
Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives
by
 , , and
, , and
Julio C. Osorio
1 ,
,
Rancés Blanco
2,
Alejandro H. Corvalán
3,
Juan P. Muñoz
4,
Gloria M. Calaf
4,5 and
Francisco Aguayo
6,* 1
Population Registry of Cali, Department of Pathology, Universidad del Valle, Cali 760042, Colombia
2
Laboratorio de Oncovirología, Programa de Virología, Instituto de Ciencias Biomédicas (ICBM), Facultad de Medicina, Universidad de Chile, Santiago 8380000, Chile
3
Advanced Center for Chronic Diseases (ACCDiS), Pontificia Universidad Católica de Chile, Santiago 8320000, Chile
4
Instituto de Alta Investigación, Universidad de Tarapacá, Arica 1000000, Chile
5
Center for Radiological Research, Columbia University Medical Center, New York, NY 10032, USA
6
Universidad de Tarapacá, Arica 1000000, Chile
*
Author to whom correspondence should be addressed.
Pathogens 2022, 11(2), 132; https://doi.org/10.3390/pathogens11020132
Submission received: 15 December 2021
/
Revised: 17 January 2022
/
Accepted: 17 January 2022
/
Published: 21 January 2022
(This article belongs to the Collection Feature Papers in Viral Pathogens)
Abstract
:Lung cancer (LC) is the leading cause of cancer death worldwide. Tobacco smoke is the most frequent risk factor etiologically associated with LC, although exposures to other environmental factors such as arsenic, radon or asbestos are also involved. Additionally, the involvement of some viral infections such as high-risk human papillomaviruses (HR-HPVs), Merkel cell polyomavirus (MCPyV), Jaagsiekte Sheep Retrovirus (JSRV), John Cunningham Virus (JCV), and Epstein–Barr virus (EBV) has been suggested in LC, though an etiological relationship has not yet been established. EBV is a ubiquitous gamma herpesvirus causing persistent infections and some lymphoid and epithelial tumors. Since EBV is heterogeneously detected in LCs from different parts of the world, in this review we address the epidemiological and experimental evidence of a potential role of EBV. Considering this evidence, we propose mechanisms potentially involved in EBV-associated lung carcinogenesis. Additional studies are warranted to dissect the role of EBV in this very frequent malignancy.
1. Introduction
Lung cancer (LC) is the leading cause of cancer death worldwide, with 2.21 million new cases and 1.80 million deaths in 2020 [1]. LC is a heterogeneous disease, broadly classified into two main groups: small cell lung carcinoma (SCLC), and non-small cell lung carcinoma (NSCLC) [2]. SCLC is a type of LC that shows a neuroendocrine origin, with a very aggressive behavior and poor prognosis [3]. NSCLC is the most prevalent type, including squamous cell carcinoma (SQC), adenocarcinoma (AdC), and large cell carcinoma (LCC) [4]. SQC shows a bronchial origin and is characterized by keratinization, intercellular bridges, tumor bulk, expression of cytokeratins 5/6, and diffuse expression of p63 [5]. AdC exhibits an alveolar origin and is characterized by thyroid transcription factor 1 (TTF1) and keratin 7 expression (KRT7) [6]. LCC is typically generated in the outer regions of the lungs and is characterized by KRT7 and epithelial membrane antigen (EMA) expressions, with a lack of TTF-1, p63, and keratin 5/6 expressions [7]. Tobacco smoke is the most frequent and well-known risk factor associated with LC [8]. Indeed, secondary metabolites from tobacco smoke promote mutations in genes such as TP53, KRAS, NRAS, HRAS, MYC, cyclin-dependent kinase inhibitor 2A (CDKN2A), and retinoblastoma (RB1) [9,10]. All of the aforementioned genes are involved in the increase in genome instability and LC progression [11]. However, additional environmental factors also play a role in LC development, such as radon, arsenic, and environmental pollution [12]. Tobacco smoke harbors more than 7000 chemicals [13] and approximately 70 of them are considered carcinogens [14], including polycyclic aromatic hydrocarbons (PAH), nitrosamines, acetaldehyde, arsenic, beryllium, cadmium, 1,3-butadiene, and azarenes, among many others [8]. Tobacco smoke chemicals can also be found inside the air we breathe as well as in our food and water [15]. In particular, large amounts of benzo[a]pyrene (BaP), a type of PAH, are released into the air through industry, agriculture, and emissions from combusting fossil fuels [16]. Other sources of BaP are produced when coal, oil, gas, wood, and garbage are burned [17].
Viral infections have been proposed to be involved in LC, namely, high-risk human papillomaviruses (HR-HPVs) [18], Merkel cell polyomavirus (MCPyV) [19], Jaagsiekte Sheep Retrovirus (JSRV) [20], John Cunningham Virus (JCV) [21], and Epstein–Barr virus (EBV) [22]. EBV is an exclusive human virus (without an animal reservoir) present in ~95% of the human population and establishing persistent infections during the lifetime [23]. EBV preferentially infects both B cells and epithelial cells [24] and is characterized as a class I carcinogen [25]. Epidemiological evidence suggests that EBV is present in a relatively small subset of LCs [22], with demographic variations, though an etiological relationship remains inconclusive. In this review, we address the epidemiological and mechanistic evidence regarding a potential role of EBV in this malignancy. Taking into consideration that previous reported data suggested a cooperation between EBV and xenobiotics for carcinogenesis [26], here we propose potential mechanisms that involve an interplay between environmental factors such as tobacco smoke and EBV for developing lung cancer. However, additional experimental and epidemiological studies are warranted to confirm these possibilities.
2. Epstein–Barr Virus: Structure, Replication, and Role in Cancer
2.1. Epstein–Barr Virus: Structure and Replication Cycle
EBV is an enveloped gamma herpesvirus and a member of the Lymphocryptovirus genus, subfamily Gammaherpesvirinae [27]. The EBV core contains a double-stranded linear DNA virus with approximately 172-kpb, enclosed by a 162-capsomer icosahedral capsid (Figure 1). The core is surrounded by the tegument, which is composed of enzymes and proteins involved in viral replication [28]. Primary EBV infection during childhood is frequently asymptomatic, though infection during adolescence can lead to symptomatic infectious mononucleosis (IM) [29]. After EBV entry in the Waldeyer’s ring cells, EBV infects naïve B cells, leading to the establishment of a latent infection in memory B cells that then enter the peripheral circulation [30]. It is not clear if EBV replicates in epithelial cells from the Waldeyer’s ring before B-cell infection. Eventually, during EBV reactivation, the virus can re-enter Waldeyer’s ring structures for transmission through saliva [31]. EBV gH/gL and gB glycoproteins are involved in the viral entry into epithelial cells by the fusion machinery [32], while glycoprotein 42 (gp42) is only required during infection of B cells [33]. Additionally, depending on the CR2 (Complement receptor type 2, CD21) expression levels, gp350 can be used as a ligand for attachment to CD21(+) epithelial cells or the gH/gL complex for attachment to CD21 (−) epithelial cells [34]. Importantly, the Ephrin receptor tyrosine kinase A2 (EphA2) has been recently proposed to be the epithelial EBV receptor [35]. After viral attachment, entry occurs by cell fusion, allowing the entry of the tegumented capsid into the cytoplasm [36]. Next, the capsid with the genome is transported to the periphery of the nucleus [37] and the viral genome crosses the nuclear pore complexes to enter the nucleus [38]. The Epstein–Barr nuclear antigen 1 (EBNA1) gene is expressed, leading to the EBV genome tethering to the host genome and is maintained as an episome [39,40]. During lytic cycle activation, the immediate-early (IE) proteins are first expressed. Indeed, the IE Zta and Rta proteins act as both key transactivators and master regulators of the lytic cycle allowing the expression of the viral DNA polymerase (BALF5) for viral DNA replication by a rolling circle mechanism [41]. Indeed, the recruitment and assembly of core replication proteins at the oriLyt are a requisite for EBV genome replication. The involved viral proteins are BALF5, BMRF1, BALF2, BBLF4 BSLF1, and BBLF2/3. The synthetized linear EBV genome is cleaved at the terminal repeats and packaged into viral capsids. The serine/threonine-protein kinase BFRF1 and BFLF2 genes allow intranuclear nucleocapsids to move towards the inner leaflet of the nuclear membrane and sprout from the nuclear envelope [38]. Next, the access of nucleocapsids to the nuclear membrane is mediated by the viral BGLF4 protein [38]. The nucleocapsids can then be transported into the juxtanuclear viral assembly compartment. Finally, the virions are released from the infected cells by exocytosis [42].
2.2. Role of EBV in Epithelial Cancers
EBV is the causal agent of undifferentiated nasopharyngeal carcinoma (NPC) [43] and a subset of gastric carcinomas (GCs) [44]. Additionally, EBV has been detected in other tumors such as those arising in the breast [45], colon [46], prostate [47], and lung, though an etiological association has not yet been established. Knowledge of the role of EBV in carcinomas has arisen from studying this infection in NPC and GC. Indeed, undifferentiated NPC is a recognized model of EBV-driven epithelial carcinogenesis [48]. Type II latency is the predominant form of latency found in undifferentiated NPC [48], in which LMP1, LMP2A, EBNA1, BARF1, EBER, BARTS, and BamHI-H rightward fragment 1-derived miRNAs (BHRF1) are detected [43]. The BHRF1 miRNA cluster encodes four miRNAs, and BART generates 28 miRNAs encoded by two miRNA clusters [49]. Viral miRNAs can regulate viral gene expression related to cancer development and progression [50].
Interestingly, evidence suggests that these latent EBV-expressed products show oncogenic properties [30]. Indeed, EBV oncoproteins activate signaling pathways such as Wnt/β-catenin, NF-κB, JNK, JAK/STAT, EGFR/MAPK, and PI3K/AKT (Figure 2) [51]. Regarding GCs, a recent meta-analysis reported that EBV prevalence among patients with GC was 8.77% worldwide [52]. The mechanisms by which EBV is involved in GC are not clear, but it is suggested that an EBV latency II profile promotes changes in the host genome methylation pattern, with subsequent increases in genomic instability [53]. Since latent EBV infection is generally not found in healthy nasopharyngeal tissue, it is plausible that previous genomic alterations are a condition for establishing EBV latency [54]. Importantly, both NPCs and GCs show genetic alterations (germ, somatic, and environmental mutations) that can predispose the tissue to latent EBV infection [55]. Regardless of the type of genetic alteration, CDKN2A (p16) loss and cyclin D1 dysregulation are key alterations that increase the genetic instability of cells. P16 is a negative regulator of cyclin D1 signaling, and its blocking or deregulation allows the progression of the cell cycle from the G1/S transition, which is related to increased cell proliferation, genomic instability, and cancer progression [56]. Increased cell proliferation due to the ectopic expression of cyclin D1 has been evaluated in tissues that have reported high efficiency in supporting latent EBV infection mediated by enhanced EBNA1 expression [57]. EBNA3C is related to cyclin A and drives a hyperphosphorylated form of retinoblastoma protein (Rb), allowing S phase entry. EBNA3 is also involved in Rb and p27 ubiquitination and subsequent degradation, which facilitates G1/S phase transition [58]. Additional genetic alterations, epigenetic modifications, and cofactors such as chronic inflammation, immunosuppression, and environmental mutagens are also required for this multistep oncogenic process.
2.3. EBV Latency and Lytic Cycle in Cancer
The establishment of EBV latency in epithelial tissue is a critical hit during EBV-driven carcinogenesis [59]. It is well-known that during latency, the EBV genome remains within the nucleus in an episomal form, and this is maintained at a constant number of copies after cell division [60]. This event occurs in B cells and in tumor epithelial cells, although evidence shows that latency is not established in normal epithelial cells, which per default activate the lytic cycle [61]. However, in EBV-driven NPCs and EBV-associated GCs (EBVaGCs), the EBV genome is always found in latency. Four latency forms (latency 0, I, II, and III) have been defined based on the pattern of expressed EBV products [62]. Latency 0 occurs in resting memory B cells, with only EBERs being detected [59]. Latency I is detected in Burkitt’s lymphoma (BL) with only EBERs and EBNA1 expression. Latency II is found in Hodgkin’s disease (HD), GC, and NPC with EBERs, BARTs, EBNA1, and three LMP genes being expressed [63]. Latency III is the least restricted latency type and is found in immortalized lymphoblastoid cell lines with EBERs, BARTs, six EBNA, and the three LMPs genes being expressed. It has been hypothesized that previous cell alterations, including p16 loss and cyclin D1 overexpression, are necessary for EBV latency establishment during NPC development [57]. However, the mechanisms and factors involved in latency establishment in epithelial cells are unknown.
Although latency establishment is a requisite for EBV-driven cancer, evidence shows that lytic cycle activation is important during EBV-driven carcinogenesis as well. Indeed, the abortive lytic cycle (ALC) involves the expression of latent genes and a restricted set of lytic genes, though without EBV maturation [64]. This is based on diverse studies in which some lytic proteins are consistently detected in cancers including Zta, Rta, and BARF1, among others [65]. In particular, BARF1 is an oncogenic early EBV protein with functions in immune evasion [66]. In latently infected B cells, lytic EBV promoters are methylated while they remain hypomethylated in normal oral keratinocytes (NOK) and Akata cells [67]. Additionally, EBV-infected human telomerase-immortalized NOK have shown CpG island hypermethylation in the transcription factor lymphoid enhancer factor 1 (LEF1) promoter region, which explains the metastatic phenotype of EBV-associated carcinomas [68]. Latent EBV infection causes genome-wide DNA methylation in GC through increased expression of LMP1 on DNA methyltransferases [69]. Global DNA hypermethylation of both viral and host DNA is frequently found in EBVaGCs [70]. The Zta protein preferentially binds to CpG-methylated motifs to reverse epigenetic silencing of viral DNA, leading to lytic activation [71].
