.; Mincheva, A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Lichter, P.; Bach, I. Functional characterization of the gene encoding RLIM, the corepressor of LIM homeodomain factors. Genomics 2000, 69, 120–130. [Google Scholar] [CrossRef]
- Ostendorff, H.P.; Peirano, R.I.; Peters, M.A.; Schluter, A.; Bossenz, M.; Scheffner, M.; Bach, I. Ubiquitination-dependent cofactor exchange on LIM homeodomain transcription factors. Nature 2002, 416, 99–103. [Google Scholar] [CrossRef]
- Shin, J.; Bossenz, M.; Chung, Y.; Ma, H.; Byron, M.; Taniguchi-Ishigaki, N.; Zhu, X.C.; Jiao, B.W.; Hall, L.L.; Green, M.R.; et al. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 2010, 467, 977–981. [Google Scholar] [CrossRef]
- Wang, F.; Shin, J.D.; Shea, J.M.; Yu, J.; Boskovic, A.; Byron, M.; Zhu, X.; Shalek, A.K.; Regev, A.; Lawrence, J.B.; et al. Regulation of X-linked gene expression during early mouse development by Rlim. eLife 2016, 5, e19127. [Google Scholar] [CrossRef]
- Fukuda, A.; Mitani, A.; Miyashita, T.; Sado, T.; Umezawa, A.; Akutsu, H. Maintenance of Xist Imprinting Depends on Chromatin Condensation State and Rnf12 Dosage in Mice. PLoS Genet. 2016, 12, e1006375. [Google Scholar] [CrossRef]
- de Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef]
- Silva, J.; Mak, W.; Zvetkova, I.; Appanah, R.; Nesterova, T.B.; Webster, Z.; Peters, A.H.; Jenuwein, T.; Otte, A.P.; Brockdorff, N. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 2003, 4, 481–495. [Google Scholar] [CrossRef]
- Mak, W.; Baxter, J.; Silva, J.; Newall, A.E.; Otte, A.P.; Brockdorff, N. Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr. Biol. 2002, 12, 1016–1020. [Google Scholar] [CrossRef]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, T.P.; Chadwick, B.; Li, E.; Zhang, Y. Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J. Biol. Chem. 2004, 279, 52812–52815. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.E.; Gendrel, A.V.; Pang, Z.Y.; Sparrow, D.B.; Whitelaw, N.; Craig, J.M.; Apedaile, A.; Hilton, D.J.; Dunwoodie, S.L.; Brockdorff, N.; et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008, 40, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Nagao, K.; Blewitt, M.; Sasaki, H.; Obuse, C.; Sado, T. Role of SmcHD1 in establishment of epigenetic states required for the maintenance of the X-inactivated state in mice. Development 2018, 145, dev166462. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Hassig, C.A.; Schreiber, S.L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr. Opin. Chem. Biol. 1997, 1, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Dossin, F.; Pinheiro, I.; Zylicz, J.J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Kuroda, K.; Han, H.; Tani, S.; Tanigaki, K.; Tun, T.; Furukawa, T.; Taniguchi, Y.; Kurooka, H.; Hamada, Y.; Toyokuni, S.; et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity 2003, 18, 301–312. [Google Scholar] [CrossRef]
- Andergassen, D.; Smith, Z.D.; Kretzmer, H.; Rinn, J.L.; Meissner, A. Diverse epigenetic mechanisms maintain parental imprints within the embryonic and extraembryonic lineages. Dev. Cell 2021, 56, 2995–3005. [Google Scholar] [CrossRef]
- Grosswendt, S.; Kretzmer, H.; Smith, Z.D.; Kumar, A.S.; Hetzel, S.; Wittler, L.; Klages, S.; Timmermann, B.; Mukherji, S.; Meissner, A. Epigenetic regulator function through mouse gastrulation. Nature 2020, 584, 102–108. [Google Scholar] [CrossRef]
- Kalantry, S.; Mills, K.C.; Yee, D.; Otte, A.P.; Panning, B.; Magnuson, T. The Polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat. Cell Biol. 2006, 8, 195–202. [Google Scholar] [CrossRef]
- Wang, J.; Mager, J.; Chen, Y.; Schneider, E.; Cross, J.C.; Nagy, A.; Magnuson, T. Imprinted X inactivation maintained by a mouse Polycomb group gene. Nat. Genet. 2001, 28, 371–375. [Google Scholar] [CrossRef]
- Masui, O.; Corbel, C.; Nagao, K.; Endo, T.A.; Kezuka, F.; Diabangouaya, P.; Nakayama, M.; Kumon, M.; Koseki, Y.; Obuse, C.; et al. Polycomb repressive complexes 1 and 2 are each essential for maintenance of X inactivation in extra-embryonic lineages. Nat. Cell Biol. 2023, 25, 134–144. [Google Scholar] [CrossRef]
