
Immunological and Genetic Investigation of SARS-CoV-2 Reinfection in an Otherwise Healthy, Young Marine Recruit

 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Results
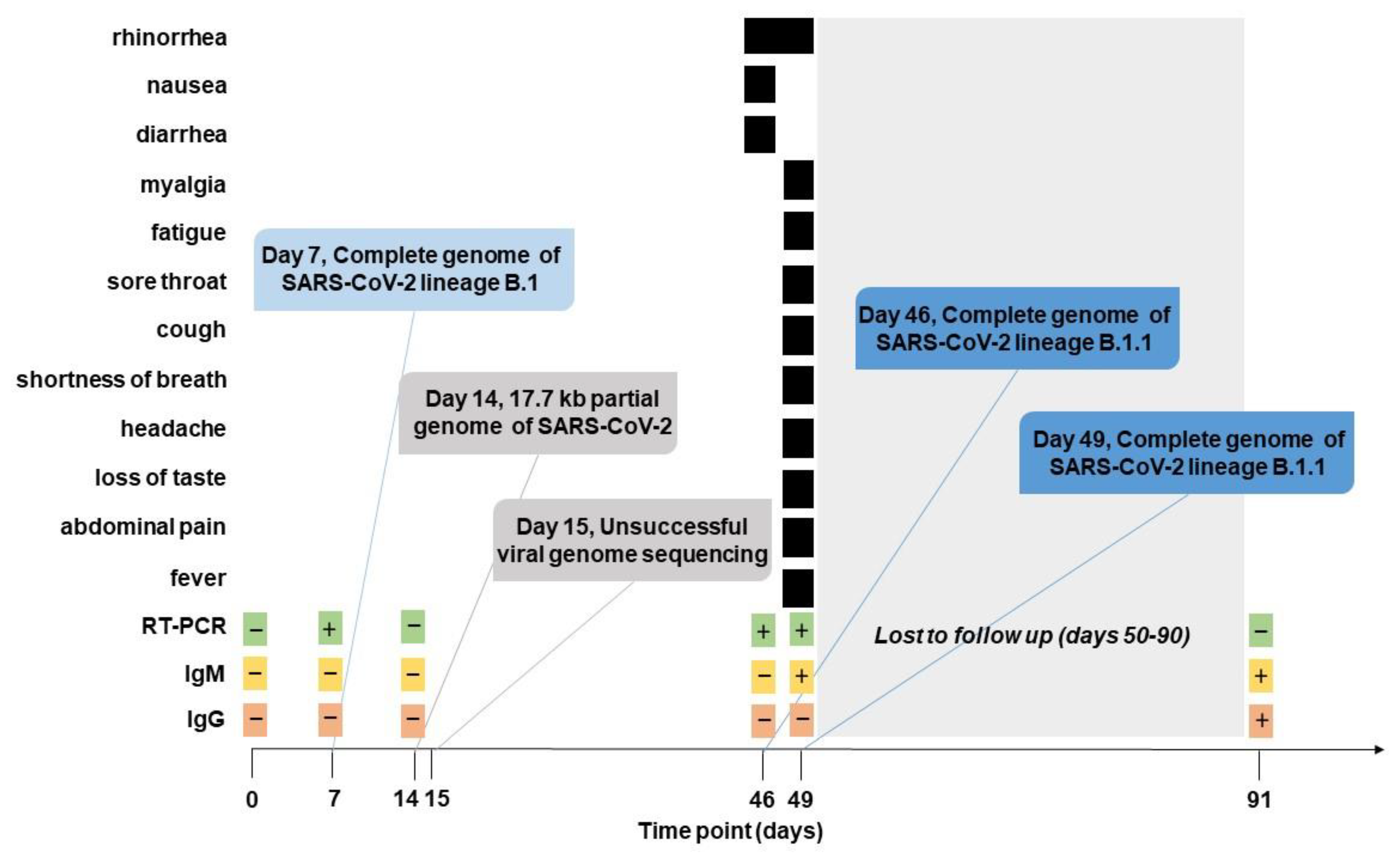
2.1. Clinical Presentation and SARS-CoV-2 Sampling
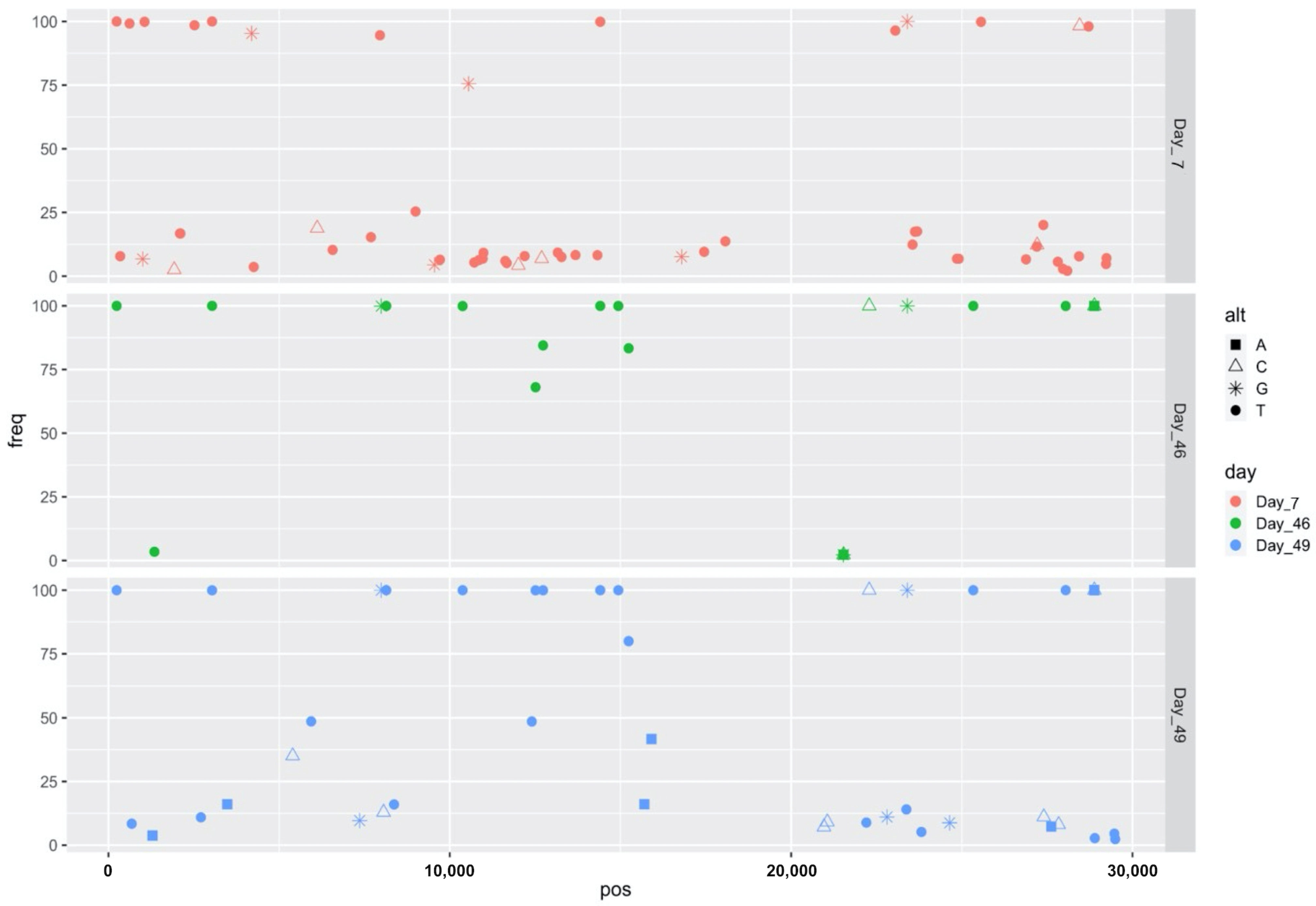
2.2. Virus Characterization
3. Discussion
4. Materials and Methods
4.1. The Study
4.2. SARS-CoV-2 Whole Genome Sequencing
4.3. Bioinformatic Analyses
4.4. SARS-CoV-2 Enzyme-Linked Immunosorbent Assay (ELISA)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Dyrdak, R.; Hodcroft, E.B.; Wahlund, M.; Neher, R.A.; Albert, J. Interactions between seasonal human coronaviruses and implications for the SARS-CoV-2 pandemic: A retrospective study in Stockholm, Sweden, 2009–2020. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2021, 136, 104754. [Google Scholar] [CrossRef]
- Callow, K.A.; Parry, H.F.; Sergeant, M.; Tyrrell, D.A. The time course of the immune response to experimental coronavirus infection of man. Epidemiol. Infect. 1990, 105, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Kiyuka, P.K.; Agoti, C.N.; Munywoki, P.K.; Njeru, R.; Bett, A.; Otieno, J.R.; Otieno, G.P.; Kamau, E.; Clark, T.G.; van der Hoek, L.; et al. Human Coronavirus NL63 Molecular Epidemiology and Evolutionary Patterns in Rural Coastal Kenya. J. Infect. Dis. 2018, 217, 1728–1739. [Google Scholar] [CrossRef] [Green Version]
- Investigative Criteria for Suspected Cases of SARS-CoV-2 Reinfection (ICR). Available online: https://www.cdc.gov/coronavirus/2019-ncov/php/invest-criteria.html (accessed on 1 November 2021).