3. EBV Infection in Lung Cancer
3.1. Pathogenesis of Lung Cancer
The LC pathogenesis is characterized by the accumulation of diverse genetic abnormalities, including chromosomal instability (CIN), loss or gain of whole chromosomes (aneuploidy), loss of heterozygosity (LOH), chromosomal deletions and rearrangements, as well as loss-of-function mutations in TSGs while activating point mutations of oncogenes. Additionally, epigenetic alterations including aberrant DNA-methylation patterns, among others, are involved in LC development (reviewed in [72,73]). The occurrence of CIN during cell division, which leads to other chromosomal aberrations, is considered one of the most important events associated with cancer development [74]. In bronchial epithelium, aneuploidy increased from moderate to severe dysplasia and carcinoma in situ lesions and was also associated with smoking status [75,76]. For instance, the percentage of cells displaying gain of whole chromosome 7 was reported to be increased from preinvasive to invasive lung lesions [75]. Chromosome 7 contains some oncogenes whose function is related to cell division, including the epidermal growth factor receptor (EGFR) (7p11.2) and MET (7q31.2) [77]. In addition, aberrations in the number of whole chromosomes have been evidenced in 51.8% to 84.8% of NSCLCs, suggesting a crucial role in LC progression [78,79,80]. In fact, a meta-analysis conducted by Choma et al., 2001 demonstrated the association of aneuploidy with poor prognosis of NSCLC patients [81]. LOH was found at chromosomes containing TSGs such as 17p13 (TP53), 9p21 (CDKN2A), and 3p14 (FHIT) in bronchial epithelium of smokers [82]. An increased LOH at chromosomes 3p21, 5q21, and 9p21 was also detected in normal bronchial mucosa adjacent to NSCLC compared to normal bronchial cells obtained from non-cancer patients [83]. Moreover, LOH was significantly related to the occurrence of carcinoma in situ [84]. Chromosomal rearrangements such as echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) fusion gene represent an important genetic alteration in LC. The aberrant fusion of the N-terminal domain of EML4 and the C-terminal region of ALK encodes a cytoplasmic chimeric protein with constitutive tyrosine kinase activity [85]. The expression of the EML4-ALK fusion gene is more frequent in lung AdCs and non-smoker patients, as well as being exclusive to the K-ras and EGFR mutations [86]. The EML4-ALK fusion protein increased the phosphorylation of STAT3 and AKT in human bronchial cells [87]. Increased tumorigenic properties in vivo of mouse 3T3 fibroblasts expressing EML4-ALK fusion protein were evidenced when compared with EML4- or ALK-transfected cells [85]. Transfection of human bronchial cells with the EML4-ALK fusion gene resulted in increased cell proliferation rates and enhanced anchorage-independent growth, though an absence of tumor formation was observed in vivo [87].
Mutations in TP53 are more frequent in SCLCs and SQCs compared to AdCs [88]. A high frequency of deletions was found in the chromosomal region 17p13, which contains the TP53 gene, in lung tumors [89]. In addition, TP53 mutations occur in about 50% of NSCLCs, mostly characterized by GC > TA transversions [90]. Loss of function of p16 was evidenced in NSCLCs, including promoter hypermethylation, point mutations, and loss of heterozygosity in the 9p21 region [91]. A low expression level was reported as well as CDKN2A gene promoter hypermethylation in alveolar atypical hyperplasia, the precursor lesion of lung AdC [92,93]. Loss of RB expression was significantly increased in SCLCs when compared with NSCLCs [94]. RB knockdown using shRNA vectors increased the proliferation rates of NSCLC cells as well as tumor formation in vivo [95]. Moreover, the somatic inactivation of both RB1 and P53 alleles in mouse lung epithelial cells led to neuroendocrine tumor formation, resembling the human SCLC [96]. Interestingly, loss of RB was associated with the EGFR mutant NSCLC that transformed into SCLC, the most aggressive form of LC disease [97]. Wild-type EGFR was found overexpressed in more than 60% of NSCLCs [98,99] and was associated with a decreased overall survival and chemoradiotherapy resistance [100]. On the other hand, EGFR mutations in the tyrosine kinase domain are frequently detected in NSCLCs. In a meta-analysis conducted by Zhang et al., 2018, EGFR mutations in NSCLCs ranged from 9.6 to 82.2% [101]. Another oncogene with a role in LC progression is the human epidermal growth factor receptor 2 (HER2). The expression of HER2 was detected in approximately 25% of LCs by immunohistochemistry (IHC) and 20% of LCs by fluorescence in situ hybridization (FISH) [102]. In addition, HER2 mutations were more frequently found in AdC (3%), females, and never smokers [103]. The most common mutation in the HER2 kinase domain is a 12 bp insertion in exon 20 (YVMA) [104,105]. HER2YVMA expression results in constitutive phosphorylation of HER2 and EGFR activation in the presence of tyrosine kinase inhibitors (TKIs). HER2 mutant also displayed an increased capacity to transform bronchial epithelial cells when compared to wild-type HER2 [106]. HER2 aberrant expression is associated with a decreased overall survival of NSCLC, mainly with AdC histology [102]. K-ras mutations are also observed in about 18–40% of lung AdCs [107,108,109] and are less common in SQCs [107]. It was demonstrated that K-ras mutations led to increased cell migration of immortalized airway cells through Akt activation [110]. Moreover, K-ras mutations were found to be related to the progression from precursor lesions (e.g., alveolar atypical hyperplasia and adenoma) to lung AdC in mice [111], which suggests a role in the development of these tumors. K-ras, EGFR, and HER2 mutations are mutually exclusive [105,112]. MYC family oncogenes (c-myc, N-myc, and L-myc) are commonly amplified in LC subtypes: AdC, SQC, and SCLC [113]. Approximately 50% of NSCLCs present activator changes in the mitogenic cascade (RAS-RAF-MEK-ERK) that induces c-MYC [114]. For instance, c-myc expression was evidenced in 35.7–58.6% of NSCLCs although an increased expression was reported in SQC compared with AdC [115,116,117]. Myc amplification and overexpression have been observed in SCLC [118]. In surgically resected SCLC, L-myc and c-myc protein expressions were detected in 6.5 and 8.7% of samples, respectively [119]. MYC family suppression induced cell cycle arrest and apoptosis inhibition in SCLC cells displaying both TP53 and RB1 inactivation [120]. Moreover, MYC inhibition induced tumor regression in K-Ras-driven LC in mice as well as in p53-deficient animals [121], which suggests the contribution of MYC for the progression of these tumors. hTERT expression has been reported (33% to 94%) in NSCLCs with poorer overall survival at three years [122] and it was significantly higher in NSCLC when compared with non-tumor tissues [123]. Among NSCLCs, AdCs display an increased expression of hTERT when compared with SQCs [122]. Furthermore, hTERT mRNA expression increased from atypical adenomatous hyperplasia to bronchioloalveolar carcinoma [124]. hTERT has been correlated with lymph node metastasis, tumor cell differentiation, and LC aggressiveness [125]. The expression of hTERT was also related to both decreased overall survival and disease-free survival of NSCLC patients [126,127]. hTERT has been associated with changes in EGFR expression that can affect NSCLC proliferation, and hTERT promoter methylation can enhance SCLC progression and metastasis [128].
3.2. Epidemiology of EBV in Lung Cancer
The relationship between LC and EBV, analyzed in diverse studies, remains unclear. Indeed, EBV is detected at highly variable frequencies in clinical specimens of LC. These variations can be explained by sociodemographic factors, type of clinical specimens (i.e., paraffin embedded tissues, fresh tissue, etc.), histological type of LC, and even the sensitivity and specificity of the molecular assays (i.e., polymerase chain reaction (PCR), ISH, IHC). It is important to point out that EBER ISH is the gold standard method with the capacity to detect and localize latent EBV in tissue samples [129]. That said, other EBV detection methods such as LMP1 IHC are commonly used, despite the fact that this protein is often undetectable even when EBER ISH is clearly positive [130]. IHC is another technique reported to detect targets such as EBNA1, EBNA2, LMP2A, BARF1, and BZLF1 (Zta). Zta is a useful marker of lytic viral replication [131], and BARF1 can be used as a marker of epithelial malignancies [132]. Additionally, PCR-derived methods and nucleic acid sequence-based amplification (NASBA) assays are also used at the research level but rarely in clinical laboratories [130].
Pulmonary lymphoepithelioma-like carcinoma (PLELC) is a rare form of NSCLC closely related to EBV infection. PLELC is characterized by an undifferentiated carcinoma accompanied by an intense lymphocytic infiltrate, which histologically resembles NPC [133]. The genomic landscape of PLELC differs from the most frequent NSCLC subtypes but shows similarity with EBV-driven NPCs [134]. These tumors are most frequent in the Asian population [135], which is also an endemic area of NPC. In southern China, EBV was reported in 30 (93.8%) of 32 LELCs by EBER ISH. Furthermore, 53.3% of EBER positive specimens expressed LMP1, and 23.3% also expressed the viral capsid antigen (VCA) [136]. Other studies conducted in China and Taiwan found EBV positivity in 100% of pulmonary LELC [137,138]. The expression of the anti-apoptotic Bcl-2 was significantly increased in five EBV-positive PLELCs when compared with other lung tumors such as SQC, AdC, and LCC [137]. In addition, 91 lung LELCs from China associated with EBV showed molecular alterations in NF-κB, JAK/STAT, and cell cycle signaling pathways [138]. Recently, another type of EBV-related NSCLC was described, displaying low lymphocytic infiltration, but similar patient features and molecular profile to classic PLELC [139,140]. Overall, the presence of EBV was significantly increased in PLELC when compared with non-PLELC [136,137]. Among the different studies carried out to detect EBV in other LCs, we can highlight the following: In North China, EBV was detected in 33.3% of LC patients [141]. In southern China, EBV was reported in 30 (58.8%) of 51 lymphoepithelioma-like carcinomas (LELCs) by EBER ISH. Furthermore, 56% of EBER positive specimens expressed LMPs, and 23% also expressed the viral capsid antigen (VCA) [136]. In China (Henan), 108 cases of LC were evaluated by EBER ISH. Additionally, expressions of LMP1 and Bcl-2 were evaluated by IHC. EBV was reported in 33.3% (EBER positive) and LMP1 was expressed in 6%. Bcl-2 was higher in EBV (+) LC than that in EBV (−) [142]. In (Beijing) China, EBV was detected in 37.9% of 87 SQCs by IHC [143]. In Hong Kong, EBV was reported in 5.4% of 167 NSCLCs by EBER ISH [144]. In Taiwan, EBV was detected in 11 (8.2%) of the 127 NSCLC cases by EBER1 ISH [137]. In Japan, 81 LCs were examined with five cases being EBER1 positive (three SQC, and two AdC) [145]. In Spain, 19 AdC and SQC cases were evaluated by EBER ISH, and 12 (63.2%) were positive [146]. Other studies have used molecular methods such as PCR or qPCR for EBV detection in LCs. In Iran, EBV was detected in 10.4% of 48 LCs by using an EBV-Eph PCR kit and IHC for LMP1 [147]. In India, EBV was detected in 2.76% of 75 cytological specimens from LC patients [148]. In Italy, EBV was detected in 26% of 65 (21/65) NSCLCs and 20% of five (1/5) SCLC in exhaled breath condensate by using real-time PCR assay [149]. In China (Wuhan), EBV DNA was detected by PCR in 52% of 48 LC fresh tissues [150].
In recent years, new technologies such as next-generation sequencing (NGS), whole exome sequencing (WES), and microRNA (miRNA) analysis have been used to characterize EBV presence in LC. Accordingly, EBV was detected in 6.1% (4/66) of NSCLCs. Additionally, the LC strains were classified as V-val and China 1 subtypes according to the amino acid sequence of EBNA1 and LMP1 proteins, respectively [151]. In addition, 91 lung LELCs from China associated with EBV showed molecular alterations in NF-κB, JAK/STAT, and cell cycle signaling pathways [138]. In the United States, data sets from 1017 LCs and 110 paired adjacent normal lung specimens revealed EBV transcripts in three LCs and one normal lung sample. In the sample with the highest EBV coverage, BamHI-A rightward transcripts (BARTs) accounted for the most EBV reads by RNA-seq. Additionally, EBNA1, LMP1 and LMP2 expression was observed. Interestingly, several viral circular RNA candidates were also detected. Thus, this study reported for the first time a type II latency-like viral transcriptome in LC in vivo [152]. Interestingly, expression of lytic genes was also detected. Among them, BZLF1 transcripts, encoding for the Zta protein, were also found in EBV-positive cases, suggesting the importance of lytic activation in EBV-driven tumors, as previously mentioned in chapter 2.3 [152,153]. Conversely, other studies have shown the absence of EBV in different histological types of LC. Indeed, a study conducted in lung AdC and mesotheliomas did not detect EBV by ISH [154]. In another study, 23 Asian SCLCs were analyzed by EBER-ISH without detecting EBV gene expression. In this study, eight SCLCs showed focal positivity for EBNA1 by IHC with only one case positive for LMP-1 by PCR [155]. Another study in Singapore showed no detection of EBV by ISH in 110 lung AdCs [156]. In 2012, the first analysis of EBV miRNAs in LC was carried out in 290 cases. Differences in EBV miRNAs between AdCs and SQCs were determined by microarrays (p < 0.01 for nine out of 16 EBV miRNAs), though such differences were not found for BART1, BART2, and BHRF1–3 expressions (p = 0.53, 0.94, and 0.47, respectively) [157]. In Korea (Busan), EBV-miR-BART19 upregulation was detected in four AdC samples by microarrays, and the results were validated by qPCR and ISH [158]. In (Shandong) China, EBV-miR-BART14-5p downregulation was detected by miRNA microarrays in plasma from patients with NSCLC harboring EGFR-activating mutations [159].
