The Role of κ-Carbides as Hydrogen Traps in High-Mn Steels



Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
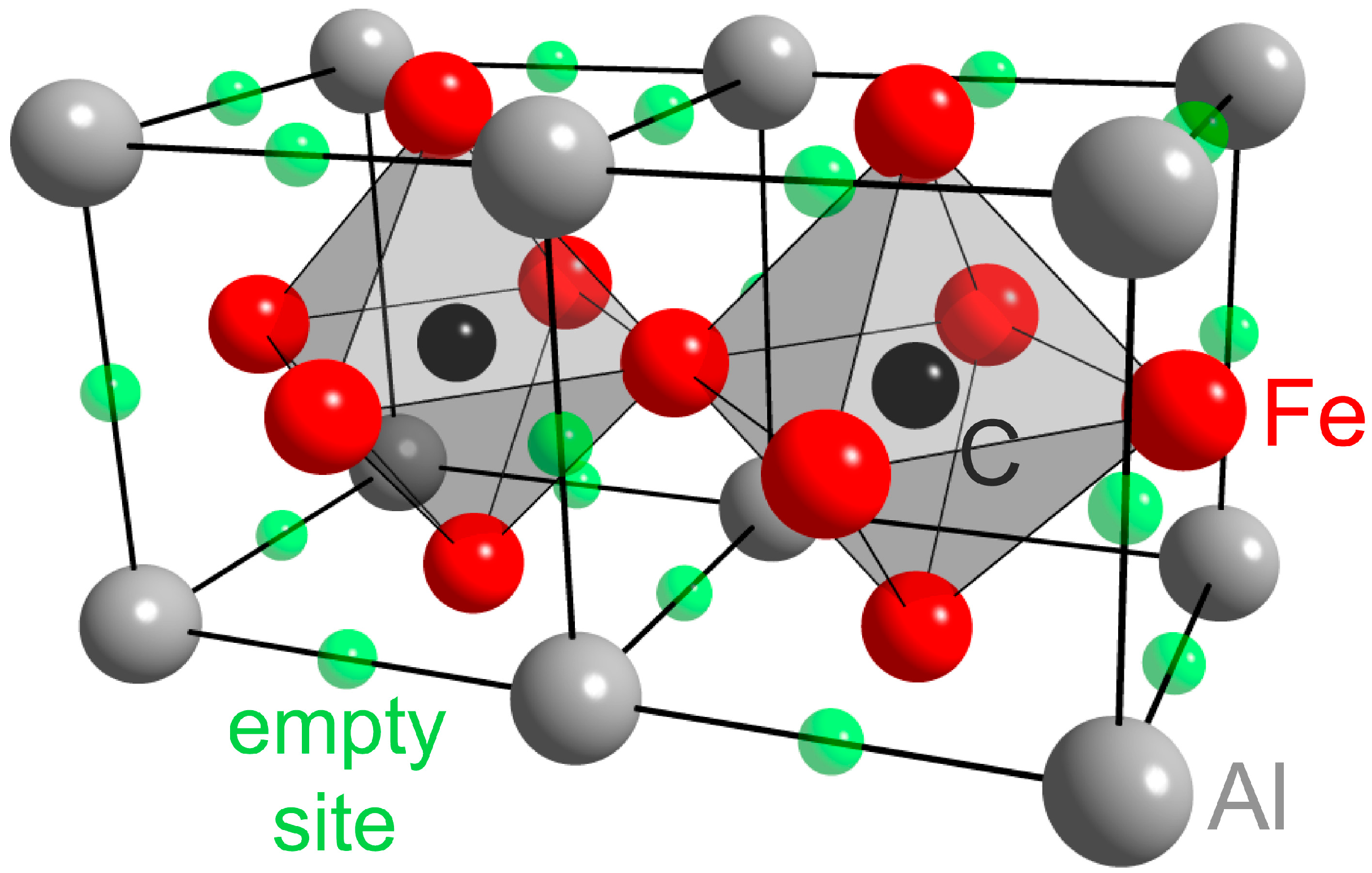
3.1. Stoichiometric Phases of L12 and E21 Symmetry
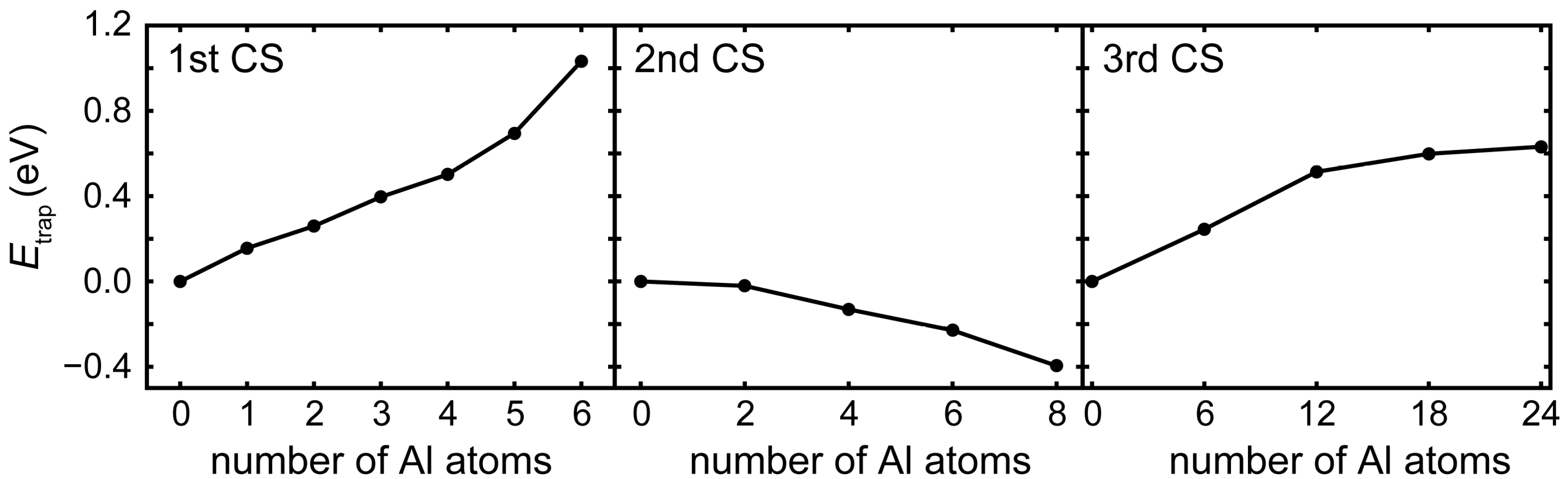
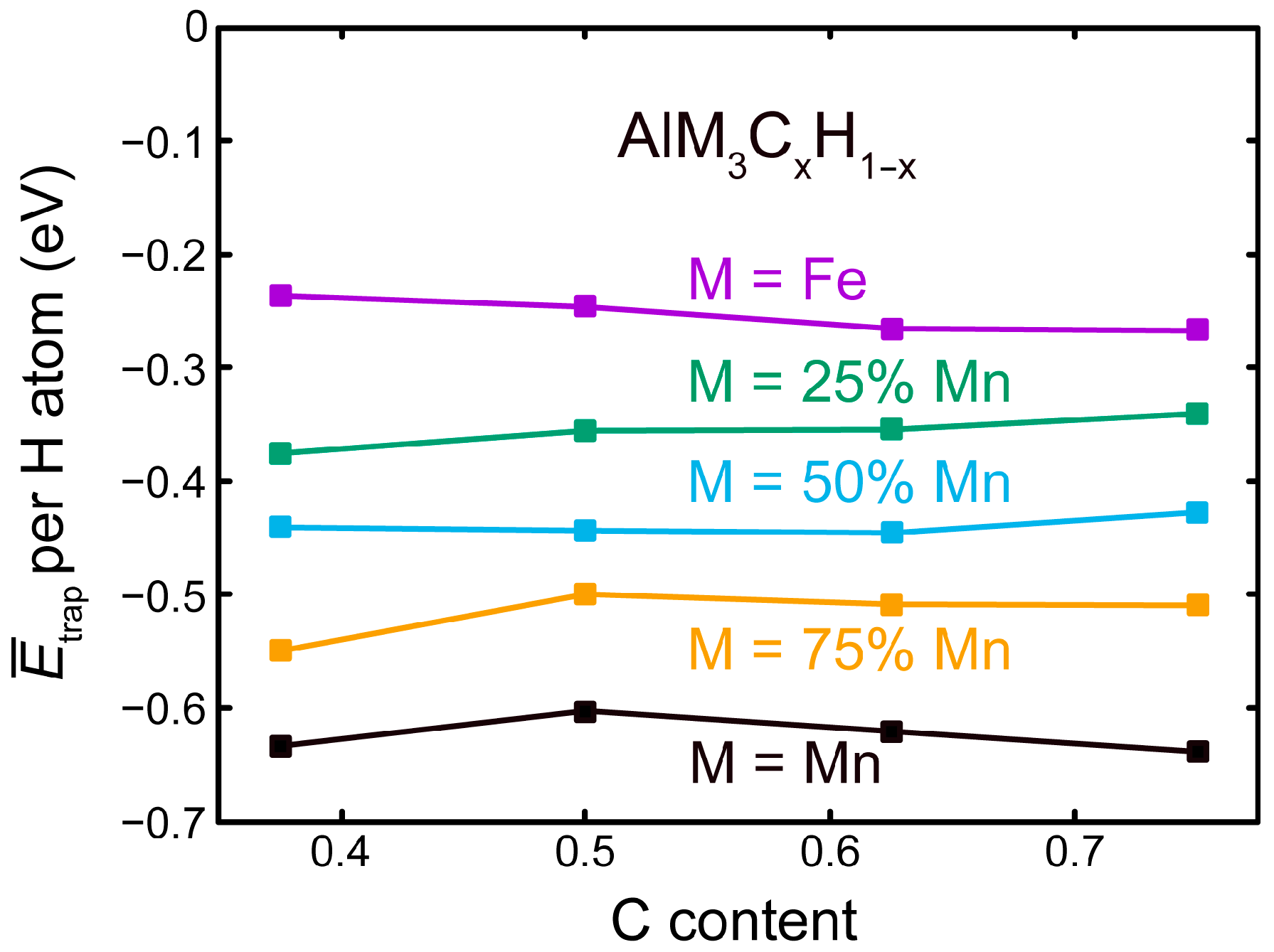
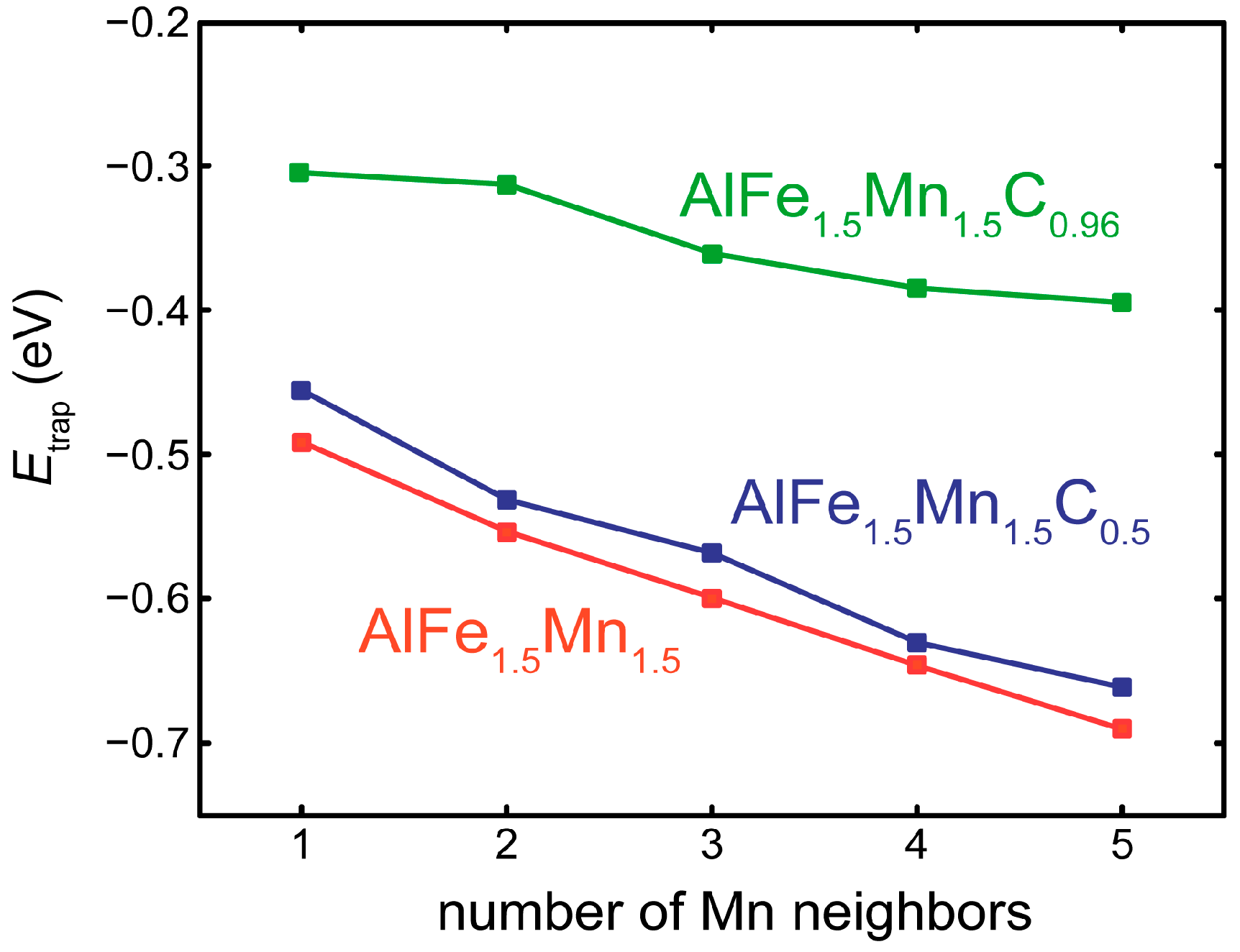
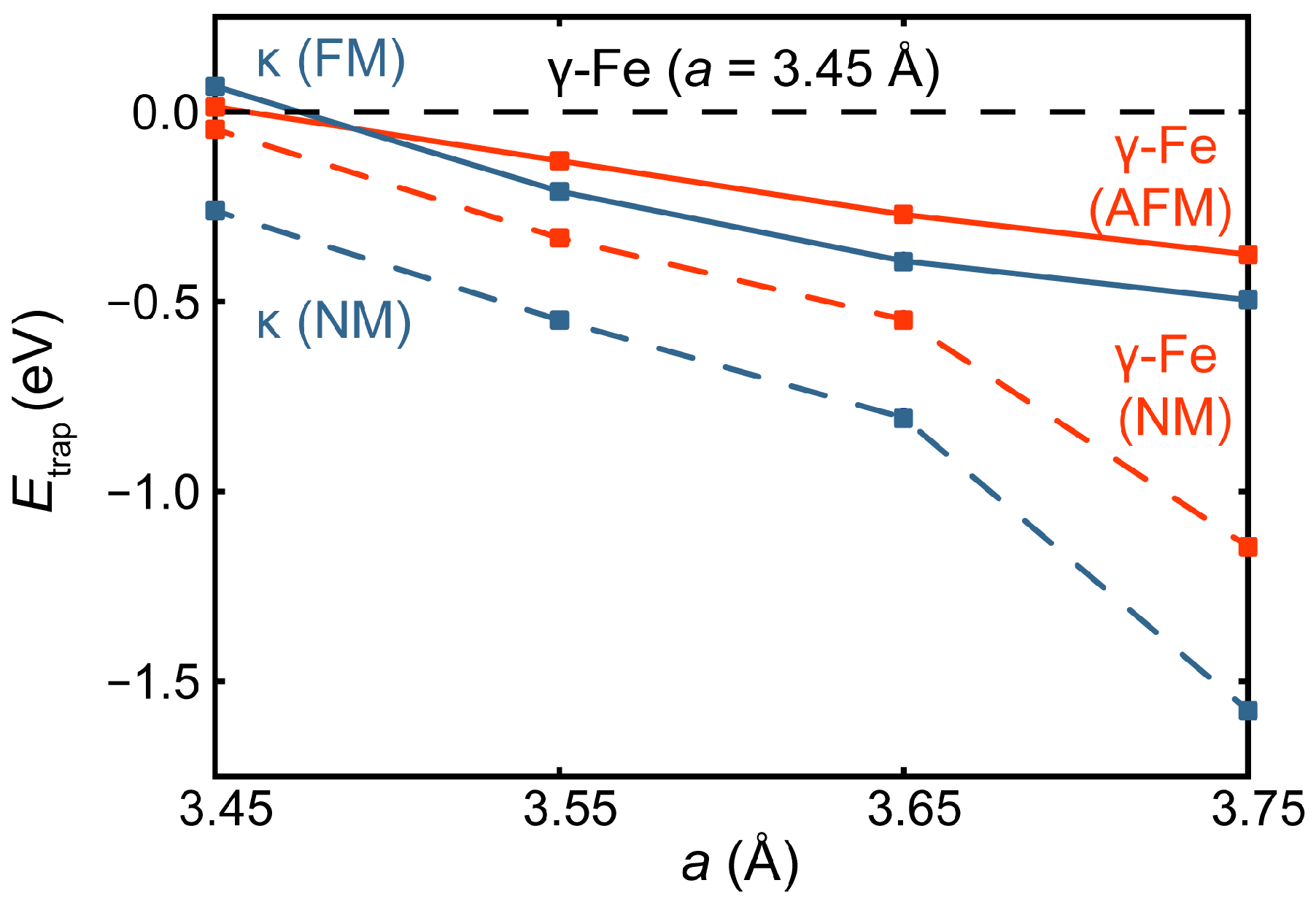
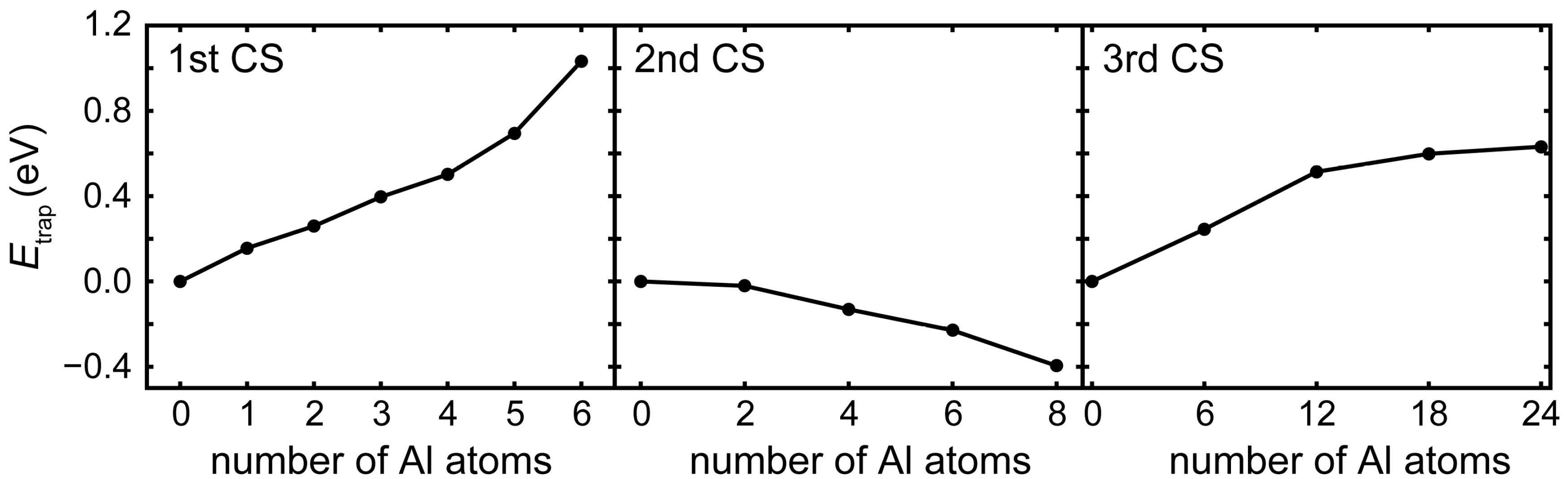
3.2. Competition of H and C in the L12 and E21 Structure
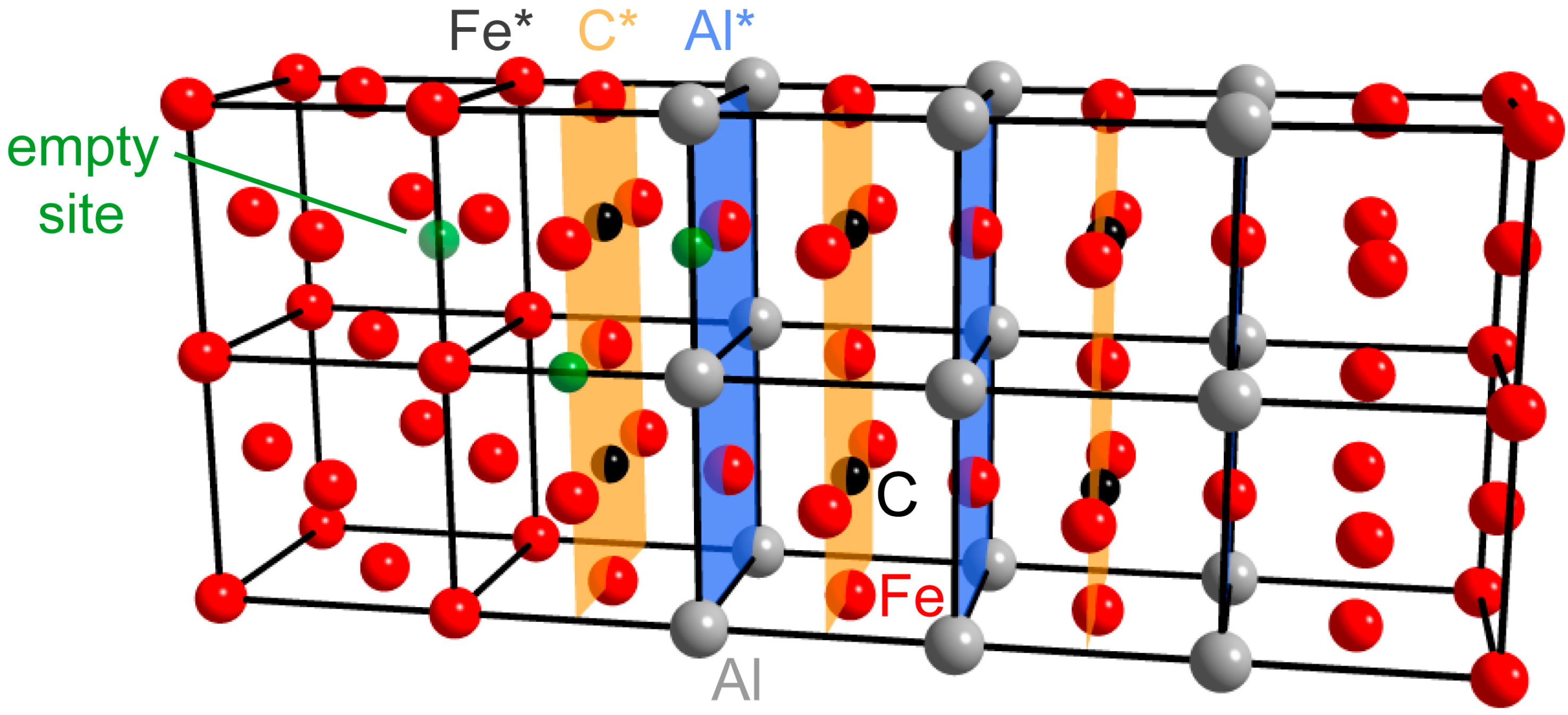
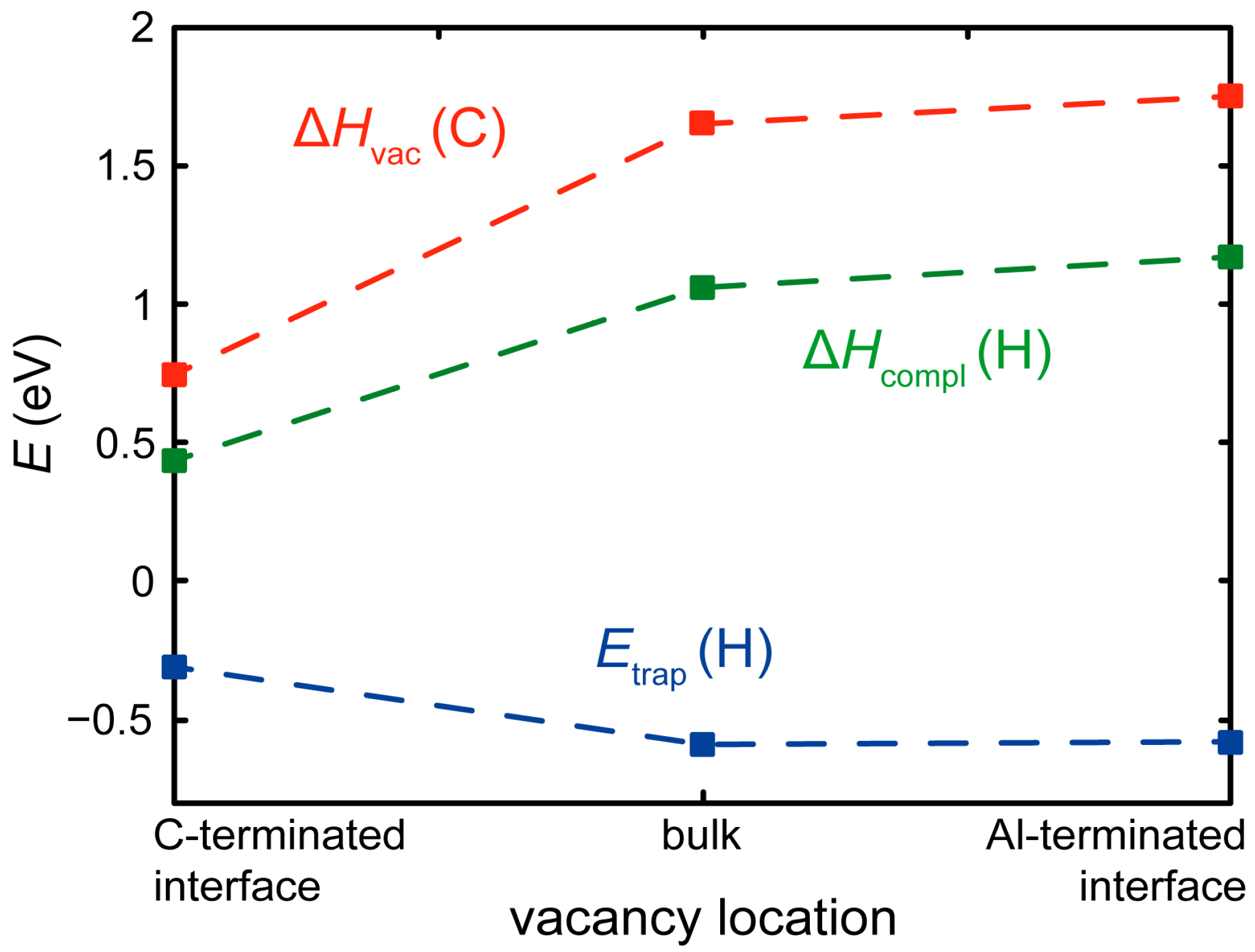
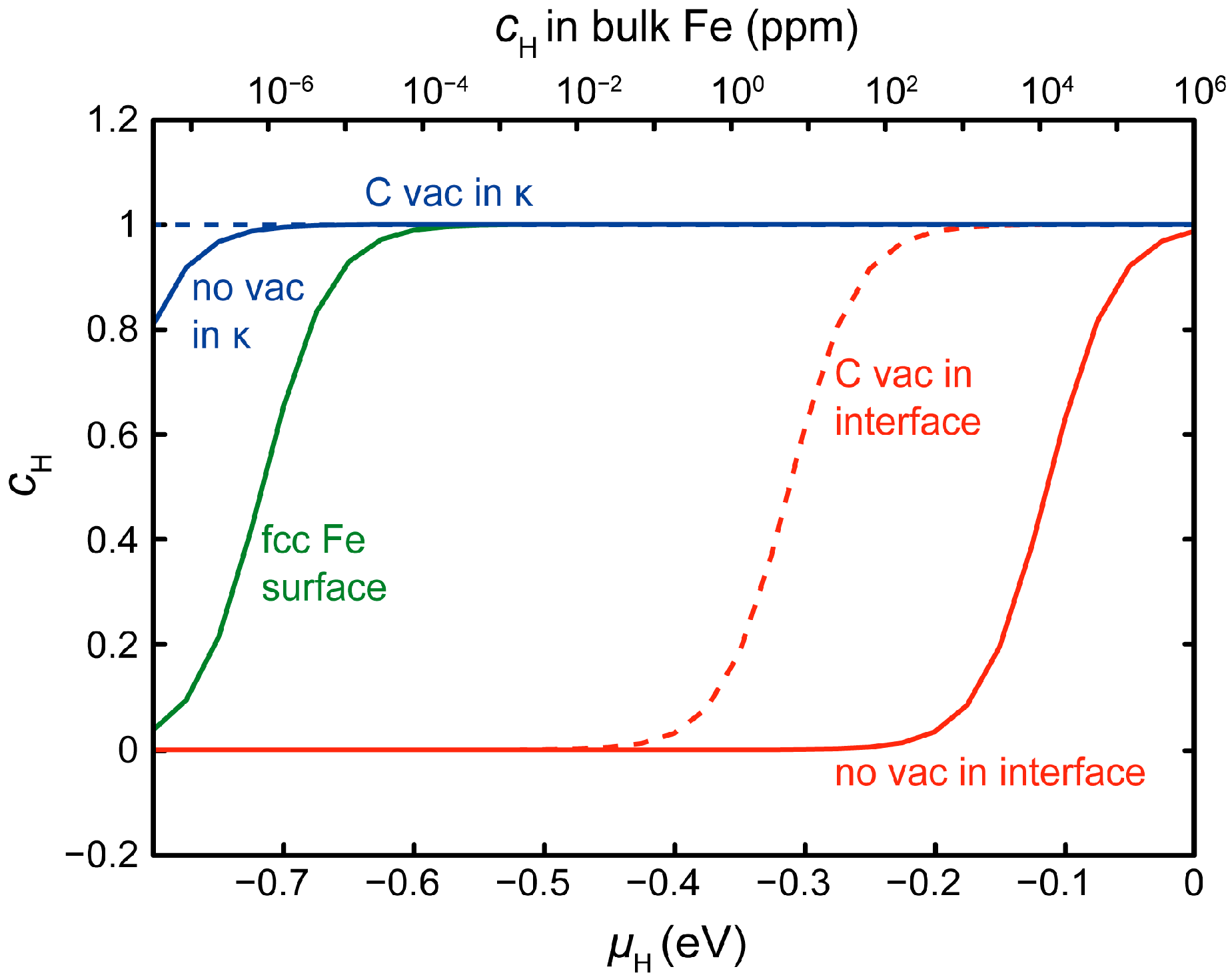
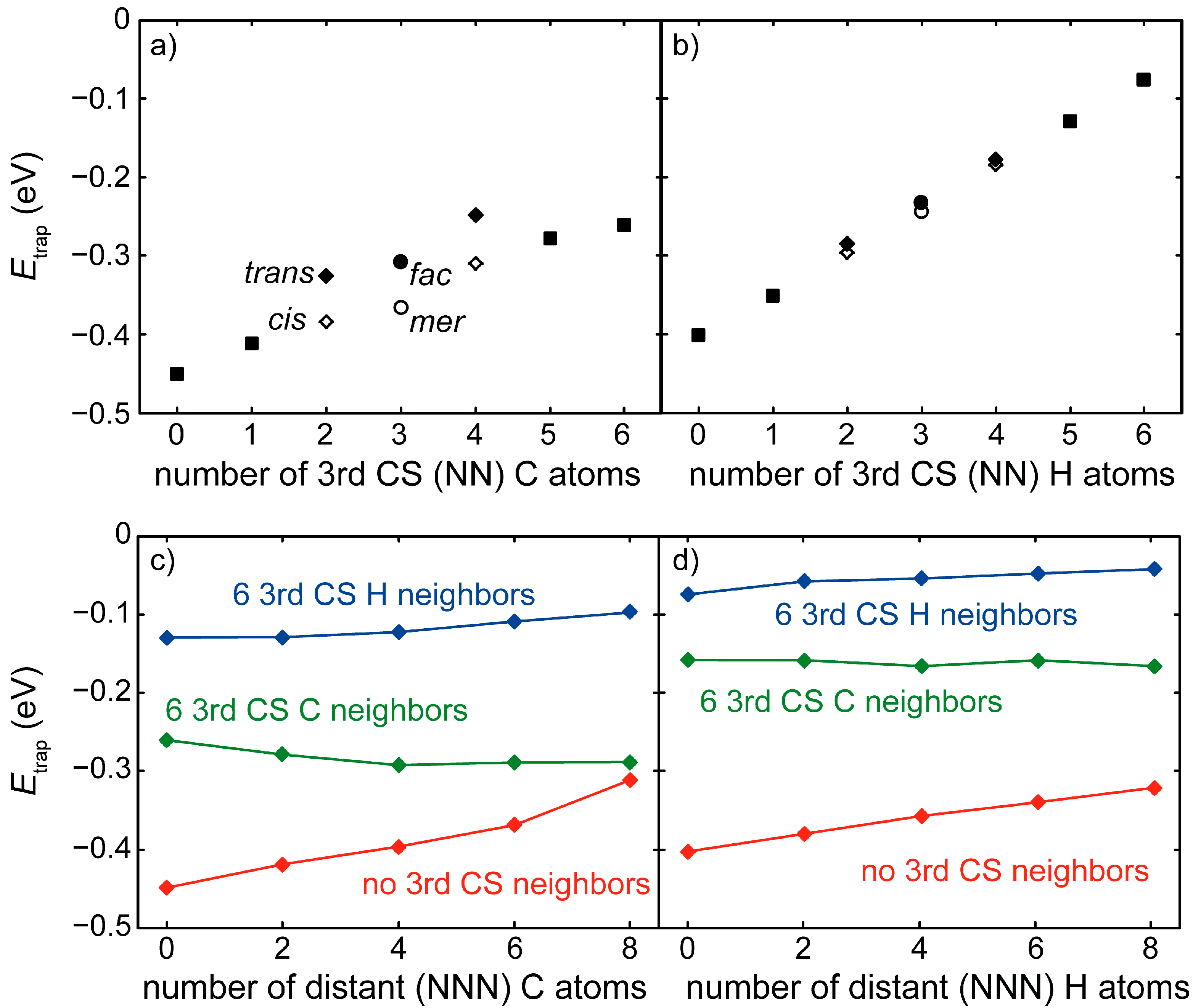
3.3. H at Interfaces between κ-Carbide and the fcc-Fe Matrix
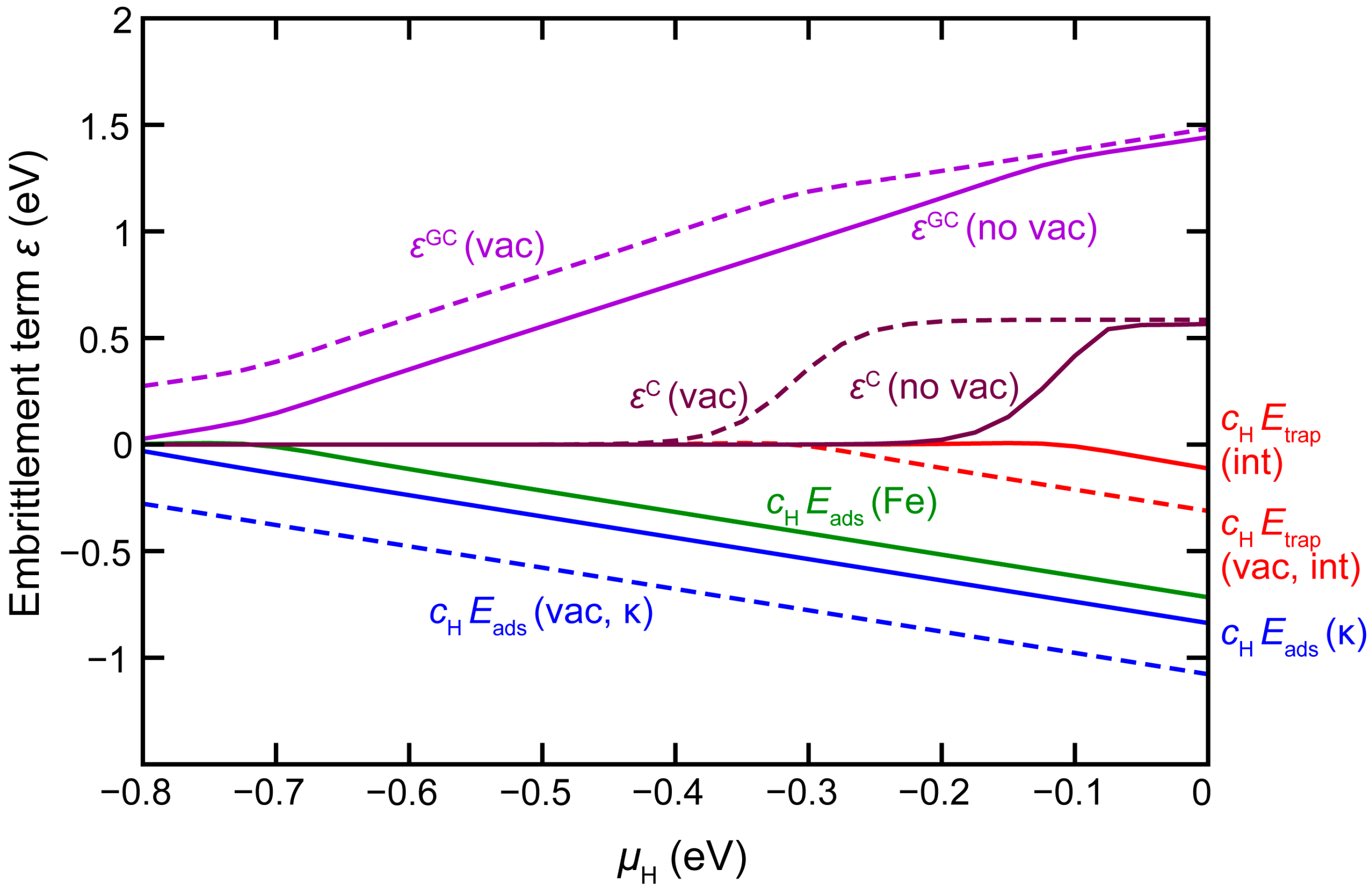
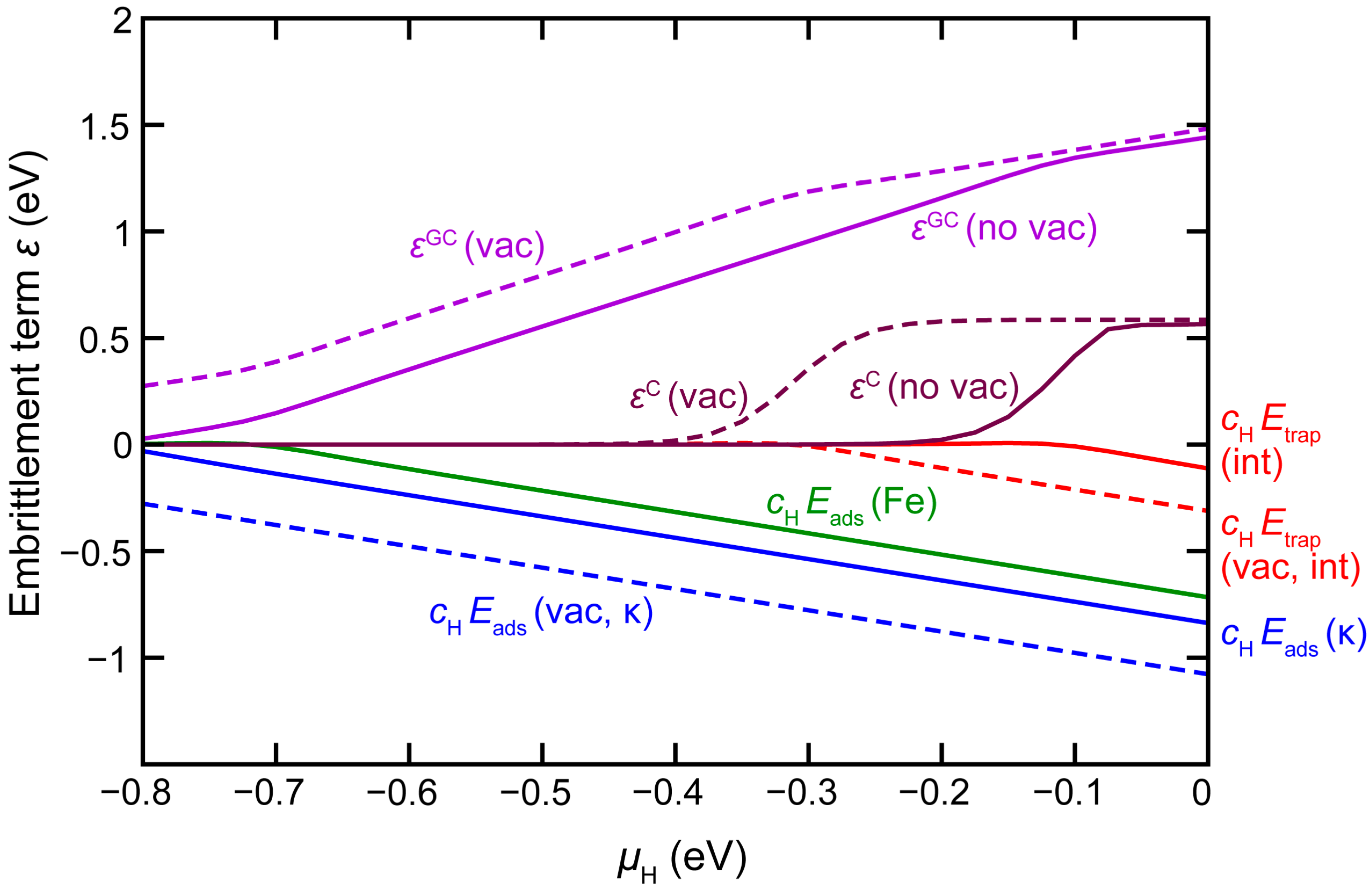
3.4. H Enhanced Decohesion of Interfaces
+ [cH(int) Etrap(int) − cH(κ) Eads(κ-surf) − cH(Fe) Eads(Fe-surf)]
Eads(X-surf) = Eel(H@X-surf) – Eel(X-surf) − μH
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gutierrez-Urrutia, I.; Raabe, D. Influence of Al content and precipitation state on the mechanical behavior of austenitic high-Mn low-density steels. Scr. Mater. 2013, 68, 343–347. [Google Scholar] [CrossRef]
