
The Role of Nuclear Factor Kappa B (NF-κB) in the Immune Response against Parasites


1
Division of Pharmacology and Toxicology, Department of Preclinical Sciences, Institute of Veterinary Medicine, Warsaw University of Life Sciences-SGGW, 02-786 Warsaw, Poland
2
Department of Biosciences and Food Technology, School of Science, STEM College, RMIT University, Bundoora, VIC 3083, Australia
*
Author to whom correspondence should be addressed.
Pathogens 2022, 11(3), 310; https://doi.org/10.3390/pathogens11030310
Submission received: 31 December 2021
/
Revised: 17 February 2022
/
Accepted: 25 February 2022
/
Published: 2 March 2022
(This article belongs to the Special Issue Immune Response of the Host and Vaccine Development)
Abstract
:The immune system consists of various cells, organs, and processes that interact in a sophisticated manner to defend against pathogens. Upon initial exposure to an invader, nonspecific mechanisms are raised through the activation of macrophages, monocytes, basophils, mast cells, eosinophils, innate lymphoid cells, or natural killer cells. During the course of an infection, more specific responses develop (adaptive immune responses) whose hallmarks include the expansion of B and T cells that specifically recognize foreign antigens. Cell to cell communication takes place through physical interactions as well as through the release of mediators (cytokines, chemokines) that modify cell activity and control and regulate the immune response. One regulator of cell states is the transcription factor Nuclear Factor kappa B (NF-κB) which mediates responses to various stimuli and is involved in a variety of processes (cell cycle, development, apoptosis, carcinogenesis, innate and adaptive immune responses). It consists of two protein classes with NF-κB1 (p105/50) and NF-κB2 (p100/52) belonging to class I, and RelA (p65), RelB and c-Rel belonging to class II. The active transcription factor consists of a dimer, usually comprised of both class I and class II proteins conjugated to Inhibitor of κB (IκB). Through various stimuli, IκB is phosphorylated and detached, allowing dimer migration to the nucleus and binding of DNA. NF-κB is crucial in regulating the immune response and maintaining a balance between suppression, effective response, and immunopathologies. Parasites are a diverse group of organisms comprised of three major groups: protozoa, helminths, and ectoparasites. Each group induces distinct effector immune mechanisms and is susceptible to different types of immune responses (Th1, Th2, Th17). This review describes the role of NF-κB and its activity during parasite infections and its contribution to inducing protective responses or immunopathologies.
1. Introduction
Parasites are a diverse group of organisms comprised of three major groups: protozoa, helminths, and ectoparasites. During the course of their evolution, they have gained the ability to live in or on hosts and gather food at the hosts’ expense. They may feed on both humans and animals; up to 2.0 billion [1] people may suffer from parasitic infections, and losses in animal production are estimated at many US$ billions per year [2]. It is not in the interest of the parasite to kill the host, consequently chronic, long-lasting infections can be common.
The persistency of many infections occurs in part due to the immunomodulatory abilities of the parasites [3]. During the thousands of years of co-evolution with host organisms, parasites have learned to trick, evade and suppress the host immune system. Parasites’ mechanisms of immunomodulation are very effective and diverse, and this has earned them the nickname “masters of regulation” [4]. Parasites can cleave antibodies [5,6,7], induce apoptosis in macrophages [8] and eosinophils [9], interfere with cytokine signaling networks [10,11,12] and cytokine release [13,14,15,16], and induce regulatory B cells [17]. However, exploring the diversity and range of parasite immunomodulation is outside the scope of this article; this topic has been reviewed elsewhere [18,19,20,21,22,23].
NF-κB plays a critical role in mediating responses to a remarkable diversity of external stimuli, and thus is a pivotal element in multiple physiological and pathological processes and is a powerful orchestrator of the immune response [24,25,26]. There are many reports in the literature regarding the role of NF-κB during specific parasite infections. The goal of this article is to explore parasites’ interplay with NF-κB. In this manuscript, we review the existing knowledge regarding NF-κB expression and activity during infections with Plasmodium spp., Trypanosoma spp., Leishmania spp., Toxoplasma spp., cestodes, nematodes, and flukes in the context of the parasite life cycle, occupied niche, and impact on the immune response. By bringing these disparate reports together and reviewing the topic in one publication we hope to shed new light on this issue, highlighting the role NF-κB plays during parasite infections and why it represents an important target for parasite manipulation.