- Sousa, E.J.; Stuart, H.T.; Bates, L.E.; Ghorbani, M.; Nichols, J.; Dietmann, S.; Silva, J.C.R. Exit from Naive Pluripotency Induces a Transient X Chromosome Inactivation-like State in Males. Cell Stem Cell 2018, 22, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Pacini, G.; Dunkel, I.; Mages, N.; Mutzel, V.; Timmermann, B.; Marsico, A.; Schulz, E.G. Integrated analysis of Xist upregulation and X-chromosome inactivation with single-cell and single-allele resolution. Nat. Commun. 2021, 12, 3638. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Pei, Y.; He, L.Q.; Peng, G.D.; Reinius, B.; Tam, P.P.L.; Jing, N.H.; Deng, Q.L. Single-Cell RNA-Seq Reveals Cellular Heterogeneity of Pluripotency Transition and X Chromosome Dynamics during Early Mouse Development. Cell Rep. 2019, 26, 2593–2607. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Rastan, S. Non-random X-chromosome inactivation in mouse X-autosome translocation embryos-Location of the inactivation centre. J. Embryol. Exp. Morphol. 1983, 78, 1–22. [Google Scholar] [CrossRef]
- Rastan, S.; Robertson, E.J. X-chromosome deletions in embryo-derived (EK) cell lines associated with lack of X-chromosome inactivation. J. Embryol. Exp. Morphol. 1985, 90, 379–388. [Google Scholar] [CrossRef]
- Clerc, P.; Avner, P. Role of the region 3’ to Xist exon 6 in the counting process of X-chromosome inactivation. Nat. Genet. 1998, 19, 249–253. [Google Scholar] [CrossRef]
- Starmer, J.; Magnuson, T. A new model for random X chromosome inactivation. Development 2009, 136, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Augui, S.; Nora, E.P.; Heard, E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 2011, 12, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Mlynarczyk-Evans, S.; Nusinow, D.A.; Panning, B. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet. 2002, 36, 233–278. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Regulation of X-chromosome counting by Tsix and Xite sequences. Science 2005, 309, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Sado, T.; Li, E.; Sasaki, H. Effect of TSIX disruption on XIST expression in male ES cells. Cytogenet. Genome Res. 2002, 99, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Gayen, S.; Maclary, E.; Buttigieg, E.; Hinten, M.; Kalantry, S. A Primary Role for the Tsix lncRNA in Maintaining Random X-Chromosome Inactivation. Cell Rep. 2015, 11, 1251–1265. [Google Scholar] [CrossRef] [PubMed]
- Gayen, S.; Maclary, E.; Hinten, M.; Kalantry, S. Sex-specific silencing of X-linked genes by Xist RNA. Proc. Natl. Acad. Sci. USA 2016, 113, E309–E318. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The Long Noncoding RNA, Jpx, Is a Molecular Switch for X Chromosome Inactivation. Cell 2010, 143, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx RNA Activates Xist by Evicting CTCF. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Barakat, T.S.; Loos, F.; van Staveren, S.; Myronova, E.; Ghazvini, M.; Grootegoed, J.A.; Gribnau, J. The Trans-Activator RNF12 and Cis-Acting Elements Effectuate X Chromosome Inactivation Independent of X-Pairing. Mol. Cell 2014, 53, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Barakat, T.S.; Achame, E.M.; Monkhorst, K.; Kenter, A.; Rentmeester, E.; Grosveld, F.; Grootegoed, J.A.; Gribnau, J. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell 2009, 139, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Collombet, S.; Ranisavljevic, N.; Nagano, T.; Varnai, C.; Shisode, T.; Leung, W.; Piolot, T.; Galupa, R.; Borensztein, M.; Servant, N.; et al. Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature 2020, 580, 142–146. [Google Scholar] [CrossRef]
- Galupa, R.; Nora, E.P.; Worsley-Hunt, R.; Picard, C.; Gard, C.; van Bemmel, J.G.; Servant, N.; Zhan, Y.X.; El Marjou, F.; Johanneau, C.; et al. A Conserved Noncoding Locus Regulates Random Monoallelic Xist Expression across a Topological Boundary. Mol. Cell 2020, 77, 352–367. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Hierholzer, A.; Chureau, C.; Liverziani, A.; Ruiz, N.B.; Cattanach, B.M.; Young, A.N.; Kumar, M.; Cerase, A.; Avner, P.A. A long noncoding RNA influences the choice of the X chromosome to be inactivated. Proc. Natl. Acad. Sci. USA 2022, 119, e2118182119. [Google Scholar] [CrossRef]