- Cruz, C.D.; Torre, A.; Troncos, G.; Lambrechts, L.; Leguia, M. Targeted full-genome amplification and sequencing of dengue virus types 1–4 from South America. J. Virol. Methods 2016, 235, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Jacot, D.; Pillonel, T.; Greub, G.; Bertelli, C. Assessment of SARS-CoV-2 Genome Sequencing: Quality Criteria and Low-Frequency Variants. J. Clin. Microbiol. 2021, 59, e0094421. [Google Scholar] [CrossRef]
- Pedro, N.; Silva, C.N.; Magalhaes, A.C.; Cavadas, B.; Rocha, A.M.; Moreira, A.C.; Gomes, M.S.; Silva, D.; Sobrinho-Simoes, J.; Ramos, A.; et al. Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms 2021, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Oved, K.; Olmer, L.; Shemer-Avni, Y.; Wolf, T.; Supino-Rosin, L.; Prajgrod, G.; Shenhar, Y.; Payorsky, I.; Cohen, Y.; Kohn, Y.; et al. Multi-center nationwide comparison of seven serology assays reveals a SARS-CoV-2 non-responding seronegative subpopulation. EClinicalMedicine 2020, 29, 100651. [Google Scholar] [CrossRef]
- Liu, W.; Russell, R.M.; Bibollet-Ruche, F.; Skelly, A.N.; Sherrill-Mix, S.; Freeman, D.A.; Stoltz, R.; Lindemuth, E.; Lee, F.H.; Sterrett, S.; et al. Predictors of Nonseroconversion after SARS-CoV-2 Infection. Emerg. Infect. Dis. 2021, 27, 2454–2458. [Google Scholar] [CrossRef] [PubMed]
- Pathela, P.; Crawley, A.; Weiss, D.; Maldin, B.; Cornell, J.; Purdin, J.; Schumacher, P.K.; Marovich, S.; Li, J.; Daskalakis, D.; et al. Seroprevalence of Severe Acute Respiratory Syndrome Coronavirus 2 Following the Largest Initial Epidemic Wave in the United States: Findings From New York City, 13 May to 21 July 2020. J. Infect. Dis. 2021, 224, 196–206. [Google Scholar] [CrossRef]
- Borremans, B.; Gamble, A.; Prager, K.C.; Helman, S.K.; McClain, A.M.; Cox, C.; Savage, V.; Lloyd-Smith, J.O. Quantifying antibody kinetics and RNA detection during early-phase SARS-CoV-2 infection by time since symptom onset. eLife 2020, 9, e60122. [Google Scholar] [CrossRef]
- Sun, B.; Feng, Y.; Mo, X.; Zheng, P.; Wang, Q.; Li, P.; Peng, P.; Liu, X.; Chen, Z.; Huang, H.; et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 940–948. [Google Scholar] [CrossRef]