- Park, K.-T.; Kim, G.; Kim, S.K.; Lee, S.W.; Hwang, S.W.; Lee, C.S. On the transitions of deformation modes of fully austenitic steels at room temperature. Met. Mater. Int. 2010, 16, 1–6. [Google Scholar] [CrossRef]
- Gutierrez-Urrutia, I.; Raabe, D. High strength and ductile low density austenitic FeMnAlC steels: Simplex and alloys strengthened by nanoscale ordered carbides. Mater. Sci. Technol. 2014, 30, 1099–1104. [Google Scholar] [CrossRef]
- Zambrano, O.A.; Valdés, J.; Aguilar, Y.; Coronado, J.J.; Rodríguez, S.A.; Logé, R.E. Hot deformation of a Fe-Mn-Al-C steel susceptible of κ-carbide precipitation. Mater. Sci. Eng. A 2017, 689, 269–285. [Google Scholar] [CrossRef]
- Haase, C.; Zehnder, C.; Ingendahl, T.; Bikar, A.; Tang, F.; Hallstedt, B.; Hu, W.; Bleck, W.; Molodov, D.A. On the deformation behavior of κ-carbide-free and κ-carbide-containing high-Mn light-weight steel. Acta Mater. 2017, 122, 332–343. [Google Scholar] [CrossRef]
- Hirth, J.P. Effects of hydrogen on the properties of iron and steel. MTA 1980, 11, 861–890. [Google Scholar] [CrossRef]
- Hojo, T.; Sugimoto, K.-I.; Mukai, Y.; Ikeda, S. Effects of Aluminum on Delayed Fracture Properties of Ultra High Strength Low Alloy TRIP-aided Steels. ISIJ Int. 2008, 48, 824–829. [Google Scholar] [CrossRef]