3.3. Potential EBV-Mediated Lung Cancer Mechanisms
3.3.1. Interactions with Tobacco Components
Tobacco Smoke May Facilitate EBV Latency Establishment in Lung Epithelial Cells
Normal epithelial cells are assumed to be unable to sustain a latent EBV infection, which is a requisite for EBV-driven cancer [54]. However, tumor epithelial cells can support EBV latency as demonstrated in NPC and EBVaGC, where EBV is generally found in a latency II status, as previously stated [48]. Thus, it is plausible that previous cell alterations are a required condition (or pre-condition) that allows EBV latency establishment in epithelial cells [61]. Tsang et al. proposed that cell alterations including p16 loss and Cyclin D1 overexpression are among the alterations necessary for EBV latency in nasopharyngeal cells, leading to NPC [61]. Indeed, in vivo experiments have determined that p16 expression is a major barrier to proliferation during EBV-driven B-cell transformation [161]. Cyclin D1 gene dysregulation has been detected in smoking patients and could be associated with the spread of preneoplastic clones across the bronchial epithelial surface [162]. Dysregulated expression of cyclin D1 has been reported in association with persistent EBV infection in NPC [57]. This notion may be applicable to other models of cancer in which EBV plays a potential oncogenic role, including LC. The fact that tobacco smoke components can provide such alterations, including p16 loss [163] and cyclin D1 overexpression [164], suggests that EBV latency establishment may be favored in the lung cells of smoker subjects. This possibility is compatible with the model established by Becnel et al., who proposed that pro-tumor signals such as chronic inflammation result in dysplastic lesions, supporting EBV infection and maintenance of type II latency in the lung epithelia [22]. Considering that tobacco smoke is a frequent cause of the chronic obstructive pulmonary disease (COPD) that is caused by chronic inflammation [165], we suggest that tobacco smoke is a factor potentially leading to EBV latency establishment in lung cells. Of note, we consider that independent of chronic inflammation, normal lung epithelial cells are susceptible to EBV infection, though this is per default lytic. Thus, the EBV latency establishment probably requires the involvement of cofactors in lung epithelial cells. However, additional studies are necessary to confirm these possibilities.
Tobacco Smoke Impairs Immune Response Facilitating EBV Infection in Lung Epithelial Cells
Studies have shown that tobacco smoke alters the immune responses in epithelial lung diseases and can induce activation of an inflammatory response [166]. In addition, tobacco smoking can increase the frequency of memory B cells and reduce the production of IgA, IgG, and IgM [167]. Furthermore, it is possible for tobacco smoke to affect different types of lymphocytes such as T helper cells and regulatory T cells. It is possible for tobacco smoke to affect B lymphocyte development through down-regulation of transcription factor 3 (E2A immunoglobulin enhancer-binding factors E12/E47) (TCF3), which is associated with a decline in the percentage of memory B cells [168]. Additionally, experimental data suggested that tobacco smoking reduces the transporter associated with antigen processing 1 (TAP1) protein abundance and HLA class I levels. This immune change can promote subsequent malignant cell transformation [169]. Loss of the antigen-presenting molecules (including both MHC class I alleles and TAP-1) is frequent in LC [170]. Tobacco smoke also leads to viral lung infections with an increased incidence and severity due to airway epithelium oxidative damage [171]. Furthermore, tobacco smoking increases the oxidative stress that has the potential to promote the maintenance of viral genomes and EBV lytic reactivation [26]. EBV-positive BL cell lines studies have shown an increased ROS expression in both type I and III latency [172]. Additionally, chemicals in tobacco smoke induce activation of NF-κB and ROS pathways, which in turn promote chronic inflammation, autoimmune damage, and cancer progression [173]. In comparison, LMP1 induces NF-κB activation, increasing its transcriptional activity in EBV-infected MOLT4-DL cells [174]. Importantly, NF-κB activation suppresses the expression of DNA damage-binding protein (DDB1), which increases genomic instability in epithelial cells [43,175]. The EBNA gene activates NF-κB to drive oncogene expression (MYC, BCL2) and increases resistance to apoptosis in lymphoblastoid cell lines [176]. In addition, EBNA1 promotes DNA damage, inducing ROS pathways (SOD1, Prx1) in NPC cells [177]. The change in the host’s immune response can create pressure for EBV to be pushed into latency, although over time EBV can reactivate [178,179]. Pressure on the immune system can be caused by metabolic changes in memory B cells after EBV infection due to the increase in glucose uptake and the activation of immunometabolic functions [180]. LMP1 expression can affect glycolysis due to the energy increase for NPC pathogenesis and progression. Indeed, LMP1-mediated metastasis involves IGF1-mTORC2 signaling activation and nuclear acetylation of the Snail promoter by the pyruvate dehydrogenase E1 component subunit alpha (PDHE1α) [181]. EBV is a down-modulator of the human leukocyte antigen (HLA) class I pathway with a close relationship to impaired glucose uptake. Studies have shown that EBVaGC presents extreme DNA hypermethylation [182], and tobacco smoke increases the aberrant methylation in CpG islands [183]. Some genetic alterations in cellular genes such as indoleamine 2,3-dioxygenase (IDO), PD-L1, cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), and V-domain immunoglobulin suppressor of T cell activation (VISTA) have been implicated as supporting EBV infection in the lung epithelia [152], and VISTA is a new target for the immunotherapies against cancer [184]. Tobacco smoke causes lowered IDO activity with immunosuppression [185], and EBV infection increases the expression level of IDO and other immune molecules such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) [186]. Clinical assays have shown that enhanced IDO activity is associated with poor prognosis in patients with LC [187]. Additionally, tobacco smoke can suppress the normal function of B cells and change the standard immune functions [168,188].
Tobacco Smoke Can Activate the EBV Lytic Switch
Epidemiological evidence suggests that tobacco smoke is associated with EBV lytic cycle activation [189]. Furthermore, a dose–response relationship with EBNA1-IgA and Zta-IgA seropositivity [190] can be used as a predictive biomarker of NPC [191]. Thus, tobacco smoke can promote the activation of the EBV lytic cycle, which can spread the virus from B cells to epithelial cells. This effect of tobacco smoke may occur through the promotion of oxidative stress, hypoxia, and inflammation [41,192]. EBV reactivation by tobacco smoke components has been shown in Akata and B95-8 lymphoblastic cell lines [191] and can be mediated by reactive oxygen signaling and high oxidative stress [193]. Additionally, EBV detection is 2.4 times more frequent in current smokers with GC when compared to non-smokers [194]. Interestingly, tobacco smoke and BaP induce PD-1/CTLA-4 in lung epithelial cells [195,196] and can block the adaptive immune response [195]. PD-1/CTLA-4 can affect the latent/lytic proportion of EBV-infected B cells [197].
3.3.2. Lung Cancer Cells May Facilitate EBV Entry through Increased CD21 Expression
CD21 expression facilitates EBV infection, and experimental data demonstrated that ectopic CD21 expression increases EBV infection [198]. Additionally, a low level of CD21 expression was considered sufficient for EBV infection [199]. Taking these findings into consideration, we evaluated the relationship between the CD21 (CR2) transcript expression in 1325 lung cancers vs. normal tissue. CD21 differences between SQC and AdC were also compared. This bioinformatic approach showed that CD21 expression was increased 1.8-fold in malignant tumors compared to normal tissue and increased 1.3-fold in lung AdC when compared to lung SQC (Figure 3). These analyses suggest the possibility that lung tumor cells expressing CD21 may favor EBV entry, although experimental confirmation is necessary.
3.3.3. EBV Interactions with other Environmental Contaminants in Lung Cancer
Airborne pollutants have been etiologically related to LC. Indeed, it has been demonstrated that PM2.5 exposure is associated with inflammation through transforming the growth factor-β/Smad signaling pathway with increased risk for lung AdC [201]. Even at a low concentration, PM2.5 was significantly correlated with the incidence of SQC and AdC [202]. Importantly, arsenic trioxide (As2O3) is another environmental contaminant (present in water, air, and food) related to LC that can activate EBV lytic gene expression in epithelial cells [203]. However, As2O3 is also able to decrease cell proliferation, promote apoptosis, and exhibit anti-angiogenesis properties in the lungs, inhibiting tumor metastasis [204]. Radon is an environmental carcinogen and the first risk factor for LC in nonsmokers [205]. Radioactive radon particles that can become lodged in the lung tissue and eventually lead to LC [206]. Radon exposure can lead to pulmonary inflammation, and some IL variants in never smokers have shown an increased LC risk [207]. Interestingly, EBV DNA and RNA sequences have been reported in SQC and AdC with exposure to radon [146]. Other environmental factors that potentially interact with EBV include pesticide/herbicides. Among these, chlorpyrifos (CPF) stands out since it can interact with EBV through the activation of oxidative stress (OS). Furthermore, CPF can increase EBV early antigens (EA) and promote lytic infection. CPF is a type of organophosphate that can increase leukemia, lymphohematopoietic, brain, colorectal, lung, and breast cancer risk [208]. It has been reported that polychlorinated biphenyls (PCBs) are immunosuppressive agents that increase NHL risk [209]. Additionally, new evidence shows that PCBs and persistent organic pollutants (POP) are associated with LC risk in the general population, even at low doses [210,211]. To investigate the associations between pesticide exposures and HL risk, three population-based studies determined that terbufos (organophosphate) was significantly associated with HL (OR: 2.53, 95% CI 1.04–6.17) (95). Additionally, occupational terbufos exposure was associated with LC, leukemia, and non-Hodgkin lymphoma [212]. Other chemicals that activated the EBV lytic cycle have been discovered from food metabolism. One of these chemicals is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which can interact with B cells through aryl hydrocarbon receptor (AHR). The latter, in turn, can directly interact with EBNA-3, affecting viral latency [213]. The activation of the EBV lytic cycle by TCDD leads to a new viral infection and EBV-associated cellular transformation; it also induces EBV reactivation in both activated B cells and salivary epithelial cells [214]. the AHR activator TCDD boosts interleukin 1 (IL-1), IL-6, and IL-8 gene expression through the NF-κB pathway in NSCLC patients [215]. Other chemical carcinogens that can act synergistically with EBV are volatile N-nitrosamines. There are reports of contamination of Chinese salted fish with volatile N-nitrosamines, resulting in increased risk of developing NPC [216] and LC [217]. A hypothetical mechanism of EBV-tobacco/environmental contaminant interaction is shown in Figure 4.
4. Conclusions and Remarks
LC is a highly prevalent malignancy worldwide with tobacco smoke being the most important risk factor. Additional risk factors are environmental pollution, radon, and arsenic, among others. The etiological role of viral infections such as EBV has not been established, although epidemiological evidence suggests that this virus may be involved in a very restricted subset of cases. Considering the evidence established in lung and additional models of epithelial cancers, we suggest that EBV can cooperate with tobacco smoke at different levels for lung carcinogenesis. The identification of clonal EBV in LC specimens will contribute to elucidation whether the infection occurs early during the neoplastic development. Thus, further knowledge of these mechanisms is necessary for the development of new therapies to improve the treatments and the diagnosis of diseases related to EBV biology.