2. Structure and Function of NF-κB
NF-κB is a transcription factor; a family of five proteins can dimerize to form NF-κB complexes: NF-κB1 (p105), NF-κB2 (p100), RelA (p65), RelB and c-Rel [27]. NF-κB1 and NF-κB2 undergo processing into mature forms, p50 and p52, respectively. All the NF-κB proteins share the Rel homology domain (RHD)—which allows them to form homo- or heterodimers [27] and bind DNA [28]—additionally p65, RelB and c-Rel contain TAD (transcriptional activation domain), enabling activation of gene expression [28]. A number of heterodimers may be formed, but the most commonly present in most cell types is p50/p65 [29], while c-Rel containing dimers are constrained predominately to hematopoietic origin cells [30].
Prior to activation, latent NF-κB dimers are coupled to inhibitors (IκB) which sequester the transcription factor in the cytoplasm. In most cells, NF-κB is associated with IκBα, IκBβ or IκBε; additionally, p105 and p100 contain an inhibitor within their sequence, thus also functioning as inhibitors (while p50 and p52 homo- and hetero-dimers repress NF-κB-dependent transcription) [31]. Translocation of NF-κB to the nucleus and activation requires dissociation from the inhibitor, which takes place upon phosphorylation of the inhibitor by an IκB kinase (IKK). In the canonical activation pathway, IKK is composed of catalytically active IKKα, IKKβ, and the regulatory subunit IKKγ (NEMO) [31], and its activity promotes p50, RelA, and c-Rel activation through IκB phosphorylation [32]. In the non-canonical pathway, NIK (Nuclear Factor κB-inducing Kinase) [33] activates IKKα which phosphorylates the p100 subunit of p100/RelB leading to migration of p52/RelB to the nucleus [32]. Both pathways regulate the expression of a distinct and overlapping set of genes [34], regulating innate and adaptive immune responses [27].
NF-κB dimers bind to sequences known as κB sites with the general consensus GGGRNWYYCC (R–purine, W–adenine or thymine, Y–pyrimidine, N–any base); however, different dimers may show differing specificity for target sequences [35]. Moreover, each particular NF-κB protein may undergo a variety of post-translational modifications [36] which impacts their stability, degradation, affinity to binding sites, and interactions within the dimer and with other transcription factors [37].
3. Role of NF-κB in the Immune Response
NF-κB is crucial for appropriate immune system functioning at all stages, from the development of primary and secondary lymphoid tissues, through hematopoiesis, to recognizing Danger-Associated Molecular Patterns (DAMPs) or Pathogen-Associated Molecular Patterns (PAMPs) and regulating effector mechanisms of immune cells [38,39]. There are two main pathways of NF-κB activation: canonical and noncanonical [40] (Figure 1).
Activation of the canonical pathway is mediated through TAK1 and has rapid and transient effects, whereas the noncanonical pathway is slow, persistent, and with the hallmark of NIK activation [32]. Moreover, the canonical pathway is activated by various stimuli including PAMPs, DAMPs, and numerous receptors, while the noncanonical pathways are induced by a specific set of receptors, e.g., B-cell-activating factor belonging to TNF family receptor (BAFFR), lymphotoxin β-receptor (LTβR), receptor activator for nuclear factor κB (RANK), TNFR2, Fn14, CD30, and CD27 [41]. Both pathways are indispensable for the appropriate regulation of the balance between immune tolerance and inflammation. Due to the number of potential NF-κB dimers [42], and interactions with other factors [43] the role of NF-κB during the course of infections is very complicated. Both positive and negative implications during parasite infections are reviewed below and depicted in Figure 2 and Figure 3.