- Song, S.W.; Kwon, Y.J.; Lee, T.; Lee, C.S. Effect of Al addition on low-cycle fatigue properties of hydrogen-charged high-Mn TWIP steels. Mater. Sci. Eng. A 2016, 677, 421–430. [Google Scholar] [CrossRef]
- Takahashi, J.; Kawakami, K.; Kobayashi, Y.; Tarui, T. The first direct observation of hydrogen trapping sites in TiC precipitation-hardening steel through atom probe tomography. Scr. Mater. 2010, 63, 261–264. [Google Scholar] [CrossRef]
- Di Stefano, D.; Nazarov, R.; Hickel, T.; Neugebauer, J.; Mrovec, M.; Elsässer, C. First-principles investigation of hydrogen interaction with TiC precipitates in α-Fe. Phys. Rev. B 2016, 93, 184108. [Google Scholar] [CrossRef]
- Dierkes, H.; van Leusen, J.; Bogdanovski, D.; Dronskowski, R. Synthesis, Crystal Structure, Magnetic Properties, and Stability of the Manganese-Rich “Mn3AlC” κ Phase. Inorg. Chem. 2017, 56, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Andryushchenko, V.A.; Gavrilyuk, V.G.; Nadutov, V.M. Atomic and magnetic ordering in k-phase of Fe-Al-C alloys. Phys. Met. Metallogr. 1985, 60, 50–55. [Google Scholar]