- Galupa, R. Lppnx lncRNA: The new kid on the block or an old friend in X-inactivation choice? Proc. Natl. Acad. Sci. USA 2023, 120, e2218989120. [Google Scholar] [CrossRef]
- Hierholzer, A.; Cerase, A.; Avner, P. Reply to Galupa et al.: Discussing the role of Lppnx in the complexity of the X controlling element, Xce. Proc. Natl. Acad. Sci. USA 2023, 120, e2219685120. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Tsai, C.L.; Lee, J.T. Transient homologous chromosome pairing marks the onset of X inactivation. Science 2006, 311, 1149–1152. [Google Scholar] [CrossRef]
- Augui, S.; Filion, G.J.; Huart, S.; Nora, E.; Guggiari, M.; Maresca, M.; Steward, A.F.; Heard, E. Sensing X chromosome pairs before X inactivation via a novel X-pairing region of the. Science 2007, 318, 1632–1636. [Google Scholar] [CrossRef]
- Marahrens, Y. X-inactivation by chromosomal pairing events. Genes Dev. 1999, 13, 2624–2632. [Google Scholar] [CrossRef]
- Bacher, C.P.; Guggiari, M.; Brors, B.; Augui, S.; Clerc, P.; Avner, P.; Eils, R.; Heard, E. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 2006, 8, 293–299. [Google Scholar] [CrossRef]
- Ogawa, Y.; Lee, J.T. Xite, X-inactivation Intergenic transcription elements that regulate the probability of choice. Mol. Cell 2003, 259, 731–743. [Google Scholar] [CrossRef]
- Stavropoulos, N.; Rowntree, R.K.; Lee, J.T. Identification of developmentally specific enhancers for Tsix in the regulation of X chromosome inactivation. Mol. Cell Biol. 2005, 25, 2757–2769. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fukue, Y.; Nolen, L.; Sadreyev, R.I.; Lee, J.T. Characterization of Xpr (Xpct) reveals instability but no effects on X-chromosome pairing or expression. Transcription 2010, 1, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Pollex, T.; Heard, E. Nuclear positioning and pairing of X-chromosome inactivation centers are not primary determinants during initiation of random X-inactivation. Nat. Genet. 2019, 51, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Shiura, H.; Abe, K. Xist/Tsix expression dynamics during mouse peri-implantation development revealed by whole-mount 3D RNA-FISH. Sci. Rep. 2019, 9, 3637. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, K.; Jonkers, I.; Rentmeester, E.; Grosveld, F.; Gribnau, J. X inactivation counting and choice is a stochastic process: Evidence for involvement of an X-linked activator. Cell 2008, 132, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Schwammle, T.; Schulz, E.G. Regulatory principles and mechanisms governing the onset of random X-chromosome inactivation. Curr. Opin. Genet. Dev. 2023, 81, 102063. [Google Scholar] [CrossRef] [PubMed]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Wallingford, M.C.; Gallant, J.; Marcho, C.; Jiao, B.; Byron, M.; Bossenz, M.; Lawrence, J.B.; Jones, S.N.; Mager, J.; et al. RLIM is dispensable for X-chromosome inactivation in the mouse embryonic epiblast. Nature 2014, 511, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Agger, K.; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef]
- Samanta, M.K.; Gayen, S.; Harris, C.; Maclary, E.; Murata-Nakamura, Y.; Malcore, R.M.; Porter, R.S.; Garay, P.M.; Vallianatos, C.N.; Samollow, P.B.; et al. Activation of Xist by an evolutionarily conserved function of KDM5C demethylase. Nat. Commun. 2022, 13, 2602. [Google Scholar] [CrossRef]
- Iwase, S.; Brookes, E.; Agarwal, S.; Badeaux, A.I.; Ito, H.; Vallianatos, C.N.; Tomassy, G.S.; Kasza, T.; Lin, G.; Thompson, A.; et al. A Mouse Model of X-linked Intellectual Disability Associated with Impaired Removal of Histone Methylation. Cell Rep. 2016, 14, 1000–1009. [Google Scholar] [CrossRef]