4. Plasmodium spp.
Malaria is a disease caused by Plasmodium spp. Over 40% of the human population is estimated to be at risk of infection [44]. A vast majority of cases occur in Africa, and despite a global trend towards reductions in the number of infections over recent decades, over 241 million malaria cases and 627,000 deaths were estimated to have occurred globally in 2020 [45]. Whether host responses contribute to pathology or protection can be hard to determine, and while proinflammatory responses to malaria infections are critical to controlling the disease, excessive inflammation is linked to pathology (Table 1). NF-κB is a highly important orchestrator of the immune response during Plasmodium infections [46,47,48]. In general, during an infection NF-κB is increased and it has pivotal and specific roles in pathogenesis and the immune response, being implicated in several processes. Cerebral malaria is considered the most severe clinical complication of infection and proinflammatory cytokines, cytoadherence, and endothelial activation are considered to have roles in pathogenesis [49]. Exposure to P. falciparum-infected erythrocytes has been shown to induce nuclear translocation of NF-κB p65 in human brain microvascular endothelial cells in vitro, along with upregulation of the NF-κB activation cascade, of NF-κB subunits (p100, p105, c-REL, RELB) and NF-κB inhibitory proteins (IκBα, IκBε)—this results in increased proinflammatory chemokine/cytokine release (CCL20 and TNF-α) and intercellular adhesion molecule 1 (ICAM-1) surface expression [48,50]. ICAM-1 is linked with the adhesion of infected erythrocytes to epithelial cells and the associated disease pathology. While this can occur in the brain, it can also occur in other organs. Upon exposure to extracellular vesicles containing P. vivax proteins, human spleen fibroblasts show upregulation of ICAM-1, linked to nuclear translocation of NF-κB [51]. This has been postulated to facilitate the formation of hidden parasite populations in the spleen relatively safe from control measures. Patients with cerebral malaria also show increased p65 translocation to the nucleus in neurons, glial cells, epithelial cells, and leukocytes [46]. These changes correlate with histopathological changes in the brain, with NF-κB p65 modulating apoptosis in brain endothelial cells and intravascular leukocytes during cerebral malaria [46]. NF-κB has also been shown to regulate apoptosis in Kupffer cells and lymphocytes in the liver during severe P. falciparum infection [52]. Other cell types also show upregulation of NF-κB pathways during infection. Increased translocation of p65 and p50 to the nucleus and degradation of IκBα occurs in monocytes following exposure to trophozoites or hemozoin. This coincides with enhanced activity of monocyte matrix metalloproteinase-9 and increased proinflammatory cytokine production, effects also important to malaria pathogenesis [53]. Interestingly, it has been reported that while significantly elevated active p65 levels occur in peripheral blood mononuclear cells (PBMCs) of P. vivax patients and P. falciparum patients with mild symptoms, those with severe P. falciparum malaria only showed increased levels of active p65 after treatment, with NF-κB expression being negatively correlated with IL-10 levels [54]. During infections, the malaria parasite can induce upregulation of NF-κB pathways in a variety of cell types. The full role of NF-κB during malaria infection is dependent on cell type and likely influenced by parasite strain and the genetics of the host. While further study is needed to fully elucidate NF-κB’s roles, it is evident that during severe infections there is a link between NF-κB upregulation and several effects that play a role in malaria pathogenesis.
5. Trypanosoma spp.
The Trypanosoma genus is comprised of several species, but only Trypanosoma cruzi and T. brucei (T. b. gambiense and T. b. rhodesiense) cause disease in humans, Chagas disease and sleeping sickness, respectively. Other species cause disease in animals and may also be models for human infection. Both Chagas disease and sleeping sickness are significant problems in sub-Saharan and tropical climates. Trypomastigotes can infect a range of cell types (Table 2). Several trypanosome species have been shown to activate NF-κB in epithelial cells, endothelial cells, and fibroblasts [55,56]. This involves a rapid increase in p65 and p65 translocation to the nucleus, as well as phosphorylation of IKKα/β, resulting in induction of a pro-inflammatory response through NF-κB -dependent expression of TNF-α, IL-1β, and IL-6 cytokines, nitric oxide, and adhesion molecules (E-selectin, VCAM-1, and ICAM-1) [55,56,57,58] Activation of NF-κB is a determinator of T. cruzi intracellular survival and tissue specificity. The ability for T. cruzi to induce NF-κB activation is highly dependent on cell type and appears to be inversely correlated with a cell’s susceptibility to infection. The high susceptibility of muscle cells to infection by T. cruzi has been proposed to be in part due to a failure to induce NF-κB activation [55]. Activation of endothelial cells via the NF-κB pathway occurs with intact trypomastigotes, however, trypanosome trans-sialidases have also been shown to mediate this activity via α-2,3 sialylated receptors [56]. Toll-like receptors (TLRs), likely through recognition of parasite glycoisnositolphospholipids and glycosylphosphatidyl inositol, have also been shown to be involved in T. cruzi-mediated increased NF-κB and the resultant pro-inflammatory response [57,59,60]. Other receptors that recognize T. cruzi and are involved in subsequent activation of NF-κB have been identified; macrophage galactose-C type lectin (MGL1) receptor is critical for the optimal activation of macrophages during T. cruzi infection. MGL1 is proposed to recognize soluble T. cruzi Lysate Antigen (TcAg) [61]. A trypanosome antigen that can disrupt the protective host response via modulation of NF-κB is cruzain from T. cruzi. Cruzain cleaves p65, this prevents macrophage activation during early infection, facilitating parasite survival and the spread of infection [62]. For efficient parasite removal, the immune system must maintain a balance between effective Th1 and Threg immune responses. A Th1 response, mediated mainly by TNF-α may be efficient at clearing the parasite, but without appropriate regulating mechanisms (mainly through IL-10) exacerbated inflammation may lead to increased pathology and disease severity. This is evident in mice with impaired IL-10 production, which are less resistant to T. congolense and T. brucei infection and show higher degrees of inflammation and immunopathologies due to dysregulated immunoregulation [63]. A major orchestrator of this regulation is NF-κB, with p65/p50 and p50/p50 complexes important players in regulating pro- and anti-inflammatory pathways, respectively [64]. p50 plays a role in down-regulation of the inflammatory response [65], and p50−/− mice fail to maintain an appropriate balance between Threg/Th1, leading to increased liver injury during T. congolense infection, associated with TNF-α overproduction [66]. While induction of NF-κB pathways to promote proinflammatory responses can confer protection against trypanosomes as outlined above, a prolonged response can confer pathogenicity. T. cruzi-induced proinflammatory responses in human colonic epithelial cells have been linked to Megacolon, a major pathology of Chagas disease [57]. Additionally, activation of NF-κB -dependent inflammatory responses in cardiomyocytes, and vascular endothelial cells by T. cruzi may contribute to vascular dysfunction and injury, and chronic cardiomyopathy [58,67,68].