Author Contributions
Conceptualization, J.C.O. and F.A.; writing—original draft preparation, J.C.O., R.B. and F.A.; writing—review and editing, J.C.O., F.A., R.B., A.H.C., J.P.M. and G.M.C.; supervision, F.A.; project administration, F.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ANID-FONDECYT Postdoctoral Grant 3190723 (R.B.) and 3190744 (J.P.M.), FONDECYT Grant 1200656 (G.M.C.), and CONICYT-FONDAP Grant 15130011 (A.H.C.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Brianna Johnson for English grammar revision.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Semenova, E.A.; Nagel, R.; Berns, A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015, 29, 1447–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Reis-Filho, J.S.; Simpson, P.T.; Martins, A.; Preto, A.; Gärtner, F.; Schmitt, F.C.; Archiv, V. Distribution of p63, cytokeratins 5/6 and cytokeratin 14 in 51 normal and 400 neoplastic human tissue samples using TARP-4 multi-tumor tissue microarray. Virchows Arch. 2003, 443, 122–132. [Google Scholar] [PubMed] [Green Version]
- Su, Y.-C.; Hsu, Y.-C.; Chai, C.-Y. Role of TTF-1, CK20, and CK7 Immunohistochemistry for Diagnosis of Primary and Secondary Lung Adenocarcinoma. Kaohsiung J. Med Sci. 2006, 22, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M. Large-cell carcinoma of the lung: A diagnostic category redefined by immunohistochemistry and genomics. Curr. Opin. Pulm. Med. 2014, 20, 324–331. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.E.; Minna, J.D. Molecular Biology of Lung Cancer: Clinical Implications. Clin. Chest Med. 2011, 32, 703–740. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Hubaux, R.; Becker-Santos, D.D.; Enfield, K.S.; Lam, S.; Lam, W.L.; Martinez, V.D. Arsenic, asbestos and radon: Emerging players in lung tumorigenesis. Environ. Health 2012, 11, 89. [Google Scholar] [CrossRef]
- Czoli, C.D.; Hammond, D. TSNA Exposure: Levels of NNAL among Canadian Tobacco Users. Nicotine Tob. Res. 2015, 17, 825–830. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 92, 1–853. [Google Scholar]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, A. Source Apportionment of Particle Bound Polycyclic Aromatic Hydrocarbons at an Industrial Location in Agra, India. Sci. World J. 2012, 2012, 781291. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Li, Y.; Liu, Z.; Liu, T.; Gai, C. Emission, distribution and toxicity of polycyclic aromatic hydrocarbons (PAHs) during municipal solid waste (MSW) and coal co-combustion. Sci. Total Environ. 2016, 565, 1201–1207. [Google Scholar] [CrossRef]
- Syrjänen, K.J. HPV infections and lung cancer. J. Clin. Pathol. 2002, 55, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Joh, J.; Jenson, A.B.; Moore, G.D.; Rezazedeh, A.; Slone, S.P.; Ghim, S.-J.; Kloecker, G.H. Human papillomavirus (HPV) and Merkel cell polyomavirus (MCPyV) in non small cell lung cancer. Exp. Mol. Pathol. 2010, 89, 222–226. [Google Scholar] [CrossRef]
- Palmarini, M.; Sharp, J.M.; de las Heras, M.; Fan, H. Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. J. Virol. 1999, 73, 6964–6972. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Aziz, H.A.; Nakanishi, Y.; Masuda, S.; Saito, H.; Tsuneyama, K.; Takano, Y. Oncogenic role of JC virus in lung cancer. J. Pathol. 2007, 212, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Becnel, D.; Abdelghani, R.; Nanbo, A.; Avilala, J.; Kahn, J.; Li, L.; Lin, Z. Pathogenic Role of Epstein—Barr Virus in Lung Cancers. Viruses 2021, 13, 877. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.B.; Delgado-Eckert, E.; Thorley-Lawson, D.A.; Shapiro, M. The Cycle of EBV Infection Explains Persistence, the Sizes of the Infected Cell Populations and Which Come under CTL Regulation. PLoS Pathog. 2013, 9, e1003685. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.-J.; Rickinson, A.B.; Rowe, M. Features Distinguishing Epstein-Barr Virus Infections of Epithelial Cells and B Cells: Viral Genome Expression, Genome Maintenance, and Genome Amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-Barr virus infection and human malignancies. Int. J. Exp. Pathol. 2001, 82, 149–170. [Google Scholar] [CrossRef]
- Aguayo, F.; Boccardo, E.; Corvalán, A.; Calaf, G.M.; Blanco, R. Interplay between Epstein-Barr virus infection and environmental xenobiotic exposure in cancer. Infect. Agents Cancer 2021, 16, 50. [Google Scholar] [CrossRef]
- Nanbo, A.; Noda, T.; Ohba, Y. Epstein–Barr Virus Acquires Its Final Envelope on Intracellular Compartments With Golgi Markers. Front. Microbiol. 2018, 9, 454. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein–Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, e00382-17. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Kirschner, A.N.; Jardetzky, T.S.; Longnecker, R. Characteristics of Epstein–Barr virus envelope protein gp42. Virus Genes 2010, 40, 307–319. [Google Scholar] [CrossRef]
- Jiang, R.; Gu, X.; Nathan, C.-A.; Hutt-Fletcher, L. Laser-capture microdissection of oropharyngeal epithelium indicates restriction of Epstein-Barr virus receptor/CD21 mRNA to tonsil epithelial cells. J. Oral Pathol. Med. 2008, 37, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. Epstein-Barr Virus Entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [Green Version]
- Döhner, K.; Cornelius, A.; Serrero, M.C.; Sodeik, B. The journey of herpesvirus capsids and genomes to the host cell nucleus. Curr. Opin. Virol. 2021, 50, 147–158. [Google Scholar] [CrossRef]
- Lee, C.-P.; Chen, M.-R. Conquering the Nuclear Envelope Barriers by EBV Lytic Replication. Viruses 2021, 13, 702. [Google Scholar] [CrossRef]
- Aydin, I.; Schelhaas, M. Viral Genome Tethering to Host Cell Chromatin: Cause and Consequences. Traffic 2016, 17, 327–340. [Google Scholar] [CrossRef]
- Frappier, L. The Epstein-Barr Virus EBNA1 Protein. Scientifica 2012, 2012, 438204. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; Bode, A.M.; Cao, Y. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int. J. Biol. Sci. 2016, 12, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Feederle, R.; Shannon-Lowe, C.; Baldwin, G.; Delecluse, H.J. Defective Infectious Particles and Rare Packaged Genomes Produced by Cells Carrying Terminal-Repeat-Negative Epstein-Barr Virus. J. Virol. 2005, 79, 7641–7647. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein—Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Tokunaga, M.; Land, C.E.; Uemura, Y.; Tokudome, T.; Tanaka, S.; Sato, E. Epstein-Barr virus in gastric carcinoma. Am. J. Pathol. 1993, 143, 1250–1254. [Google Scholar]
- Arbach, H.; Viglasky, V.; Lefeu, F.; Guinebretière, J.-M.; Ramirez, V.; Bride, N.; Boualaga, N.; Bauchet, T.; Peyrat, J.-P.; Mathieu, M.-C.; et al. Epstein-Barr Virus (EBV) Genome and Expression in Breast Cancer Tissue: Effect of EBV Infection of Breast Cancer Cells on Resistance to Paclitaxel (Taxol). J. Virol. 2006, 80, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Bedri, S.; Sultan, A.A.; Alkhalaf, M.; Al Moustafa, A.-E.; Vranic, S. Epstein-Barr virus (EBV) status in colorectal cancer: A mini review. Hum. Vaccines Immunother. 2018, 15, 603–610. [Google Scholar] [CrossRef]
- Whitaker, N.J.; Glenn, W.K.; Sahrudin, A.; Orde, M.M.; Delprado, W.; Lawson, J.S. Human papillomavirus and Epstein Barr virus in prostate cancer: Koilocytes indicate potential oncogenic influences of human papillomavirus in prostate cancer. Prostate 2013, 73, 236–241. [Google Scholar] [CrossRef]
- Young, L.S.; Dawson, C.W. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Iizasa, H.; Kim, H.; Kartika, A.V.; Kanehiro, Y.; Yoshiyama, H. Corrigendum: Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front. Immunol. 2020, 11, 498. [Google Scholar] [CrossRef] [Green Version]
- Richardo, T.; Prattapong, P.; Ngernsombat, C.; Wisetyaningsih, N.; Iizasa, H.; Yoshiyama, H.; Janvilisri, T. Epstein-Barr Virus Mediated Signaling in Nasopharyngeal Carcinoma Carcinogenesis. Cancers 2020, 12, 2441. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis Shows That Prevalence of Epstein–Barr Virus-Positive Gastric Cancer Differs Based on Sex and Anatomic Location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woellmer, A.; Hammerschmidt, W. Epstein–Barr virus and host cell methylation: Regulation of latency, replication and virus reactivation. Curr. Opin. Virol. 2013, 3, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, R.-J.; Han, B.-W.; Cai, Q.-Q.; Zuo, X.-Y.; Xia, T.; Chen, J.-R.; Feng, L.-N.; Lim, J.Q.; Chen, S.-W.; Zeng, M.; et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019, 33, 1451–1462. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.Y.; Takada, K.; Lui, V.W.Y.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Murakami, M.; Kaul, R.; Saha, A.; Cai, Q.; Robertson, E.S. Deregulation of the cell cycle machinery by Epstein—Barr virus nuclear antigen 3C. Future Virol. 2009, 4, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Frappier, L. Epstein-Barr virus: Current questions and challenges. Tumour Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef]
- Hirai, K.; Shirakata, M. Replication Licensing of the EBV oriP Minichromosome. Curr. Top. Microbiol. Immunol. 2001, 258, 13–33. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar]
- Klein, G. Viral latency and transformation: The strategy of Epstein-Barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, W.; Liu, J.; Wang, J.; Luo, B. The effect of EBV on WIF1, NLK, and APC gene methylation and expression in gastric carcinoma and nasopharyngeal cancer. J. Med. Virol. 2017, 89, 1844–1851. [Google Scholar] [CrossRef]
- Hsu, M.; Wu, S.-Y.; Chang, S.-S.; Su, I.-J.; Tsai, C.-H.; Lai, S.-J.; Shiau, A.-L.; Takada, K.; Chang, Y. Epstein-Barr Virus Lytic Transactivator Zta Enhances Chemotactic Activity through Induction of Interleukin-8 in Nasopharyngeal Carcinoma Cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Blanco, R.; Aguayo, F. Role of BamHI—A Rightward Frame 1 in Epstein-Barr Virus-Associated Epithelial Malignancies. Biology 2020, 9, 461. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein–Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Birdwell, C.E.; Prasai, K.; Dykes, S.; Jia, Y.; Munroe, T.G.; Bienkowska-Haba, M.; Scott, R.S. Epstein—Barr virus stably confers an invasive phenotype to epithelial cells through reprogramming of the WNT pathway. Oncotarget 2018, 9, 10417–10435. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, K.; Funata, S.; Fukuyo, M.; Seto, Y.; Aburatani, H.; Fukayama, M.; Kaneda, A. Epstein—Barr virus infection induces genome-wide de novo DNA methylation in non-neoplastic gastric epithelial cells. J. Pathol. 2017, 242, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Uozaki, H.; Fukayama, M. Epstein-Barr Virus and Gastric Carcinoma—Viral Carcinogenesis through Epigenetic Mechanisms. Int. J. Clin. Exp. Pathol. 2008, 1, 198–216. [Google Scholar]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein—Barr Virus reversing epigenetic repression. PLoS Pathog 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.P.; Carbone, D.P. The molecular basis of lung cancer: Molecular abnormalities and therapeutic implications. Respir. Res. 2003, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Cooper, W.A.; Lam, D.C.L.; O’Toole, S.A.; Minna, J.D. Molecular biology of lung cancer. J. Thorac. Dis. 2013, 5, S479–S490. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Swanton, C. The Role of Aneuploidy in Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zojer, N.; Dekan, G.; Ackermann, J.; Fiegl, M.; Kaufmann, H.; Drach, J.; Huber, H. Aneuploidy of chromosome 7 can be detected in invasive lung cancer and associated premalignant lesions of the lung by fluorescence in situ hybridisation. Lung Cancer 2000, 28, 225–235. [Google Scholar] [CrossRef]