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes that escape from X inactivation. Hum. Genet. 2011, 130, 237–245. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, J.; Ma, L.; Fang, H.; Ma, R.; Groneck, C.; Filippova, G.N.; Deng, X.; Ma, W.; Disteche, C.M.; et al. KDM6A facilitates Xist upregulation at the onset of X inactivation. bioRxiv 2023. bioRxiv:2023.08.16.553617. [Google Scholar]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.Z.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E.; et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef]
- Robert-Finestra, T.; Tan, B.F.; Mira-Bontenbal, H.; Timmers, E.; Gontan, C.; Merzouk, S.; Giaimo, B.D.; Dossin, F.; van IJcken, W.F.J.; Martens, J.W.M.; et al. SPEN is required for Xist upregulation during initiation of X chromosome inactivation. Nat. Commun. 2021, 12, 7000. [Google Scholar] [CrossRef] [PubMed]
- Jachowicz, J.W.; Strehle, M.; Banerjee, A.K.; Blanco, M.R.; Thai, J.; Guttman, M. Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat. Struct. Mol. Biol. 2022, 29, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.F.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.H. Systematic Discovery of Xist RNA Binding Proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhail, M.; Haas, W.; et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015, 349, aab2276. [Google Scholar] [CrossRef]
- Monfort, A.; Di Minin, G.; Postlmayr, A.; Freimann, R.; Arieti, F.; Thore, S.; Wutz, A. Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep. 2015, 12, 554–561. [Google Scholar] [CrossRef]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef]
- Nesterova, T.B.; Wei, G.F.; Coker, H.; Pintacuda, G.; Bowness, J.S.; Zhang, T.Y.; Almedia, M.; Bloech, B.; Moindrot, B.; Carter, E.J.; et al. Systematic allelic analysis defines the interplay of key pathways in X chromosome inactivation. Nat. Commun. 2019, 10, 3129. [Google Scholar] [CrossRef] [PubMed]
- Zylicz, J.J.; Bousard, A.; Zumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef]
- Bowness, J.S.; Nesterova, T.B.; Wei, G.F.; Rodermund, L.; Almeida, M.; Coker, H.; Carter, E.J.; Kadaster, A.; Brockdorff, N. Xist-mediated silencing requires additive functions of SPEN and Polycomb together with differentiation-dependent recruitment of SmcHD1. Cell Rep. 2022, 39, 110830. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef]
- Pintacuda, G.; Wei, G.F.; Roustan, C.; Kirmizitas, B.A.; Solcan, N.; Cerase, A.; Castello, A.; Mohammed, S.; Moindrot, B.; Nesterova, T.B.; et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 2017, 68, 955–969. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Bousard, A.; Raposo, A.C.; Zylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E.; et al. The role of Xist-mediated Polycomb recruitment in the initiation of X-chromosome inactivation. EMBO Rep. 2019, 20, e48019. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef]
- Jansz, N.; Nesterova, T.; Keniry, A.; Iminitoff, M.; Hickey, P.F.; Pintacuda, G.; Masui, O.; Kobelke, S.; Geoghegan, N.; Breslin, K.A.; et al. Smchd1 Targeting to the Inactive X Is Dependent on the Xist-HnrnpK-PRC1 Pathway. Cell Rep. 2018, 25, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Kalantry, S.; Magnuson, T. The polycomb group protein EED is dispensable for the initiation of random X-chromosome inactivation. PLoS Genet. 2006, 2, 656–664. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Induction of Xist RNA and establishment of gene silencing on inactive-X during imprinted X-chromosome inactivation in the early mouse embryo. Top: Diagram of early 2-, 4-, 8-, or 16-cell female mouse embryos. E, embryonic day. Middle: Depiction of the expression status of the two X chromosomes in each cell. Xm, maternal-X; Xp, paternal-X. Bottom: Schematic of the Xist locus or X-linked genes subject to silencing at different stages of X-inactivation. The Xm and Xp are both active prior to the initiation of imprinted X-inactivation in the early embryo. Between the 2- and 4-cell stages, the XistAR lncRNA and RNF12 protein promote Xp-Xist expression, whereas the Xm-Xist locus is silenced by PRC2-catalyzed H3K27me3 and PRC1-catalyzed H2AK119ub1. Following induction, Xp-Xist RNA directly or indirectly recruits gene silencing proteins and complexes, including PRC1, PRC2, SPEN, and SMCHD1. These proteins and complexes establish and maintain gene silencing on the Xp.