- Seol, J.-B.; Raabe, D.; Choi, P.; Park, H.S.; Kwak, J.H.; Park, C.G. Direct evidence for the formation of ordered carbides in a ferrite based low-density Fe–Mn–Al–C alloy studied by transmission electron microscopy and atom probe tomography. Scr. Mater. 2013, 68, 348–353. [Google Scholar] [CrossRef]
- Yao, M.J.; Dey, P.; Seol, J -B.; Choi, P.; Herbig, M.; Marceau, R.K.W.; Hickel, T.; Neugebauer, J.; Raabe, D. Combined atom probe tomography and density functional theory investigation of the Al off-stoichiometry of kappa-carbides in an austenitic Fe-Mn-Al-C low density steel. Acta Mater. 2016, 106, 229–238. [Google Scholar]
- Dey, P.; Nazarov, R.; Dutta, B.; Yao, M.; Herbig, M.; Friák, M.; Hickel, T.; Raabe, D.; Neugebauer, J. Ab initio explanation of disorder and off-stoichiometry in Fe-Mn-Al-C κ carbides. Phys. Rev. B 2017, 95, 104108. [Google Scholar] [CrossRef]
- Palm, M.; Inden, G. Experimental determination of phase equilibria in the Fe-Al-C system. Intermetallics 1995, 3, 443–454. [Google Scholar]
- Desai, S.K.; Neeraj, T.; Gordon, P.A. Atomistic mechanism of hydrogen trapping in bcc Fe–Y solid solution: A first principles study. Acta Mater. 2010, 58, 5363–5369. [Google Scholar] [CrossRef]