6. Toxoplasma spp.
Toxoplasma gondii is an obligate intracellular Apicomplexan parasite able to inhabit a broad spectrum of mammals [69]. It has been estimated that up to one-third of the world’s population may be seropositive to this parasite [70]. The host–pathogen interplay and molecular basis of host resistance are complicated, but a pattern showing a key role for a balance between IL-1β, TNF-α, IL-12, and IFN-γ exists [71,72,73,74,75]. Upon disruption of this balance the protective but over-expressed Th1 cytokines (IFN-γ, IL-1β, TNF-α, and IL-12) lead to immunopathologies [76]. NF-κB is involved in the positive and negative regulation of these cytokines [77,78,79] and is a crucial factor required for eliciting a protective response (Table 3). This is demonstrated by the fact that RelB−/− mice develop impaired innate and adaptive immune responses and do not survive T. gondii infection [80]. T. gondii infection in mice results in a global increase in NF-κB activity [81]. However, most data indicate T. gondii induces a suppressing NF-κB phenotype in infected macrophages in mice leading to delayed IL-12 production and failure to produce TNF-α. Despite increased p65 phosphorylation [82], and IκBα phosphorylation [83], translocation of p65 and c-Rel is not observed [81,83,84]. This process of NF-κB suppression requires active invasion by the parasite [84], and recently downregulation of miR-187 by the parasite has been implicated in p65 phosphorylation and mediation of IL-12 expression [82]. On the other hand, infected human monocytes show an activated phenotype with an increase in both p65 phosphorylation [85] and p65 translocation [74]. This leads to IL-1β induction [86], and also requires active invasion by the parasite, with dense granule protein GRA15 playing a role, and parasite strain lineage also important [74]. LPS-induced IL-1β levels also appear unaffected in T. gondii infected monocytes [86].
Of course, it is worth remembering that the use of different models may produce different results. A well-established model to investigate the immune response is based on simultaneous stimulation of cells with lipopolysaccharide (LPS) and the factor under investigation, to observe if there is an abrogation of the LPS-induced proinflammatory response [93,94]. Conflicting results have been reported for mouse macrophages, some indicating T. gondii has the ability to abolish LPS-induced p65 translocation [83], whereas others indicate LPS-induced p65 translocation is unaffected, as is LPS-induced nuclear NF-κB-binding activity [92]. Nevertheless, the consensus is that T. gondii infection interferes with NF-κB activation at a step downstream of these processes and effectively dampens the host immune response, facilitating parasite survival and multiplication [82,83,84,92]. Another LPS-activated immune cell population—neutrophils—also skew their phenotype towards suppression when infected with T. gondii; dampened inflammasome activity and IL-1β release is associated with reduced p65 (Ser536) phosphorylation and inhibition of IκBα degradation–classical hallmarks of reduced NF-κB activity [86]. Data from non-immune cells also confirms T. gondii’s abilities to deactivate NF-κB. Despite human fibroblasts showing some signs of increased NF-κB activity upon T. gondii exposure, like IKK-dependent degradation of IκB, the phenotype is actually one of reduced transcription due to altered downstream processes: reduced phosphorylation of p65 and no p65, p50, or c-Rel nucleus accumulation [89]. Phosphorylation of p65 at Ser468 by virulence factor ROP18, targeting it for degradation, and blocking nuclear translocation has also been demonstrated as a means to inhibiting the host NF-κB pathway in fibroblasts and macrophages [91]. These results highlight the importance of phosphorylation as a means by which the parasite can mediate NF-κB pathway inhibition [37].