Figure 1.
Induction of Xist RNA and establishment of gene silencing on inactive-X during imprinted X-chromosome inactivation in the early mouse embryo. Top: Diagram of early 2-, 4-, 8-, or 16-cell female mouse embryos. E, embryonic day. Middle: Depiction of the expression status of the two X chromosomes in each cell. Xm, maternal-X; Xp, paternal-X. Bottom: Schematic of the Xist locus or X-linked genes subject to silencing at different stages of X-inactivation. The Xm and Xp are both active prior to the initiation of imprinted X-inactivation in the early embryo. Between the 2- and 4-cell stages, the XistAR lncRNA and RNF12 protein promote Xp-Xist expression, whereas the Xm-Xist locus is silenced by PRC2-catalyzed H3K27me3 and PRC1-catalyzed H2AK119ub1. Following induction, Xp-Xist RNA directly or indirectly recruits gene silencing proteins and complexes, including PRC1, PRC2, SPEN, and SMCHD1. These proteins and complexes establish and maintain gene silencing on the Xp.

Figure 2.
Stochastic Xist induction during random X-chromosome inactivation in the mouse embryo. The Xp is reactivated by embryonic day (E) ~4.5 in the female embryo. At this stage, both the Xm and Xp express activators of Xist. Increased expression of X-linked activators stochastically induces Xist on the Xm and/or Xp between E5.0 to E5.5. Individual embryonic epiblast cells transiently exhibit distinct patterns of Xist induction. In cases where both X chromosomes induce Xist, both X chromosomes may initiate silencing, which in turn results in a reduction of expression of the X-linked Xist activators. Reduced expression of Xist activators leads to a loss of Xist induction. This feedback loop resolves only when Xist RNA is expressed from a single X chromosome and that single X chromosome is transcriptionally silenced in female cells. Upon inactivation of a single X chromosome, the expression of X-linked activator(s) of Xist reaches an intermediate level, thereby preventing induction of Xist from the active X chromosome in female cells.
Figure 2.
Stochastic Xist induction during random X-chromosome inactivation in the mouse embryo. The Xp is reactivated by embryonic day (E) ~4.5 in the female embryo. At this stage, both the Xm and Xp express activators of Xist. Increased expression of X-linked activators stochastically induces Xist on the Xm and/or Xp between E5.0 to E5.5. Individual embryonic epiblast cells transiently exhibit distinct patterns of Xist induction. In cases where both X chromosomes induce Xist, both X chromosomes may initiate silencing, which in turn results in a reduction of expression of the X-linked Xist activators. Reduced expression of Xist activators leads to a loss of Xist induction. This feedback loop resolves only when Xist RNA is expressed from a single X chromosome and that single X chromosome is transcriptionally silenced in female cells. Upon inactivation of a single X chromosome, the expression of X-linked activator(s) of Xist reaches an intermediate level, thereby preventing induction of Xist from the active X chromosome in female cells.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Malcore, R.M.; Kalantry, S. A Comparative Analysis of Mouse Imprinted and Random X-Chromosome Inactivation. Epigenomes 2024, 8, 8. https://doi.org/10.3390/epigenomes8010008
AMA Style
Malcore RM, Kalantry S. A Comparative Analysis of Mouse Imprinted and Random X-Chromosome Inactivation. Epigenomes. 2024; 8(1):8. https://doi.org/10.3390/epigenomes8010008
Chicago/Turabian StyleMalcore, Rebecca M., and Sundeep Kalantry. 2024. "A Comparative Analysis of Mouse Imprinted and Random X-Chromosome Inactivation" Epigenomes 8, no. 1: 8. https://doi.org/10.3390/epigenomes8010008