- Vetter, P.; Cordey, S.; Schibler, M.; Vieux, L.; Despres, L.; Laubscher, F.; Andrey, D.O.; Martischang, R.; Harbarth, S.; Cuvelier, C.; et al. Clinical, virological and immunological features of a mild case of SARS-CoV-2 reinfection. Clin. Microbiol. Infect. 2021, 27, 791.e1–791.e4. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.; Brodniak, S.L.; Voegtly, L.J.; Cer, R.Z.; Glang, L.A.; Malagon, F.J.; Long, K.A.; Potocki, R.; Smith, D.R.; Lanteri, C.; et al. A Case of Early Re-infection with SARS-CoV-2. Clin. Infect. Dis. 2021, 73, e2827–e2828. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Hesse, E.M.; Paulin, H.N.; Datta, D.; Katz, L.S.; Talwar, A.; Chang, G.; Galang, R.R.; Harcourt, J.L.; Tamin, A.; et al. Clinical and Laboratory Findings in Patients with Potential SARS-CoV-2 Reinfection, May–July 2020. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Letizia, A.G.; Ge, Y.; Vangeti, S.; Goforth, C.; Weir, D.L.; Kuzmina, N.A.; Balinsky, C.A.; Chen, H.W.; Ewing, D.; Soares-Schanoski, A.; et al. SARS-CoV-2 seropositivity and subsequent infection risk in healthy young adults: A prospective cohort study. Lancet Respir. Med. 2021, 9, 712–720. [Google Scholar] [CrossRef]
- Hansen, C.H.; Michlmayr, D.; Gubbels, S.M.; Molbak, K.; Ethelberg, S. Assessment of protection against reinfection with SARS-CoV-2 among 4 million PCR-tested individuals in Denmark in 2020: A population-level observational study. Lancet 2021, 397, 1204–1212. [Google Scholar] [CrossRef]
- Letizia, A.G.; Ge, Y.; Goforth, C.W.; Weir, D.L.; Lizewski, R.; Lizewski, S.; Soares-Schanoski, A.; Vangeti, S.; Marjanovic, N.; Sealfon, S.C.; et al. SARS-CoV-2 Seropositivity among US Marine Recruits Attending Basic Training, United States, Spring-Fall 2020. Emerg. Infect. Dis. 2021, 27, 1188. [Google Scholar] [CrossRef] [PubMed]
- Letizia, A.G.; Ramos, I.; Obla, A.; Goforth, C.; Weir, D.L.; Ge, Y.; Bamman, M.M.; Dutta, J.; Ellis, E.; Estrella, L.; et al. SARS-CoV-2 Transmission among Marine Recruits during Quarantine. N. Engl. J. Med. 2020, 383, 2407–2416. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol. Protocols.io. 2020. Available online: https://dx.doi.org/10.17504/protocols.io.bbmuik6w (accessed on 22 January 2020).
- Voegtly, L.; Long, K.; Rice, G.K.; Cer, R.Z.; Bishop-Lilly, K.A. Viral Amplicon Illumina Workflow (VAIW): A Custom Pipeline to Analyze the SARS-CoV-2 Genomes Prepared with an Amplicon (ARTIC (v3) and YouSeq (v2)) Based Library Protocols. Docker Hub. Available online: https://hub.docker.com/r/bdrdgenomics/viral_amplicon_illumina_workflow (accessed on 6 October 2020).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Bushnell, B. BBMAP: A Fast, Accurate, Splice-Aware Aligner. 2014. Available online: https://sourceforge.net/projects/bbmap (accessed on 1 July 2020).