This contrasts with a report of mouse fibroblasts which show a more activated phenotype, with induced p50 and p65 translocation to the nucleus, enhanced DNA binding by p50, p65, RelB, and p52, and phosphorylation (though no observed degradation) of IκB upon T. gondii infection [90]. This response was reported to result in an anti-apoptotic phenotype. NF-κB is considered an anti-apoptotic factor, therefore, despite efforts by the parasite to dampen NF-κB activity complete abrogation of activity is not likely to be in the interest of the parasite; apoptosis would destroy the niche it lives in–the cell [95]. Some Toxoplasma-infected cells appear resistant to apoptosis [96] and show NF-κB dependent anti-apoptotic gene expression patterns [90]; moreover, NF-κB deprived mice (p65−/−) are unable to prevent apoptosis hallmarks in infected cells [97]. While our understanding of the interplay between this parasite and NF-κB is still in its infancy, the findings presented here indicate T. gondii has developed sophisticated mechanisms to manipulate host NF-κB to effectively modulate the host response to facilitate parasite survival. The dampening of host responses is also likely to minimize immune-mediated host pathology.
7. Leishmania spp.
Leishmania is a protozoan parasite that inhabits phagolysosomes [98]. Data shows that 12 million people worldwide suffer from leishmaniasis [99]. Clinical manifestations of the disease depend on the Leishmania species and are classified as: cutaneous, visceral, mucocutaneous, and diffuse cutaneous leishmaniasis [100]. Leishmania resides in host macrophages, as well as neutrophils and inflammatory monocytes [99]. Like other protozoan parasites, Leishmania has developed mechanisms to survive in the host and evade the immune response. A coordinated immune response (Table 4) is required to eliminate Leishmania, and effective parasite removal is associated with proinflammatory IL-12, TNF-α, and IFN-γ, whereas IL-4, IL-10, IL-13, and TGF-β are associated with parasite survival [101]. The key cytokine believed to be involved in parasite survival is IL-10, with IL-10 knockout (KO) mice resistant to infection [102,103]. It is therefore not surprising that the parasites’ efforts at dampening the proinflammatory response, through increased IL-10 release are intense. The pivotal role of NF-κB in immune response regulation, including as a positive and negative regulator of macrophage gene expression [104], also key to Leishmania invasion, means NF-κB offers an attractive target for the parasite to manipulate. Upon contact with L. major amastigote, human monocytes preferentially induce p50/p50 and p50/c-Rel, with inhibition of p50/p65 translocation [105]. p50/p50 complexes are associated with IL-10 [64], and infected monocytes release increased concentrations of both IL-10 and TNF-α [105]. While IL-10 is expected to be beneficial for the parasite and is implicated in Leishmania survival, TNF-α is involved in inflammation and is putatively protective. Interestingly, infection of macrophages by L. major promastigotes results in inhibition of IL-12, a response beneficial to the parasite, though this is not due to inhibition of NF-κB activation [106].
Leishmania parasites may modulate the NF-κB pathway in numerous ways and for various reasons. Upon infection of human or mouse macrophages, the promastigote stage of several Leishmania species (but not L. aethiopica or L. tarentolae) can cause cleavage of p65 into p35. This is dependent on Leishmania protease gp63. p35 is involved in the expression of specific chemokines and the parasite has been proposed to induce those favorable to its survival [98,109]. Cleavage of p65 to p35 has also been reported following dendritic cell infection by L. infantum promastigotes [111]. L. aethiopica amastigotes have been shown to manipulate a variety of signaling pathways, including NF-κB, in human macrophages during spreading [109]. This results in reduced p65 expression and phosphorylation (Ser32/36) of IκB, indicating downregulation of NF-κB. This down-regulation of NF-κB leading to cell apoptosis is expected to facilitate the spread of the parasite [109]. Conversely, decreased expression of IκB was also reported which is expected to induce p65 translocation and NF-κB pathway activation and may indicate pleiotropic impacts of Leishmania on NF-κB pathways.
Leishmania species can promote silent infections under certain conditions; this can sometimes be achieved through the modulation of NF-κB pathways. It has been observed that when L. donovani promastigotes infect mouse macrophages, there is no change in the expression of cytosolic or nuclear p50 or p65 expression in response. This results from active downregulation of NF-κB, mediated through Hypoxia Inducible Factor-1α (HIF-1α) and miR-210 [110]. This leads to decreased pro-inflammatory cytokine expression and NO production by the macrophages and facilitates parasite survival. The generation of a permissive environment is also evident upon L. amazonensis amastigote infection of mouse dendritic cells. The infection leads to pleiotropic inhibition of TLR/NF-κB/NLRP3 pathways, promoting transcriptional activation of the alternative NF-κB pathway which is proposed to lead to MHC class I-restricted antigen presentation and stalling of dendritic cell maturation [107]. Stalled mouse dendritic cell maturation is also evident upon infection by L. infantum promastigotes [111]. Here the parasite impairs NF-κB through cleavage of p65, although no impact on IkBα expression was reported.