- Psiachos, D.; Hammerschmidt, T.; Drautz, R. Ab initio study of the interaction of H with substitutional solute atoms in α-Fe: Trends across the transition-metal series. Comput. Mater. Sci. 2012, 65, 235–238. [Google Scholar] [CrossRef]
- Nazarov, R.; Hickel, T.; Neugebauer, J. Ab initio study of H-vacancy interactions in fcc metals: Implications for the formation of superabundant vacancies. Phys. Rev. B 2014, 89, 144108. [Google Scholar] [CrossRef]
- Du, Y.A.; Ismer, L.; Rogal, J.; Hickel, T.; Neugebauer, J.; Drautz, R. First-principles study on the interaction of H interstitials with grain boundaries in α- and γ-Fe. Phys. Rev. B 2011, 84, 144121. [Google Scholar] [CrossRef]
- Baligidad, R.G.; Prakash, U.; Radhakrishna, A.; Ramakrishna Rao, V. Effect of carbides on embrittlement of Fe3Al based intermetallic alloys. Scr. Mater. 1997, 36, 667–671. [Google Scholar] [CrossRef]
- Sen, M.; Balasubramaniam, R. Hydrogen trapping at carbide-matrix interfaces in Fe3AlC intermetallics. Scr. Mater. 2001, 44, 619–623. [Google Scholar] [CrossRef]
- Koyama, M.; Springer, H.; Merzlikin, S.V.; Tsuzaki, K.; Akiyama, E.; Raabe, D. Hydrogen embrittlement associated with strain localization in a precipitation-hardened Fe–Mn–Al–C light weight austenitic steel. Int. J. Hydrogen Energy 2014, 39, 4634–4646. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11185. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector-augmented wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1774. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Ekholm, M.; Abrikosov, I.A. Structural and magnetic ground-state properties of γ-FeMn alloys from ab initio calculations. Phys. Rev. B 2011, 84, 104423. [Google Scholar] [CrossRef]
- Lee, K.; Yuan, M.; Wilcox, J. Understanding Deviations in Hydrogen Solubility Predictions in Transition Metals through First-Principles Calculations. J. Phys. Chem. C 2015, 119, 19642–19653. [Google Scholar] [CrossRef]
- Kellou, A.; Raulot, J.M.; Grosdidier, T. Structural and thermal properties of Fe3Al, Fe3AlC and hypothetical Fe3AlX (X = H, B, N, O) compounds: Ab initio and quasi-harmonic Debye modelling. Intermetallics 2010, 18, 1293–1296. [Google Scholar] [CrossRef]
- Connétable, D.; Maugis, P. First principle calculations of the k-Fe3AlC perovskite and iron/aluminium intermetallics. Intermetallics 2008, 16, 345–352. [Google Scholar] [CrossRef]
- Noh, J.Y.; Kim, H. Ab initio calculations on the effect of Mn substitution in the κ-carbide Fe3AlC. J. Korean Phys. Soc. 2013, 62, 481–485. [Google Scholar] [CrossRef]