- ARTIC Network Github. 2020. Available online: https://github.com/artic-network/fieldbioinformatics/blob/master/artic/align_trim.py (accessed on 20 May 2020).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, A.; McCrone, J.T.; Scher, E. Pangolin. GitHub. 2020. Available online: https://github.com/cov-lineages/pangolin (accessed on 9 November 2021).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Ct Value per Target | Patient Status | |||
|---|---|---|---|---|
| Time point (day) | S gene | N gene | ORF1ab | |
| 7 | 28.9 | 28.3 | 28.5 | Asymptomatic |
| 46 | 16.9 | 16.8 | 17.4 | Symptomatic |
| 49 | 28.0 | 27.0 | 27.3 | Symptomatic |
| SNV | Frequency (%) | SNV Type | AA Change | Gene |
|---|---|---|---|---|
| C346T | 7.83 | synonymous | - | ORF1ab/nsp1 |
| C619T | 99.19 | synonymous | - | ORF1ab/nsp1 |
| A1005G | 6.74 | nonsynonymous | K247R, K67R | ORF1ab/nsp2 |
| C1059T | 99.87 | nonsynonymous | T265I, T85I | ORF1ab/nsp2 |
| T1927C | 2.56 | synonymous | - | ORF1ab/nsp2 |
| C2096T | 16.76 | nonsynonymous | Q611 *, Q431 * | ORF1ab, nsp2 |
| C2110T | 16.76 | synonymous | - | ORF1ab, nsp2 |
| C2523T | 98.52 | nonsynonymous | T753I, T573I | ORF1ab, nsp2 |
| A4197G | 95.31 | nonsynonymous | E1311G, E493G | ORF1ab, nsp3 |
| G4257T | 3.62 | nonsynonymous | G1331V, G513V | ORF1ab, nsp3 |
| G6116C | 18.85 | nonsynonymous | A1951P, A1133P | ORF1ab, nsp3 |
| C6568T | 10.31 | synonymous | - | ORF1ab, nsp3 |
| C7691T | 15.29 | nonsynonymous | Q2476 *, Q1658 * | ORF1ab, nsp3 |
| G7954T | 94.57 | nonsynonymous | Q2563H, Q1754H | ORF1ab, nsp3 |
| G8999T | 25.39 | nonsynonymous | A2912S, A149S | ORF1ab, nsp4 |
| C9551G | 4.46 | nonsynonymous | P3096A, P333A | ORF1ab, nsp4 |
| C9712T | 6.46 | synonymous | - | ORF1ab, nsp4 |
| A10552G | 75.56 | synonymous | - | ORF1ab, nsp5 |
| C10718T | 5.37 | nonsynonymous | R3485 *, R222 * | ORF1ab, nsp5 |
| C10854T | 6.33 | nonsynonymous | S3530L, S267L | ORF1ab, nsp5 |
| C10965T | 6.86 | nonsynonymous | T3567I, T304I | ORF1ab, nsp5 |
| G10986T | 9.16 | nonsynonymous | R3574I, R5I | ORF1ab, nsp6 |
| G11625T | 5.95 | nonsynonymous | G3787V, G218V | ORF1ab, nsp6 |
| C11668T | 5.16 | - | ORF1ab, nsp6 | |
| T12009C | 4.28 | nonsynonymous | L3915P, L56P | ORF1ab, nsp7 |
| C12194T | 7.90 | nonsynonymous | L3977F, L35F | ORF1ab, nsp8 |
| G12692C | 6.92 | nonsynonymous | E4143Q, E3Q | ORF1ab, nsp9 |
| C13164T | 9.25 | nonsynonymous | T4300I, T47I | ORF1ab, nsp10 |
| C13274T | 7.53 | nonsynonymous | P4337S, P84S | ORF1ab, nsp10 |
| C13684T | 8.29 | nonsynonymous | H4474Y, H82Y | ORF1ab, nsp12 |
| C14325T | 8.23 | synonymous | - | ORF1ab, nsp12 |
| C16792G | 7.61 | nonsynonymous | R5510G, R186G | ORF1ab, nsp13 |
| C17452T | 9.55 | nonsynonymous | P5730S, P406S | ORF1ab, nsp13 |
| G18074T | 13.68 | nonsynonymous | S5937I, S12I | ORF1ab, nsp14 |
| C23053T | 96.43 | synonymous | - | s |
| C23556T | 12.38 | nonsynonymous | P665L | s |
| C23625T | 17.46 | nonsynonymous | A688V | s |
| C23692T | 17.58 | synonymous | - | s |
| G24858T | 6.83 | nonsynonymous | G1099V | s |
| C24909T | 6.82 | nonsynonymous | T1116I | s |
| G25563T | 99.87 | nonsynonymous | Q57H | ORF3a |
| C26882T | 6.60 | synonymous | - | m |
| C27196T | 11.62 | - | - | noncoding region |
| T27206C | 12.23 | nonsynonymous | F2S | ORF6 |
| C27389T | 20.09 | - | - | noncoding region |