- Jonsson, S.; Varella-Garcia, M.; Miller, Y.E.; Wolf, H.J.; Byers, T.; Braudrick, S.; Kiatsimkul, P.; Lewis, M.; Kennedy, T.C.; Keith, R.L.; et al. Chromosomal Aneusomy in Bronchial High-Grade Lesions Is Associated with Invasive Lung Cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.M.; Lockwood, W.W.; Buys, T.P.H.; Chari, R.; Coe, B.P.; Lam, S.; Lam, W.L. Integrative genomic and gene expression analysis of chromosome 7 identified novel oncogene loci in non-small cell lung cancer. Genome 2008, 51, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Gawrychowski, J.; Lackowska, B.; Gabriel, A. Prognosis of the surgical treatment of patients with non-small cell lung cancer (NSCLC)-relation to DNA ploidy. Eur. J. Cardio-Thorac. Surg. 2003, 23, 870–877. [Google Scholar] [CrossRef] [Green Version]
- Oliani, C.; Barana, D.; Cazzadori, A.; Zanolin, E.; Santo, A.; Pasini, F.; Padovani, M.; Mazzini, G.; Cetto, G.L. Cytofluorimetric evaluation of DNA ploidy in lung cancer: A bronchoscopic study. Int. J. Biol. Markers. 2005, 20, 87–92. [Google Scholar] [CrossRef]
- Hofmann, H.S.; Knolle, J.; Bahn, H.; Klapperstück, T.; Lautenschläger, C.; Neef, H. Flow cytometric analysis of DNA content and proliferation and immunohistochemical staining of Ki-67 in non-small cell lung cancer. J. Cardiovasc. Surg. 2001, 42, 555–560. [Google Scholar]
- Choma, D.; Daurès, J.-P.; Quantin, X.; Pujol, J.L. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br. J. Cancer 2001, 85, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Lee, J.S.; Kurie, J.M.; Fan, Y.H.; Lippman, S.M.; Broxson, A.; Khuri, F.R.; Hong, W.K.; Yu, R.; Ro, J.Y.; et al. Clonal Genetic Alterations in the Lungs of Current and Former Smokers. J. Natl. Cancer Inst. 1997, 89, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Ortega, J.; Saez, M.C.; Sierra, E.; Torres, A.; Balibrea, J.L.; Hernando, F.; Sanz-Esponera, J.; Merino, M.J. 3p21, 5q21, and 9p21 allelic deletions are frequently found in normal bronchial cells adjacent to non-small-cell lung cancer, while they are unusual in patients with no evidence of malignancy. J. Pathol. 2001, 195, 429–434. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Virmani, A.K.; Gazdar, A.F.; Lam, S.; Leriche, J.; Behrens, C.; Fong, K.; Samet, J.M.; Srivastava, S.; Minna, J.D. Molecular Damage in the Bronchial Epithelium of Current and Former Smokers. J. Natl. Cancer Inst. 1997, 89, 1366–1373. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Xu, S.; Qu, J.; Liu, B. Clinicopathologic Features of Patients with Non-Small Cell Lung Cancer Harboring the EML4-ALK Fusion Gene: A Meta-Analysis. PLoS ONE 2014, 9, e110617. [Google Scholar] [CrossRef]
- Miyanaga, A.; Matsumoto, M.; Beck, J.A.; Horikawa, L.; Oike, T.; Okayama, H.; Tanaka, H.; Burkett, S.S.; Robles, A.I.; Khan, M.; et al. EML4-ALK induces cellular senescence in mortal normal human cells and promotes anchorage-independent growth in hTERT-transduced normal human cells. BMC Cancer 2021, 21, 310. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Silwal-Pandit, L.; Meza-Zepeda, L.A.; Vodák, D.; Vu, P.; Sagerup, C.; Hovig, E.; Myklebost, O.; Børresen-Dale, A.-L.; Brustugun, O.T.; et al. TP53 Mutation Spectrum in Smokers and Never Smoking Lung Cancer Patients. Front. Genet. 2016, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Demirhan, O.; Taştemir, D.; Hastürk, S.; Kuleci, S.; Hanta, I. Alterations in p16 and p53 genes and chromosomal findings in patients with lung cancer: Fluorescence in situ hybridization and cytogenetic studies. Cancer Epidemiol. 2010, 34, 472–477. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; Lardon, F.; Rolfo, C.; Pauwels, P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit. Rev. Oncol. 2016, 99, 63–73. [Google Scholar] [CrossRef]
- Tanaka, R.; Wang, D.; Morishita, Y.; Inadome, Y.; Minami, Y.; Iijima, T.; Fukai, S.; Goya, T.; Noguchi, M. Loss of function of p16 gene and prognosis of pulmonary adenocarcinoma. Cancer 2005, 103, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Sueoka, N.; Irie, K.; Iwanaga, K.; Tokunaga, O.; Hayashi, S.-I.; Nakachi, K.; Sueoka, E. Detection and discrimination of preneoplastic and early stages of lung adenocarcinoma using hnRNP B1 combined with the cell cycle-related markers p16, cyclin D1, and Ki-67. Lung Cancer 2003, 40, 45–53. [Google Scholar] [CrossRef]
- Licchesi, J.D.; Westra, W.H.; Hooker, C.M.; Herman, J.G. Promoter Hypermethylation of Hallmark Cancer Genes in Atypical Adenomatous Hyperplasia of the Lung. Clin. Cancer Res. 2008, 14, 2570–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Tateishi, R. Retinoblastoma Protein Expression in Lung Cancer: An Immunohistochemical Analysis. Oncology 1994, 51, 544–551. [Google Scholar] [CrossRef]
- Zagorski, W.A.; Knudsen, E.S.; Reed, M.F. Retinoblastoma Deficiency Increases Chemosensitivity in Lung Cancer. Cancer Res. 2007, 67, 8264–8273. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Rengifo, C.E.; Blanco, R.; Blanco, D.; Cedeño, M.; Frómeta, M.; Calzado, E.R. Immunohistochemical Characterization of Three Monoclonal Antibodies Raised against the Epidermal Growth Factor and Its Receptor in Non-Small-Cell Lung Cancer: Their Potential Use in the Selection of Patients for Immunotherapy. J. Biomarkers 2013, 2013, 627845. [Google Scholar] [CrossRef]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar]
- Cuneo, K.C.; Nyati, M.K.; Ray, D.; Lawrence, T.S. EGFR targeted therapies and radiation: Optimizing efficacy by appropriate drug scheduling and patient selection. Pharmacol. Ther. 2015, 154, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.M.; Zhu, Q.G.; Ding, X.X.; Lin, S.; Zhao, J.; Guan, L.; Li, T.; He, B.; Zhang, H.Q. Prognostic value of EGFR and KRAS in resected non-small cell lung cancer: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Shao, X.; Gao, W.; Bai, J.; Wang, R.; Huang, P.; Yin, Y.; Liu, P.; Shu, Y. The Role of Human Epidermal Growth Factor Receptor 2 as a Prognostic Factor in Lung Cancer: A Meta-Analysis of Published Data. J. Thorac. Oncol. 2010, 5, 1922–1932. [Google Scholar] [CrossRef] [Green Version]
- Pillai, R.N.; Behera, M.; Berry, L.D.; Rossi, M.R.; Kris, M.G.; Johnson, B.E.; Bunn, P.A.; Ramalingam, S.S.; Khuri, F.R. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. Cancer 2017, 123, 4099–4105. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, K.; Suda, K.; Onozato, R.; Kosaka, T.; Endoh, H.; Sekido, Y.; Shigematsu, H.; Kuwano, H.; Yatabe, Y.; Mitsudomi, T. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer 2011, 74, 139–144. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.; Fang, R.; Han, X.; Luo, X.; Wang, R.; Pan, Y.; Hu, H.; Zhang, Y.; Pao, W.; et al. Lung adenocarcinomas with HER2-activating mutations are associated with distinct clinical features and HER2/EGFR copy number gains. J. Thorac. Oncol. 2012, 7, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Scoccianti, C.; Vesin, A.; Martel, G.; Olivier, M.; Brambilla, E.; Timsit, J.F.; Tavecchio, L.; Brambilla, C.; Field, J.K.; Hainaut, P.; et al. Prognostic value of TP53, KRAS and EGFR mutations in nonsmall cell lung cancer: The EUELC cohort. Eur. Respir. J. 2012, 40, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Villaruz, L.C.; Socinski, M.A.; Cunningham, D.E.; Chiosea, S.I.; Burns, T.F.; Siegfried, J.M.; Dacic, S. The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer 2013, 119, 2268–2274. [Google Scholar] [CrossRef] [Green Version]
- Martorell, P.M.; Huerta, M.; Quilis, A.C.; Abellán, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Beltrán, D.D.; Keränen, S.R.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA Mutations and ALK Rearrangement in a Comprehensive Cohort of 326 Consecutive Spanish Nonsquamous NSCLC Patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef]
- Okudela, K.; Hayashi, H.; Ito, T.; Yazawa, T.; Suzuki, T.; Nakane, Y.; Sato, H.; Ishi, H.; KeQin, X.; Masuda, A.; et al. K-ras Gene Mutation Enhances Motility of Immortalized Airway Cells and Lung Adenocarcinoma Cells via Akt Activation: Possible Contribution to Non-Invasive Expansion of Lung Adenocarcinoma. Am. J. Pathol. 2004, 164, 91–100. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Kobayashi, M.; Uehara, T.; Fujimoto, M.; Tsuruyama, T.; Date, H.; Haga, H. HER2 status in lung adenocarcinoma: A comparison of immunohistochemistry, fluorescence in situ hybridization (FISH), dual-ISH, and gene mutations. Lung Cancer 2014, 85, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Yoo, S.; Kong, R.; Sato, T.; Sinha, A.; Karam, S.; Bao, L.; Fridrikh, M.; Emoto, K.; Nudelman, G.; et al. Prototypical oncogene family Myc defines unappreciated distinct lineage states of small cell lung cancer. Sci. Adv. 2021, 7, eabc2578. [Google Scholar] [CrossRef] [PubMed]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. Myc Is a Metastasis Gene for Non-Small-Cell Lung Cancer. PLoS ONE 2009, 4, e6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef]
- Zhou, C.; Che, G.; Zheng, X.; Qiu, J.; Xie, Z.; Cong, Y.; Pei, X.; Zhang, H.; Sun, H.; Ma, H. Expression and clinical significance of PD-L1 and c-Myc in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2663–2674. [Google Scholar] [CrossRef]
- Dragoj, M.; Bankovic, J.; Podolski-Renic, A.; Stojkovic Buric, S.; Pesic, M.; Tanic, N.; Stankovic, T. Association of Overexpressed MYC Gene with Altered PHACTR3 and E2F4 Genes Contributes to Non-small Cell Lung Carcinoma Pathogenesis. J. Med. Biochem. 2019, 38, 188–195. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J.; Battey, J.F.; Sausville, E.A.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.L.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nat. Cell Biol. 1985, 318, 69–73. [Google Scholar] [CrossRef]
- Qin, J.; Xie, F.; Li, C.; Han, N.; Lu, H. MYCL1 Amplification and Expression of L-Myc and c-Myc in Surgically Resected Small-Cell Lung Carcinoma. Pathol. Oncol. Res. 2021, 27, 1609775. [Google Scholar] [CrossRef]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgün, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-Q.; Cutz, J.-C.; Liu, N.; Lau, D.; Shepherd, F.A.; Squire, J.A.; Tsao, M.-S. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Br. J. Cancer 2006, 94, 1452–1459. [Google Scholar] [CrossRef]
- Zalewska-Ziob, M.; Dobija-Kubica, K.; Biernacki, K.; Adamek, B.; Kasperczyk, J.; Bruliński, K.; Ostrowska, Z. Clinical and prognostic value of hTERT mRNA expression in patients with non-small-cell lung cancer. Acta Biochim. Pol. 2017, 64, 641–646. [Google Scholar] [CrossRef]
- Kawai, T.; Hiroi, S.; Nakanishi, K.; Meeker, A.K. Telomere length and telomerase expression in atypical adenomatous hyperplasia and small bronchioloalveolar carcinoma of the lung. Am. J. Clin. Pathol. 2007, 127, 254–262. [Google Scholar] [CrossRef]