- von Appen, J.; Dronskowski, R.; Chakrabarty, A.; Hickel, T.; Spatschek, R.; Neugebauer, J. Impact of Mn on the solution enthalpy of hydrogen in austenitic Fe-Mn alloys: A first-principles study. J. Comp. Chem. 2014, 35, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zhang, W.; von Appen, J.; Dronskowski, R.; Bleck, W. κ-phase Formation in Fe–Mn–Al–C Austenitic Steels. Steel Res. Int. 2015, 86, 1161–1169. [Google Scholar] [CrossRef]
- Timmerscheidt, T.A.; Dronskowski, R. An Ab Initio Study of Carbon-Induced Ordering in Austenitic Fe-Mn-Al-C Alloys. Steel Res. Int. 2017, 88, 1600292. [Google Scholar] [CrossRef]
- Milella, P.P. Fatigue and Corrosion in Metals, 1st ed.; Springer: Milan, Italy, 2013. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | a0 (Å) | μtheo (μB/Fe atom) | ΔEel (kJ/mol) |
|---|---|---|---|---|
| Fe3Al | BiF3, D03 | 5.74 | 2.0 | −78 |
| Fe3Al | Cu3Au, L12 | 3.65 | 2.3 | −77 |
| Fe3AlC | perovskite, E21 | 3.75 | 1.0 | −90 |
| Fe3AlH | perovskite, E21 | 3.68 | 2.1 | −85 |
| Stacking Sequence of Layers in SC | Etrap (H) Adjacent Fe Layer (eV) | Etrap (H) Interface Layer (eV) | Etrap (H) Adjacent κ Layer (eV) |
|---|---|---|---|