It is difficult to draw one clear conclusion regarding the processes and roles of NF-κB modulation during Leishmania infection based on the varied data in the literature. Moreover, there may not even be one clear conclusion. Authors have analyzed various laboratory models, cell types, and different Leishmania species, hence contrasting results may be expected. This is supported by the results of Nogueira et al. who evaluated responses to various Leishmania species (L. braziliensis, L. infantum, L. amazonensis) and showed that only L. amazonensis was able to induce p65 translocation to the nucleus and a pro-inflammatory response in a particular laboratory model [108]. The variability among species to cleave p65 to p35 should also be noted [98,109]. Species-specific and stage-specific variations, as well as the development of effective immune responses complicate the situation; nevertheless, from the known data emerges a pattern which generally indicates that Leishmania modulates the NF-κB pathway towards a dampening of TNF-α expression and production of an environment that facilitates parasite survival.
8. Helminths
Helminths are a divergent group of multicellular parasites (Cestoda, Nematoda, Trematoda). Due to their size, like protozoan parasites, they cannot be neutralized by phagocytosis; moreover, their abilities to trick and evade the host immune system allow them to efficiently regulate the immune response towards a Th2 type. This is beneficial for the parasite and also arguably the host—the parasite survives and gains reproductive success while the host does not suffer from immunopathologies. The ability to dampen Th1 responses has led to an exploration of the use of helminths (or their products) as treatments for allergies and autoimmune diseases (as reviewed elsewhere [112,113]). The high prevalence of helminth infections has also led to research regarding the development of vaccines. These have led to numerous studies regarding vaccine trials, the impacts of helminth antigens on symptoms of autoimmune diseases (or allergies), and cytokine patterns during infections, yet only a small proportion of the research has focused on characterizing the intracellular mechanisms of the immune response in the context of NF-κB and this topic seems to have been relatively neglected to date.
8.1. Cestodes
Helminth parasites excrete and secrete a range of molecules during host invasion. These molecules function at the host-parasite interface and are often how parasites modulate the host immune response (Table 5). The excretory-secretory product (ES) from Taenia crassiceps is known to skew immune responses through the downregulation of proinflammatory pathways. One way T. crassiceps can achieve this is through modulation of dendritic cells, which play a major role in the initiation of Th2 polarization [114]. T. crassiceps ES can modulate signaling in dendritic cells, including in the NF-κB pathway, leading to significantly attenuated LPS-induced p65 phosphorylation. This contributes to the blocking of dendritic cell maturation and a dampening of proinflammatory IL-12 and TNF-α, which affects eventual T-cell responses [114]. Mesocestoides corti soluble antigens also exhibit NF-κB-modulated immunosuppressive properties. M. corti helminth soluble factors (HSFs) inhibit LPS-induced p65 phosphorylation and acetylation in microglia leading to abrogation of IL-6 and TNF-α release [115]. This parasite suppression of host inflammation and immunity provides another example of how parasites may actively modulate host responses to produce asymptomatic phases of the disease. Parasite somatic antigens function to maintain the parasite’s metabolism, physiological processes and regulate homeostasis, and generally have less contact with the host immune system. While parasites actively suppress host immune responses, parasite damage or death can sometimes lead to an inflammatory response and pathology. This may occur during Taenia solium infections. T. solium larval somatic antigens activate the NF-κB pathway in human monocytes, through increased IkB-α degradation and enhanced DNA binding by p65, p50, or c-Rel. This results in a release of the chemokines CXCL8 and CCL2, which may have a role in immune cell recruitment, which may be a factor associated with inflammation and pathology [116].
8.2. Nematodes
Insights into the role of the NF-κB pathway during nematode infections come from the well-established model organism Trichuris muris. A detailed and nuanced role for NF-κB in mounting a protective response has been revealed during in vivo experiments (Table 6). Mice able to clear T. muris infections show enhanced NF-κB activity with elevated levels of IL-4 and IL-13 and lower levels of IFN-γ, demonstrating a protective Th2 response and skewing away from a susceptible Th1 response [118]. c-Rel KO mice maintain resistance to infection, whereas p105/50 (NF-κB1) and p100/52(NF-κB2) KO mice develop chronic infection, with the highest immunopathologies observed among p105/50 deficient mice, correlating with elevated IFN-γ concentrations [118]. p50/p50 complexes are associated with regulatory IL-10 [64,65,119] suggesting a role for IL-10 in controlling intestinal inflammation. The data from KO studies indicate roles for components of the NF-κB pathway and demonstrates NF-κB utilization during helminth infections as an inductor of a protective regulatory response. Colonic epithelium cells show increased p65 phosphorylation upon exposure to T. muris ES which leads to the release of proinflammatory factors [120].