- Hara, H.; Yamashita, K.; Shinada, J.; Yoshimura, H.; Kameya, T. Clinicopathologic significance of telomerase activity and hTERT mRNA expression in non-small cell lung cancer. Lung Cancer 2001, 34, 219–226. [Google Scholar] [CrossRef]
- Wang, L.; Soria, J.-C.; Kemp, B.L.; Liu, D.D.; Mao, L.; Khuri, F.R. hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 2883–2889. [Google Scholar]
- Wang, K.; Wang, R.L.; Liu, J.J.; Zhou, J.; Li, X.; Hu, W.W.; Jiang, W.J.; Hao, N.B. The prognostic significance of hTERT overexpression in cancers: A systematic review and meta-analysis. Medicine 2018, 97, e11794. [Google Scholar] [CrossRef]
- Zhai, G.; Li, J.; Zheng, J.; An, P.; Chen, X.; Wang, X.; Li, C. Expression of Concern: hTERT promoter methylation promotes small cell lung cancer progression and radiotherapy resistance. J. Radiat. Res. 2021, 62, 556. [Google Scholar]
- Ambinder, R.F.; Mann, R.B. Epstein-Barr-encoded RNA in situ hybridization: Diagnostic applications. Hum. Pathol. 1994, 25, 602–605. [Google Scholar] [CrossRef]
- Gulley, M.L. Molecular Diagnosis of Epstein-Barr Virus-Related Diseases. J. Mol. Diagn. 2001, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Grässer, F.A.; Murray, P.G.; Kremmer, E.; Klein, K.; Remberger, K.; Feiden, W.; Reynolds, G.; Niedobitek, G.; Young, L.S.; Mueller-Lantzsch, N. Monoclonal antibodies directed against the Epstein-Barr virus-encoded nuclear antigen 1 (EBNA1): Immunohistologic detection of EBNA1 in the malignant cells of Hodgkin’s disease. Blood 1994, 84, 3792–3798. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.P.; Brink, A.A.; Vervoort, M.B.; Middeldorp, J.M.; Meijer, C.J.; Brule, A.J.V.D. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. Mol. Pathol. 1999, 52, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Haba, R.; Tanizawa, J.; Katsuki, N.; Kadota, K.; Miyai, Y.; Bando, K.; Shibuya, S.; Nakano, M.; Kushida, Y. Cytopathologic features and differential diagnostic considerations of primary lymphoepithelioma-like carcinoma of the lung. Diagn. Cytopathol. 2012, 40, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Chau, S.-L.; Tong, J.H.-M.; Chow, C.; Kwan, J.S.-H.; Lung, R.W.-M.; Chung, L.-Y.; Tin, E.K.-Y.; Wong, S.S.-Y.; Cheung, A.H.-K.; Lau, R.W.-H.; et al. Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing. Cancers 2020, 12, 2065. [Google Scholar] [CrossRef]
- Tang, L.; Chen, N.; He, W.; Zhou, J.; Zhang, J.; Lin, Z.; Wang, Z.; Hao, J.; Lin, F. The clinicopathological features and prognosis of primary pulmonary lymphoepithelioma-like carcinoma: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0240729. [Google Scholar] [CrossRef]
- Han, A.-J.; Xiong, M.; Zong, Y.-S. Association of Epstein-Barr Virus With Lymphoepithelioma-Like Carcinoma of the Lung in Southern China. Am. J. Clin. Pathol. 2000, 114, 220–226. [Google Scholar] [CrossRef]
- Chen, F.F.; Yan, J.J.; Lai, W.W.; Jin, Y.T.; Su, I.J. Epstein-Barr virus-associated nonsmall cell lung carcinoma: Undifferentiated “lymphoepithelioma-like” carcinoma as a distinct entity with better prognosis. Cancer 1998, 82, 2334–2342. [Google Scholar] [CrossRef]
- Hong, S.; Liu, D.; Luo, S.; Fang, W.; Zhan, J.; Fu, S.; Zhang, Y.; Wu, X.; Zhou, H.; Chen, X.; et al. The genomic landscape of Epstein-Barr virus-associated pulmonary lymphoepithelioma-like carcinoma. Nat. Commun. 2019, 10, 3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Ho, H.L.; Lin, C.I.; Chou, T.Y.; Wang, Y.C. Whole-exome Sequencing of Epstein-Barr Virus-associated Pulmonary Carcinoma With Low Lymphocytic Infiltration Shows Molecular Features Similar to Those of Classic Pulmonary Lymphoepithelioma-like Carcinoma: Evidence to Support Grouping Together as One Disease Entity. Am. J. Surg. Pathol. 2021, 45, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Kao, H.L.; Lee, K.L.; Wu, M.H.; Ho, H.L.; Chou, T.Y. Epstein-Barr Virus-Associated Pulmonary Carcinoma: Proposing an Alternative Term and Expanding the Histologic Spectrum of Lymphoepithelioma-like Carcinoma of the Lung. Am. J. Surg. Pathol. 2019, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-M.; Han, G.-L.; Zhang, S.-J. Detection of Epstein-Barr virus in lung carcinoma tissue by in situ hybridization. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi Zhonghua Shiyan He Linchuang Bingduxue Zazhi Chin. J. Exp. Clin. Virol. 2007, 21, 288–290. [Google Scholar]
- Li, C.-M.; Zhang, S.-J.; Zhu, J.-H.; Han, G.-L. Study on EB virus infection, LMP1 and Bcl-2 expression in lung cancer patients. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi Zhonghua Shiyan He Linchuang Bingduxue Zazhi Chin. J. Exp. Clin. Virol. 2011, 25, 277–279. [Google Scholar]
- Zhang, L.; Liu, H.; Wang, Z. Detection of EB virus in pulmonary carcinoma by in situ hybridization. Zhonghua Bing Li Xue Za Zhi Chin. J. Pathol. 1996, 25, 1–3. [Google Scholar]
- Wong, M.P.; Chung, L.P.; Yuen, S.T.; Leung, S.Y.; Chan, S.Y.; Wang, E.; Fu, K.H. In situ detection of Epstein-Barr virus in non-small cell lung carcinomas. J. Pathol. 1995, 177, 233–240. [Google Scholar] [CrossRef]
- Kasai, K.; Sato, Y.; Kameya, T.; Inoue, H.; Yoshimura, H.; Kon, S.; Kikuchi, K. Incidence of latent infection of Epstein-Barr virus in lung cancers—An analysis of EBER1 expression in lung cancers byin situ hybridization. J. Pathol. 1994, 174, 257–265. [Google Scholar] [CrossRef]
- Gómez-Román, J.J.; Martínez, M.N.; Fernández, S.L.; Val-Bernal, J.F. Epstein—Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod. Pathol. 2009, 22, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Jafarian, A.H.; Omidi-Ashrafi, A.; Mohamadian-Roshan, N.; Karimi-Shahri, M.; Ghazvini, K.; Boroumand-Noughabi, S. Association of Epstein Barr virus deoxyribonucleic acid with lung carcinoma. Indian J. Pathol. Microbiol. 2013, 56, 359–364. [Google Scholar] [CrossRef]
- Gupta, P.; Haldar, D.; Naru, J.; Dey, P.; Aggarwal, A.N.; Minz, R.; Aggarwal, R. Prevalence of human papillomavirus, Epstein-Barr virus, and cytomegalovirus in fine needle aspirates from lung carcinoma: A case-control study with review of literature. Diagn. Cytopathol. 2016, 44, 987–993. [Google Scholar] [CrossRef]
- Carpagnano, G.E.; Lacedonia, D.; Natalicchio, M.I.; Cotugno, G.; Zoppo, L.; Martinelli, D.; Antonetti, R.; Foschino-Barbaro, M.P. Viral colonization in exhaled breath condensate of lung cancer patients: Possible role of EBV and CMV. Clin. Respir. J. 2016, 12, 418–424. [Google Scholar] [CrossRef]
- Xia, H.; Wu, J.; Chen, C.; Mao, Y.; Zhu, J.; Chang, Q.; Mi, K.; Zhao, J.; Zhang, M. The relationship between Epstein-Barr-virus infection and expression of p53, Bcl-2 and C-myc gene in lung cancer. Zhongguo Fei Ai Za Zhi 2000, 3, 265–268. [Google Scholar] [CrossRef]
- Wang, S.; Xiong, H.; Yan, S.; Wu, N.; Lu, Z. Identification and Characterization of Epstein-Barr Virus Genomes in Lung Carcinoma Biopsy Samples by Next-Generation Sequencing Technology. Sci. Rep. 2016, 6, 26156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.J.; Hudnall, S.D.; Lazarides, A.; Bahler, A.; Fraire, A.E.; Cagle, P.T. Absence of evidence for an etiologic role for Epstein-Barr virus in neoplasms of the lung and pleura. Mod. Pathol. 1996, 9, 491–495. [Google Scholar]
- Chu, P.G.; Cerilli, L.A.; Chen, Y.-Y.; Mills, S.E.; Weiss, L.M. Epstein–Barr virus plays no role in the tumorigenesis of small-cell carcinoma of the lung. Mod. Pathol. 2004, 17, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.-T.; Chuah, K.L.; Leong, S.S.; Tan, E.H.; Toh, C.K. Assessment of human papillomavirus and Epstein-Barr virus in lung adenocarcinoma. Oncol. Rep. 2009, 21, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Gulley, M.L.; Zhao, Y.; Rubagotti, M.; Marincola, F.M.; Rotunno, M.; Tang, W.; Bergen, A.W.; Bertazzi, P.A.; Roy, D.; et al. Epstein–Barr virus microRNAs and lung cancer. Br. J. Cancer 2011, 105, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Cho, J.S.; Kim, Y.; Lee, C.H.; Lee, M.K.; Shin, D.H. Discriminating between Terminal- and Non-Terminal Respiratory Unit-Type Lung Adenocarcinoma Based on MicroRNA Profiles. PLoS ONE 2016, 11, e0160996. [Google Scholar] [CrossRef]
- Ma, Y.; Pan, X.; Xu, P.; Mi, Y.; Wang, W.; Wu, X.; He, Q.; Liu, X.; Tang, W.; An, H.-X. Plasma microRNA alterations between EGFR-activating mutational NSCLC patients with and without primary resistance to TKI. Oncotarget 2017, 8, 88529–88536. [Google Scholar] [CrossRef] [Green Version]
- Brouchet, L.; Valmary, S.; Dahan, M.; Didier, A.; Galateau-Salle, F.; Brousset, P.; Degano, B. Detection of oncogenic virus genomes and gene products in lung carcinoma. Br. J. Cancer 2005, 92, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Ratschiller, D.; Heighway, J.; Gugger, M.; Kappeler, A.; Pirnia, F.; Schmid, R.; Borner, M.; Betticher, D. Cyclin D1 Overexpression in Bronchial Epithelia of Patients With Lung Cancer Is Associated With Smoking and Predicts Survival. J. Clin. Oncol. 2003, 21, 2085–2093. [Google Scholar] [CrossRef]
- Yanagawa, N.; Tamura, G.; Oizumi, H.; Takahashi, N.; Shimazaki, Y.; Motoyama, T. Frequent epigenetic silencing of the p16 gene in non-small cell lung cancers of tobacco smokers. Jpn. J. Cancer Res. 2002, 93, 1107–1113. [Google Scholar] [CrossRef]
- Ramos-García, P.; Gil-Montoya, J.A.; Scully, C.; Ayén, A.; González-Ruiz, L.; Navarro-Triviño, F.J.; González-Moles, M.A. An update on the implications of cyclin D1 in oral carcinogenesis. Oral Dis. 2016, 23, 897–912. [Google Scholar] [CrossRef]
- Arellano-Orden, E.; Calero-Acuña, C.; Moreno-Mata, N.; Gómez-Izquierdo, L.; Sánchez-López, V.; López-Ramírez, C.; Tobar, D.; López-Villalobos, J.L.; Gutiérrez, C.; Blanco-Orozco, A.; et al. Cigarette Smoke Decreases the Maturation of Lung Myeloid Dendritic Cells. PLoS ONE 2016, 11, e0152737. [Google Scholar] [CrossRef] [Green Version]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Liang, C.-L.; Liu, H.; Zeng, Y.-Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2017, 8, 268–284. [Google Scholar] [CrossRef] [Green Version]
- Pan, F.; Yang, T.-L.; Chen, X.-D.; Chen, Y.; Gao, G.; Liu, Y.-Z.; Pei, Y.-F.; Sha, B.-Y.; Jiang, Y.; Xu, C.; et al. Impact of female cigarette smoking on circulating B cells in vivo: The suppressed ICOSLG, TCF3, and VCAM1 gene functional network may inhibit normal cell function. Immunogenetics 2010, 62, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Fine, C.I.; Han, C.D.; Sun, X.; Liu, Y.; McCutcheon, J.A. Tobacco reduces membrane HLA class I that is restored by transfection with transporter associated with antigen processing 1 cDNA. J. Immunol. 2002, 169, 6012–6019. [Google Scholar] [CrossRef] [Green Version]
- Korkolopoulou, P.; Kaklamanis, L.; Pezzella, F.; Harris, A.; Gatter, K.C. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br. J. Cancer 1996, 73, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Chen, Q.; Xie, M. Smoking increases the risk of infectious diseases: A narrative review. Tob. Induc. Dis. 2020, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein—Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc. Natl. Acad. Sci. USA 2005, 102, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, F.; Buonaguro, L.; Caponigro, F.; Ionna, F.; Starita, N.; Annunziata, C.; Buonaguro, F.M.; Tornesello, M.L. Update on Head and Neck Cancer: Current Knowledge on Epidemiology, Risk Factors, Molecular Features and Novel Therapies. Oncology 2015, 89, 125–136. [Google Scholar] [CrossRef]