| C27813T | 5.64 | nonsynonymous | L20F | ORF7 |
| C27964T | 2.85 | nonsynonymous | S24L | ORF8 |
| G28089T | 2.11 | nonsynonymous | G66C | ORF8 |
| C28435T | 7.78 | synonymous | - | n |
| G28451C | 98.29 | nonsynonymous | G60R | n |
| A28715T | 98.05 | nonsynonymous | T148S | n |
| C29226T | 4.79 | nonsynonymous | S318L | n |
| G29239T | 7.06 | nonsynonymous | M322I | n |
| SNV | Frequency (%) | SNV Type | AA Change | Gene |
|---|---|---|---|---|
| C1348T | 3.42 | synonymous | - | ORF1ab, nsp2 |
| C21530G | 2.25 | nonsynonymous | S7089C, S291C | ORF1ab, nsp16 |
| G21535T | 2.25 | nonsynonymous | D7091Y, D293Y | ORF1ab, nsp16 |
| T21534A | 2.26 | nonsynonymous | S7090R, S292R | ORF1ab, nsp16 |
| A21536C | 2.24 | nonsynonymous | D7091A, D293A | ORF1ab, nsp16 |
| SNV | Frequency (%) | SNV Type | AA Change | Gene |
|---|---|---|---|---|
| C683T | 8.42 | synonymous | - | ORF1ab, nsp1 |
| G1289A | 3.79 | nonsynonymous | E342K, E162K | ORF1ab, nsp2 |
| C2710T | 10.92 | synonymous | - | ORF1ab, nsp2 |
| G3483A | 16.07 | nonsynonymous | G1073E, G255E | ORF1ab, nsp3 |
| G5397C | 35.07 | nonsynonymous | C1711S, C893S | ORF1ab, nsp3 |
| A5939T | 48.59 | nonsynonymous | I1892F, I1074F | ORF1ab, nsp3 |
| T7361G | 9.64 | nonsynonymous | W2366G, W1548G | ORF1ab, nsp3 |
| T8060C | 12.93 | nonsynonymous | S2599P, S1781P | ORF1ab, nsp3 |
| G8368T | 15.97 | synonymous | - | ORF1ab, nsp3 |
| C12403T | 48.55 | synonymous | - | ORF1ab, nsp8 |
| C15701A | 16.09 | nonsynonymous | S5146 *, S754 * | ORF1ab, nsp12 |
| G15907A | 41.67 | nonsynonymous | G5215S, G823S | ORF1ab, nsp12 |
| T20961C | 7.16 | synonymous | - | ORF1ab, nsp16 |
| T21060C | 9.13 | synonymous | - | ORF1ab, nsp16 |
| G22203T | 8.86 | nonsynonymous | R214L | s |
| A22810G | 11.06 | synonymous | - | s |
| C23376T | 14.03 | nonsynonymous | S605F | s |
| C23816T | 5.19 | nonsynonymous | L752F | s |
| A24644G | 8.76 | nonsynonymous | K1028E | s |
| T27402C | 11.10 | synonymous | - | ORF7a |
| G27621A | 7.30 | synonymous | - | ORF7a |
| T27837C | 8.08 | nonsynonymous | F28L | ORF7b |
| C28896T | 2.75 | nonsynonymous | A208V | n |
| A29469T | 4.50 | nonsynonymous | D399V | n |
| G29494T | 2.38 | nonsynonymous | L407F | n |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letizia, A.G.; Arnold, C.E.; Adhikari, B.N.; Voegtly, L.J.; Glang, L.; Rice, G.K.; Goforth, C.W.; Schilling, M.A.; Weir, D.L.; Malagon, F.; et al. Immunological and Genetic Investigation of SARS-CoV-2 Reinfection in an Otherwise Healthy, Young Marine Recruit. Pathogens 2021, 10, 1589. https://doi.org/10.3390/pathogens10121589
Letizia AG, Arnold CE, Adhikari BN, Voegtly LJ, Glang L, Rice GK, Goforth CW, Schilling MA, Weir DL, Malagon F, et al. Immunological and Genetic Investigation of SARS-CoV-2 Reinfection in an Otherwise Healthy, Young Marine Recruit. Pathogens. 2021; 10(12):1589. https://doi.org/10.3390/pathogens10121589
Chicago/Turabian StyleLetizia, Andrew G., Catherine E. Arnold, Bishwo N. Adhikari, Logan J. Voegtly, Lindsay Glang, Gregory K. Rice, Carl W. Goforth, Megan A. Schilling, Dawn L. Weir, Francisco Malagon, and et al. 2021. "Immunological and Genetic Investigation of SARS-CoV-2 Reinfection in an Otherwise Healthy, Young Marine Recruit" Pathogens 10, no. 12: 1589. https://doi.org/10.3390/pathogens10121589