During T. spiralis infection, NF-κB (though P2X7R-mediated NLRP3 activation) modulates the killing capacity of macrophages, with NF-κB downregulation resulting in decreased killing capacity [123]. T. spiralis ES from various life stages can reduce LPS-induced p65 expression and nuclear translocation [124,125]. The resulting inhibition of LPS-induced pro-inflammatory (TNF-α, IL-1β, IL-6, IL-12) cytokine expression, and induction of regulatory cytokines (IL-10, TGF-β) indicates a role during infections in the modulation of macrophage responses towards phenotypes conducive to worm survival and host health [124,125].
Modulation of macrophage regulation is also observed during Brugia malayi infections. Downregulation and impaired activation of NF-κB-p65 and NF-κB-p50/105 in mouse macrophages during the course of infection contributes to macrophage polarization towards M2 and Mreg phenotypes and downregulation of proinflammatory IL-12 release and increased secretion of Th2/Threg cytokines: IL-4 and IL-10, respectively [126]. IL-10 and IL-12 release is also impaired in human DCs exposed to B. malayi microfilariae [130]; however, pathologies during infection are associated with increased Th1/Th17 responses [131] and enhanced angiogenesis [132]. NF-κB is involved in both angiogenesis [133] and inflammation [134] regulation. Although the basic levels of angiogenic factors do not differ between asymptomatic and filarial lymphedema (lymphatic pathology) patients, the cells from lymphedema patients produce higher levels of vascular endothelial growth factor (VEGF)-C [127] and NF-κB activation has a role in elevated angiogenic growth factor production, and associated pathology, in these patients [127]. While several nematode derived molecules may contribute to the development of angiogenesis, Jothi et al. identified a prominent angiogenic factor from B. malayi: Asparaginyl–tRNA Synthetase [135], which acts through the NF-κB pathway [121].
Prevention of DC maturation, upon incubation with L4 larvae of Heligmosomoides polygyrus [128] and the ability of adult stage H. polygyrus somatic antigens to inhibit proliferation and apoptosis of host immune cells [129] are other examples where parasite regulation of the NF-κB pathway is utilized by nematodes to modulate host immune responses and facilitate survival.
8.3. Flukes
The formation of liver granuloma and fibrosis following egg deposition is a serious pathology that can occur during schistosomiasis. The TNF-α level, raised by parasite egg antigens, is positively correlated with parasite burden [136], and increased TNF-α mediates increased morbidity [137]. As the NF-κB pathway is involved in TNF-α expression, this indicates increased NF-κB activity. While no data are available from immune cells (Table 7), liver and colonic cells show increased p65 expression, translocation to the nucleus, and phosphorylation (Ser276) [136,138,139], with involvement of NF-κB in apoptosis. The role of NF-κB in pathology is supported by the fact that its inhibition in vivo prevents granuloma and fibrosis [140,141]. Fasciola hepatica uses a variety of molecules to modulate host immune responses to facilitate its survival and manage host pathology, driving anti-inflammatory responses and Th1 suppression; it is considered a potent immune modulator. Some, but not all of the immune-modulatory effects induced by the parasite involve interaction with the NF-κB pathway. F. hepatica tegumental antigens (Teg) prevent NF-κB activation (p65 expression) in dendritic cells. This leads to suppression of dendritic cell maturation and function—suppressing induction of proinflammatory cytokines and cell surface markers [142]. The ability of F. hepatica to modulate responses [93,143,144] has led to attempts to use its components to treat a range of diseases and conditions. An example is the use of F. hepatica extracellular vesicles (EV) to treat colitis. EV were shown to suppress induced p65 translocation in a mice colitis model, effectively reducing the release of proinflammatory cytokines TNF-α, IL-6, and IL-17A, and attenuating clinical symptoms of colitis [145]. F. hepatica EV, Teg, and ES are comprised of a variety of molecules and their action is a sum of the action of all the antigens present. Therefore, these antigens may impact NF-κB in various ways. Two ES components, glutathione S-transferases (GST) and fatty acid binding protein (FABP) have separately been shown to be able to block LPS-induced NF-κB-dependent gene expression [146,147,148]. These molecules can suppress macrophage LPS-induced pro-inflammatory cytokines, activate a suppressive dendritic cell phenotype, and induce anti-inflammatory effects that attenuate septic shock and promote survival in a mouse model [146,147,148].