- Takada, H.; Imadome, K.-I.; Shibayama, H.; Yoshimori, M.; Wang, L.; Saitoh, Y.; Uota, S.; Yamaoka, S.; Koyama, T.; Shimizu, N.; et al. EBV induces persistent NF-κB activation and contributes to survival of EBV-positive neoplastic T- or NK-cells. PLoS ONE 2017, 12, e0174136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-R.; Liu, M.-T.; Chang, Y.-T.; Wu, C.-C.; Hu, C.-Y.; Chen, J.-Y. Epstein-Barr Virus Latent Membrane Protein 1 Represses DNA Repair through the PI3K/Akt/FOXO3a Pathway in Human Epithelial Cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Cai, J.; Li, J.; Chen, S.; Zeng, Z.; Peng, Q.; Ban, Y.; Zhou, Y.; Li, Z.; Xiong, W.; et al. Rediscovery of NF-κB signaling in nasopharyngeal carcinoma: How genetic defects of NF-κB pathway interplay with EBV in driving oncogenesis? J. Cell Physiol. 2018, 233, 5537–5549. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.; Tsang, C.M.; To, K.; Lo, K. The role of Epstein–Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Racero, I.T.; Gómez, N.C.; Leal, C.R.; Tinoco, E.L. Infections by Epstein-Barr virus and cytomegalovirus. Medicine 2014, 11, 2954–2964. [Google Scholar] [CrossRef]
- Feng, W.-H.; Cohen, J.I.; Fischer, S.; Li, L.; Sneller, M.; Goldbach-Mansky, R.; Raab-Traub, N.; Delecluse, H.-J.; Kenney, S.C. Reactivation of Latent Epstein-Barr Virus by Methotrexate: A Potential Contributor to Methotrexate-Associated Lymphomas. JNCI J. Natl. Cancer Inst. 2004, 96, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. mTORC2-mediated PDHE1α nuclear translocation links EBV-LMP1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef] [Green Version]
- Usui, G.; Matsusaka, K.; Mano, Y.; Urabe, M.; Funata, S.; Fukayama, M.; Ushiku, T.; Kaneda, A. DNA Methylation and Genetic Aberrations in Gastric Cancer. Digestion 2021, 102, 25–32. [Google Scholar] [CrossRef]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [Green Version]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? ImmunoTargets Ther. 2021, 10, 185–200. [Google Scholar] [CrossRef]
- Pertovaara, M.; Heliövaara, M.; Raitala, A.; Oja, S.S.; Knekt, P.; Hurme, M. The activity of the immunoregulatory enzyme indoleamine 2,3-dioxygenase is decreased in smokers. Clin. Exp. Immunol. 2006, 145, 469–473. [Google Scholar] [CrossRef]
- Liu, W.L.; Lin, Y.H.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.D.; Zhang, G. Epstein—Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-κB pathways: Impairment in T cell functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Jiang, C.; Liu, Y.; Li, Y.; Wu, H.; Feng, J.; Xu, Y. Association between IDO activity and prognosis in patients with non-small cell lung cancer after radiotherapy. Ann. Transl. Med. 2020, 8, 1169. [Google Scholar] [CrossRef]
- Skok, M.; Grailhe, R.; Changeux, J.-P. Nicotinic receptors regulate B lymphocyte activation and immune response. Eur. J. Pharmacol. 2005, 517, 246–251. [Google Scholar] [CrossRef]
- He, Y.-Q.; Xue, W.-Q.; Xu, F.-H.; Xu, Y.-F.; Zhang, J.-B.; Yu, H.-L.; Feng, Q.-S.; Chen, L.-Z.; Cao, S.-M.; Liu, Q.; et al. The Relationship between Environmental Factors and the Profile of Epstein-Barr Virus Antibodies in the Lytic and Latent Infection Periods in Healthy Populations from Endemic and Non-Endemic Nasopharyngeal Carcinoma Areas in China. EBioMedicine 2018, 30, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-Y.; He, Y.-Q.; Xue, W.-Q.; Zhou, T.; Liao, Y.; Zheng, M.-Q.; Jia, Y.-J.; Yuan, L.-L.; Jia, W.-H. Association Between Serum Cotinine Level and Serological Markers of Epstein–Barr Virus in Healthy Subjects in South China Where Nasopharyngeal Carcinoma Is Endemic. Front. Oncol. 2019, 9, 865. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.-H.; Xiong, D.; Xu, Y.-F.; Cao, S.-M.; Xue, W.-Q.; Qin, H.-D.; Liu, W.-S.; Cao, J.-Y.; Zhang, Y.; Feng, Q.-S.; et al. An Epidemiological and Molecular Study of the Relationship between Smoking, Risk of Nasopharyngeal Carcinoma, and Epstein–Barr Virus Activation. JNCI J. Natl. Cancer Inst. 2012, 104, 1396–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Fang, C.-Y.; Wu, C.-C.; Tsai, C.-H.; Lin, S.-F.; Chen, J.-Y. Reactive Oxygen Species Mediate Epstein-Barr Virus Reactivation by N-Methyl-N’-Nitro-N-Nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, M.C.; Koriyama, C.; Matsuo, K.; Kim, W.-H.; Herrera-Goepfert, R.; Liao, L.M.; Yu, J.; Carrasquilla, G.; Sung, J.J.; Alvarado-Cabrero, I.; et al. Case-case comparison of smoking and alcohol risk associations with Epstein-Barr virus-positive gastric cancer. Int. J. Cancer 2013, 134, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-Z.; Zhang, L.; Zhao, X.-C.; Gao, S.-H.; Qu, L.-W.; Yu, H.; Fang, W.-F.; Zhou, Y.-C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- El-Fattah, E.E.A.; Abdelhamid, A.M. Benzo[a]pyrene immunogenetics and immune archetype reprogramming of lung. Toxicology 2021, 463, 152994. [Google Scholar] [CrossRef]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog 2016, 12, e1005642. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.X.; Young, L.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein–Barr virus infection and replication in a human epithelial cell system. Nat. Cell Biol. 1992, 356, 347–350. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Diamond, M.E.; Sage, D.R.; Hayman, J.; Yates, Y.L. CD21-Dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol. 1999, 73, 2115–2125. [Google Scholar] [CrossRef] [Green Version]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases (Review). Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, A.; Miller, A.B.; Weichenthal, S.A.; To, T.; Wall, C.; Van Donkelaar, A.; Martin, R.V.; Crouse, D.; Villeneuve, P.J. Long-term exposure to fine particulate matter air pollution and the risk of lung cancer among participants of the Canadian National Breast Screening Study. Int. J. Cancer 2016, 139, 1958–1966. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Stone, J.; Desai, P.; Kosowicz, J.; Liu, J.; Ambinder, R. Arsenicals, the Integrated Stress Response, and Epstein–Barr Virus Lytic Gene Expression. Viruses 2021, 13, 812. [Google Scholar] [CrossRef]
- Ren, C.; Zhou, Y.; Liu, W.; Wang, Q. Paradoxical effects of arsenic in the lungs. Environ. Health Prev. Med. 2021, 26, 1–13. [Google Scholar] [CrossRef]
- Li, C.; Wang, C.; Yu, J.; Fan, Y.; Liu, D.; Zhou, W.; Shi, T. Residential Radon and Histological Types of Lung Cancer: A Meta-Analysis of Case–Control Studies. Int. J. Environ. Res. Public Health 2020, 17, 1457. [Google Scholar] [CrossRef] [Green Version]
- Alavanja, M.C.R. Biologic damage resulting from exposure to tobacco smoke and from radon: Implication for preventive interventions. Oncogene 2002, 21, 7365–7375. [Google Scholar] [CrossRef] [Green Version]
- Leng, S.; Thomas, C.L.; Snider, A.M.; Picchi, M.A.; Chen, W.; Willis, D.G.; Carr, T.G.; Krzeminski, J.; Desai, D.; Shantu, A.; et al. Radon Exposure, IL-6 Promoter Variants, and Lung Squamous Cell Carcinoma in Former Uranium Miners. Environ. Heal Perspect. 2016, 124, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Moyano, P.; García, J.; García, J.M.; Pelayo, A.; Muñoz-Calero, P.; Frejo, M.T.; Anadon, M.J.; Lobo, M.; Del Pino, J. Chlorpyrifos-induced cell proliferation in human breast cancer cell lines differentially mediated by estrogen and aryl hydrocarbon receptors and KIAA1363 enzyme after 24 h and 14 days exposure. Chemosphere 2020, 251, 126426. [Google Scholar] [CrossRef]
- Kramer, S.; Hikel, S.M.; Adams, K.; Hinds, D.; Moon, K. Current Status of the Epidemiologic Evidence Linking Polychlorinated Biphenyls and Non-Hodgkin Lymphoma, and the Role of Immune Dysregulation. Environ. Health Perspect. 2012, 120, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Park, E.; Kim, J.; Oh, J.-K.; Kim, B.; Hong, Y.-C.; Lim, M.K. Impact of environmental exposure to persistent organic pollutants on lung cancer risk. Environ. Int. 2020, 143, 105925. [Google Scholar] [CrossRef]
- Fu, J.; Mai, B.; Sheng, G.; Zhang, G.; Wang, X.; Peng, P.; Xiao, X.; Ran, R.; Cheng, F.; Peng, X.; et al. Persistent organic pollutants in environment of the Pearl River Delta, China: An overview. Chemosphere 2003, 52, 1411–1422. [Google Scholar] [CrossRef]
- Bonner, M.R.; Williams, B.A.; Rusiecki, J.A.; Blair, A.; Freeman, L.E.B.; Hoppin, J.; Dosemeci, M.; Lubin, J.; Sandler, D.P.; Alavanja, M.C.R. Occupational exposure to terbufos and the incidence of cancer in the Agricultural Health Study. Cancer Causes Control 2010, 21, 871–877. [Google Scholar] [CrossRef]
- Kashuba, E.V.; Gradin, K.; Isaguliants, M.; Szekely, L.; Poellinger, L.; Klein, G.; Kazlauskas, A. Regulation of Transactivation Function of the Aryl Hydrocarbon Receptor by the Epstein-Barr Virus-encoded EBNA-3 Protein. J. Biol. Chem. 2006, 281, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Mishima, K.; Yamamoto-Yoshida, S.; Ushikoshi, R.; Nakagawa, Y.; Yamamoto, K.; Ryo, K.; Ide, F.; Saito, I. Aryl Hydrocarbon Receptor-Mediated Induction of EBV Reactivation as a Risk Factor for Sjögren’s Syndrome. J. Immunol. 2012, 188, 4654–4662. [Google Scholar] [CrossRef] [Green Version]
- Attafi, I.M.; Bakheet, S.A.; Korashy, H.M. The role of NF-κB and AhR transcription factors in lead-induced lung toxicity in human lung cancer A549 cells. Toxicol. Mech. Methods 2020, 30, 197–207. [Google Scholar] [CrossRef]
- Zou, X.N.; Lu, S.H.; Liu, B. Volatile N-nitrosamines and their precursors in chinese salted fish—A possible etiological factor for NPC in China. Int. J. Cancer 1994, 59, 155–158. [Google Scholar] [CrossRef]
- Wang, S.Y.; Hu, Y.L.; Wu, Y.L.; Li, X.; Chi, G.B.; Chen, Y.; Dai, W.S. A comparative study of the risk factors for lung cancer in Guangdong, China. Lung Cancer 1996, 14 (Suppl. S1), S99–S105. [Google Scholar] [CrossRef]
Figure 1.
Epstein–Barr virus genome organization. (A) EBV lineal genome. (B) EBV circular genome representation.
Figure 1.
Epstein–Barr virus genome organization. (A) EBV lineal genome. (B) EBV circular genome representation.

Figure 2.
EBV proteins in epithelial tumors and their functions in cancer progression. This model suggests initial damage that can be caused by environmental pollutants (xenobiotics). Next, the epithelial injury allows the EBV latency establishment with the subsequent EBV protein expression. Finally, the EBV protein expression allows the activation of different cell signaling pathways such as Wnt/β-catenin, NF-κB, JNK, JAK/STAT, EGFR/MAPK, and PI3K/AKT.
Figure 2.
EBV proteins in epithelial tumors and their functions in cancer progression. This model suggests initial damage that can be caused by environmental pollutants (xenobiotics). Next, the epithelial injury allows the EBV latency establishment with the subsequent EBV protein expression. Finally, the EBV protein expression allows the activation of different cell signaling pathways such as Wnt/β-catenin, NF-κB, JNK, JAK/STAT, EGFR/MAPK, and PI3K/AKT.
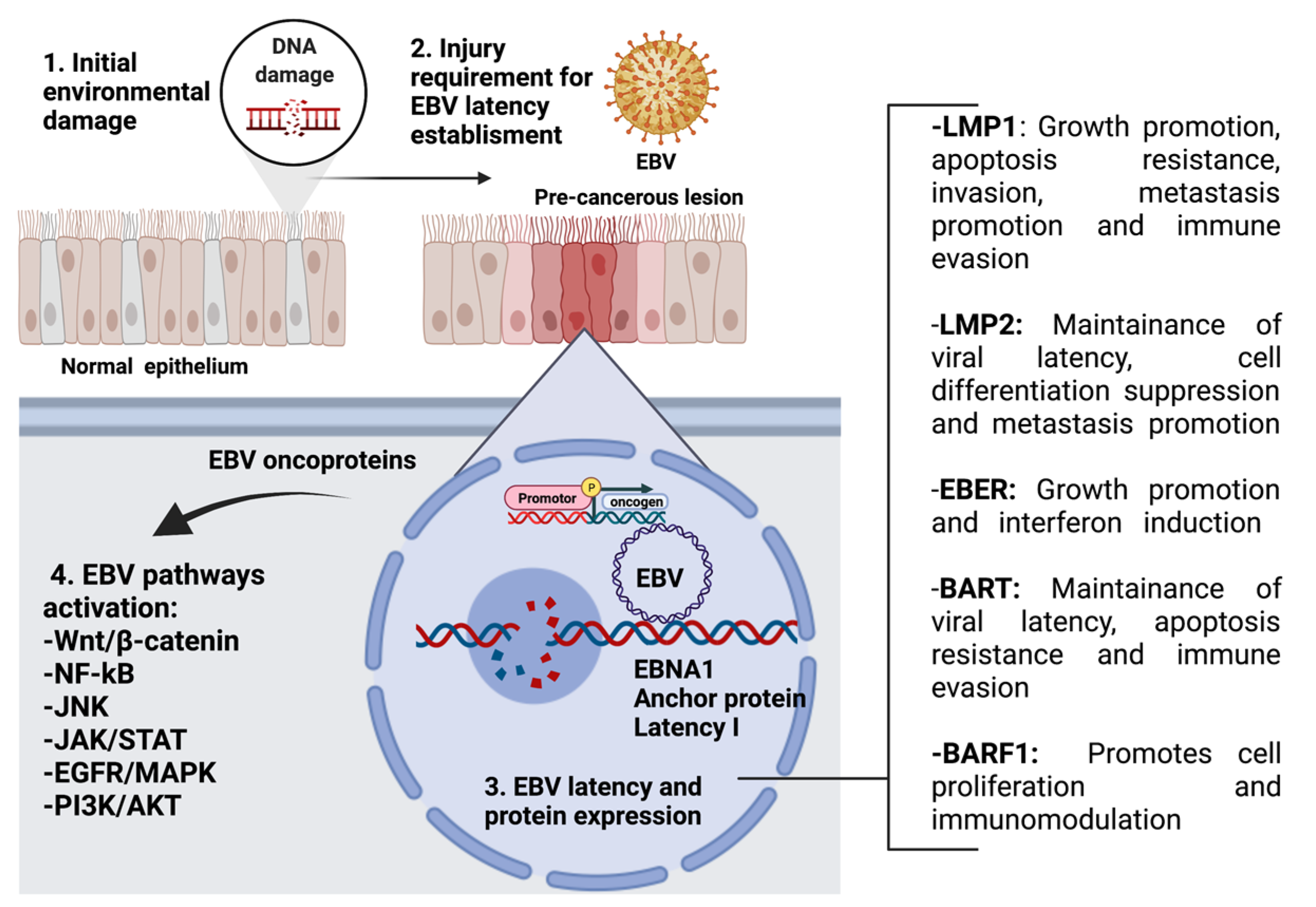
Figure 3.