| Fe*–C*–Al*–C*–Fe*–Fe*–Fe*–Fe*–Fe*–Fe* | −0.20 | −0.18 (C) | 0.04 (Al) |
| Fe*–Al*–C*–Al*–C*–Fe*–Fe*–Fe*–Fe*–Fe* | 0.01 | 0.003 (Al) | −0.05 (C) |
| Fe*–C*–Al*–C*–Al*–C*–Al*–Fe*–Fe*–Fe* | −0.20 | −0.24 (C) | 0.04 (Al) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmerscheidt, T.A.; Dey, P.; Bogdanovski, D.; Von Appen, J.; Hickel, T.; Neugebauer, J.; Dronskowski, R. The Role of κ-Carbides as Hydrogen Traps in High-Mn Steels. Metals 2017, 7, 264. https://doi.org/10.3390/met7070264
Timmerscheidt TA, Dey P, Bogdanovski D, Von Appen J, Hickel T, Neugebauer J, Dronskowski R. The Role of κ-Carbides as Hydrogen Traps in High-Mn Steels. Metals. 2017; 7(7):264. https://doi.org/10.3390/met7070264
Chicago/Turabian StyleTimmerscheidt, Tobias A., Poulumi Dey, Dimitri Bogdanovski, Jörg Von Appen, Tilmann Hickel, Jörg Neugebauer, and Richard Dronskowski. 2017. "The Role of κ-Carbides as Hydrogen Traps in High-Mn Steels" Metals 7, no. 7: 264. https://doi.org/10.3390/met7070264
APA StyleTimmerscheidt, T. A., Dey, P., Bogdanovski, D., Von Appen, J., Hickel, T., Neugebauer, J., & Dronskowski, R. (2017). The Role of κ-Carbides as Hydrogen Traps in High-Mn Steels. Metals, 7(7), 264. https://doi.org/10.3390/met7070264