CD21 (CR2) transcript expression in LC (TCGA, n = 1325) according to sample type and histological classification of tumors (***: p < 0.001 for both; Welch’s t-test). The expression of CD21 was increased 1.8-fold in malignant tumors (1323 primary tumors and two recurrences) and increased 1.3-fold in lung AdC compared to normal tissue and lung SQC. Raw data were extracted from University of California, Santa Cruz (ena.ucsc.edu). UCSC Xena functional genomics explorer (https://xenabrowser.net) [200].
Figure 3.
CD21 (CR2) transcript expression in LC (TCGA, n = 1325) according to sample type and histological classification of tumors (***: p < 0.001 for both; Welch’s t-test). The expression of CD21 was increased 1.8-fold in malignant tumors (1323 primary tumors and two recurrences) and increased 1.3-fold in lung AdC compared to normal tissue and lung SQC. Raw data were extracted from University of California, Santa Cruz (ena.ucsc.edu). UCSC Xena functional genomics explorer (https://xenabrowser.net) [200].
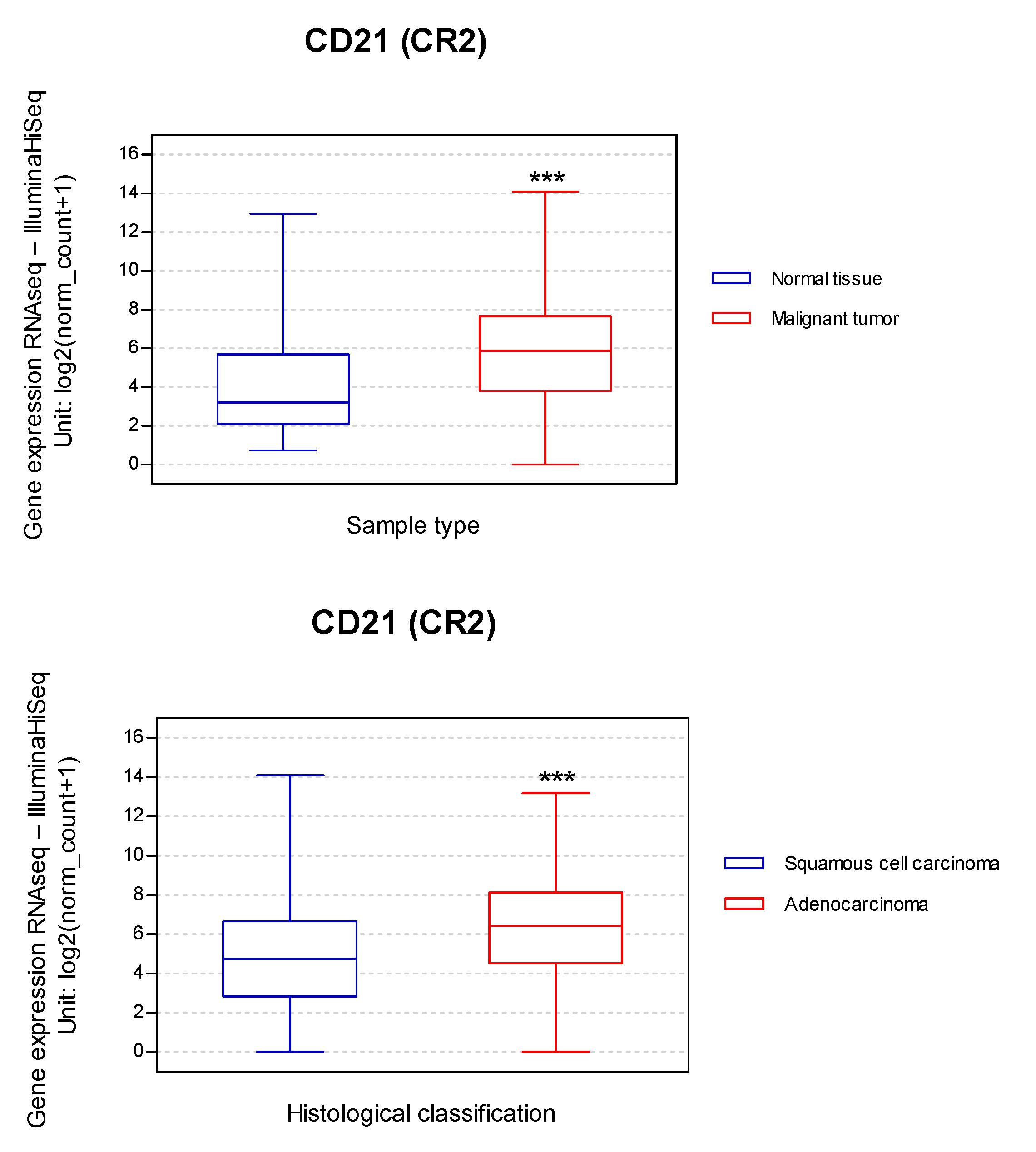
Figure 4.
Hypothetical mechanism of interaction between cigarette smoke components or environmental contaminants with EBV in lung epithelial cells. This interaction involves local immunosuppression facilitating EBV infection, latency establishment, and latent/lytic switch activation.
Figure 4.
Hypothetical mechanism of interaction between cigarette smoke components or environmental contaminants with EBV in lung epithelial cells. This interaction involves local immunosuppression facilitating EBV infection, latency establishment, and latent/lytic switch activation.


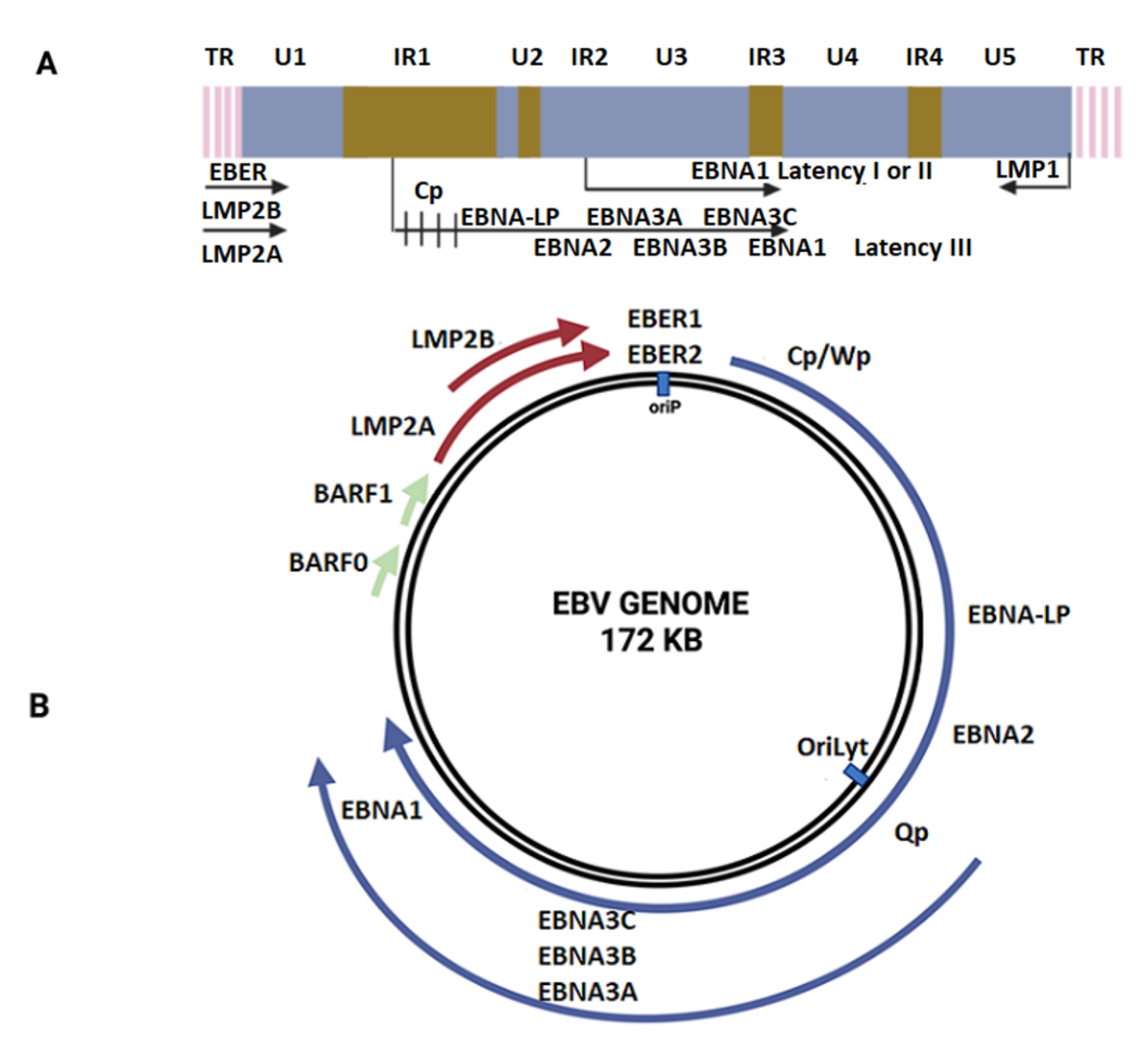{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
EBV frequency in lung cancer.
| Tumor Types | Total | EBV (+) | (%) | EBV Gene | Methods | Country | Ref |
|---|---|---|---|---|---|---|---|
| AdC, SQC, SCLC, LCC | 80 | 5 | 6.3 | EBNA | ISH, IHC, PCR | Japan | Kasai [145] |
| NSCLC | 167 | 9 | 5.4 | EBNA | ISH, Southern Blot, IHC | China | Wong [144] |
| AC, mesothelioma | 130 | 0 | 0 | Absent | ISH | United States | Conway [154] |
| SQC, AdC, LCC, LELC | 127 | 11 | 8.7 | EBNA | ISH, IH | China | Chen [137] |
| LELC, SQC, AdC, LCC, SCC | 51 | 30 | 58.8 | EBER, LMP1, VCA | ISH, IHC | China | Han [136] |
| SCC | 23 | 1 | 4.3 | LMP-1 | ISH, IHC, PCR | United States | Chu [155] |
| SQC, AC, AdC-SCC, LCLC, SCLC | 122 | 0 | 0 | Absent | ISH, IHC, PCR | France | Brouchet [160] |
| NSCLC | 108 | 36 | 33.3 | EBER | ISH | China | Li [141] |
| SQC, AdC | 19 | 12 | 63.2 | EBER | ISH, PCR, IHC | Spain | Gomez-Roman [146] |
| AdC | 110 | 0 | 0 | Absent | ISH | Singapore | Lim [156] |
| SQC, AdC | 48 | 7 | 14.6 | BART1, BART2, and BHRF1–3 | qPCR, Microarray | United States | Koshiol [157] |
| SQC, AdC, SCC | 48 | 5 | 10.4 | LMP-1 | PCR | Iran | Jafarian [147] |
| NSCLC | 66 | 4 | 6.1 | LMP1 and EBNA1 | NGS | China | Wang [151] |
| NSCLC | 1017 | 3 | 0.3 | EBNA-1, LMP-1 and LMP-2 | NGS, ISH | United States | Kheir [152] |
| SQC, AdC, SCLC | 73 | 2 | 2.7 | Absent | PCR | Unites States | Gupta [148] |
| LELC | 91 | 91 | 100.0 | EBER | ISH-WES | China | Hong [138] |
| LC | 108 | 36 | 33.3 | EBER, LMP1, BCL-2 | IHC | China | Li [142] |
| SQC | 87 | 33 | 37.9 | Absent | ISH | China | Zhang [143] |
| NSCLC, SQC | 70 | 18 | 31.4 | Absent | qPCR | Italy | Carpagnano [149] |
| LC | 48 | 25 | 52 | Absent | PCR, in situ PCR | China | Xia [150] |
| AdC | 4 | - | - | EBER | Microarray, qPCR, ISH | Korea | Kim [158] |
| NSCLC | Sample mix | - | - | Absent | Microarray, qPCR | Korea | Ma [159] |
LC, lung cancer; NSCLC, non-small cell lung carcinoma; SQC, squamous cell carcinoma; AdC, adenocarcinoma; LCC, large cell carcinoma; SCLC, small cell lung carcinoma; PLELC, pulmonary lymphoepithelioma-like carcinoma.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Osorio, J.C.; Blanco, R.; Corvalán, A.H.; Muñoz, J.P.; Calaf, G.M.; Aguayo, F. Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens 2022, 11, 132. https://doi.org/10.3390/pathogens11020132
AMA Style
Osorio JC, Blanco R, Corvalán AH, Muñoz JP, Calaf GM, Aguayo F. Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens. 2022; 11(2):132. https://doi.org/10.3390/pathogens11020132
Chicago/Turabian StyleOsorio, Julio C., Rancés Blanco, Alejandro H. Corvalán, Juan P. Muñoz, Gloria M. Calaf, and Francisco Aguayo. 2022. "Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives" Pathogens 11, no. 2: 132. https://doi.org/10.3390/pathogens11020132
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.