9. Conclusions
The NF-κB pathway is vital in regulating immune function and it plays an important role during infections by parasites, being integral to the formation of protective responses, or indeed pathologies. For example, NF-κB is pivotal to the induction of protective proinflammatory responses against trypanosomes and to the activation of macrophages during Leishmania or T. cruzi infections. Alternatively, hosts deficient in NF-κB pathway elements or unable to appropriately activate or regulate NF-κB pathways are often more susceptible to infections or pathologies. This has been demonstrated with T. gondii [80], T. muris [118], and trypanosome infections [63], while the high susceptibility of muscle cells to T. cruzi infection is proposed to be linked to a failure to induce NF-κB activation [55]. Altered NF-κB activation in certain cell types during parasite infections can also lead to increased pathology, as observed in the brain in cases of cerebral malaria [32].
The wide impact of NF-κB on various processes and in numerous cell types and its importance to mounting effective immune responses means that modulation of NF-κB pathways represents a vital strategy for many parasites to employ to facilitate survival. As such, parasites have evolved numerous strategies to either up- or down-regulate NF-κB. These include, but are not limited to, direct phosphorylation of NF-κB pathway proteins or their inhibitors [91], binding of certain receptors to activate specific pathways [56,57,61], or cleavage of NF-κB proteins via parasite proteases [62,98,111]. While each modulates NF-κB activity, the resulting outcomes of these actions are dependent on the cell types involved and whether NF-κB activity is increased or decreased.
The data presented in this review demonstrates that NF-κB has many crucial roles, including many essential to mounting an effective response against protozoan and metazoan parasites. By presenting several relevant host-parasite interactions involving parasite modulation of the NF-κB pathway we have highlighted how it is a target for parasites to create an environment within the host conducive to survival. We hope this has shed new light on this important regulator of immunity for the reader.
Author Contributions
Conceptualization, P.B.; writing—original draft preparation, P.B. and L.J.N. writing—review, L.J.N.; visualization, P.B. and L.J.N.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Subvention of Department of Preclinical Sciences, Institute of Veterinary Medicine, Warsaw University of Life Sciences (Poland)—Subvention no. 505-35-730700-FK0000-35.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were collected or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McKay, D.M.; Shute, A.; Lopes, F. Helminths and intestinal barrier function. Tissue Barriers 2017, 5, e1283385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narladkar, B.W. Projected economic losses due to vector and vector-borne parasitic diseases in livestock of india and its significance in implementing the concept of integrated practices for vector management. Vet. World 2018, 11, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Arora, N.; Nair, M.G.; Prasad, A. The interplay of helminthic neuropeptides and proteases in parasite survival and host immunomodulation. Biochem. Soc. Trans. 2022, 50, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Maizels, R.M.; Balic, A.; Gomez-Escobar, N.; Nair, M.; Taylor, M.D.; Allen, J.E. Helminth parasites—Masters of regulation. Immunol. Rev. 2004, 201, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Berasain, P.; Carmona, C.; Frangione, B.; Dalton, J.P.; Goñi, F. Fasciola hepatica: Parasite-secreted proteinases degrade all human IgG subclasses: Determination of the specific cleavage sites and identification of the immunoglobulin fragments produced. Exp. Parasitol. 2000, 94, 99–110. [Google Scholar] [CrossRef]
- Li, A.H.; Moon, S.U.; Park, Y.K.; Na, B.K.; Hwang, M.G.; Oh, C.M.; Cho, S.H.; Kong, Y.; Kim, T.S.; Chung, P.R. Identification and characterization of a cathepsin L-like cysteine protease from Taenia solium metacestode. Vet. Parasitol. 2006, 141, 251–259. [Google Scholar] [CrossRef]
- Rhoads, M.L.; Fetterer, R.H. Developmentally regulated secretion of cathepsin L-like cysteine proteases by Haemonchus contortus. J. Parasitol. 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Guasconi, L.; Serradell, M.C.; Masih, D.T. Fasciola hepatica products induce apoptosis of peritoneal macrophages. Vet. Immunol. Immunopathol. 2012, 148, 359–363. [Google Scholar] [CrossRef]
- Serradell, M.C.; Guasconi, L.; Cervi, L.; Chiapello, L.S.; Masih, D.T. Excretory-secretory products from Fasciola hepatica induce eosinophil apoptosis by a caspase-dependent mechanism. Vet. Immunol. Immunopathol. 2007, 117, 197–208. [Google Scholar] [CrossRef]
- Sun, X.; Yang, F.; Shen, J.; Liu, Z.; Liang, J.; Zheng, H.; Fung, M.; Wu, Z. Recombinant Sj16 from Schistosoma japonicum contains a functional N-terminal nuclear localization signal necessary for nuclear translocation in dendritic cells and interleukin-10 production. Parasitol. Res. 2016, 115, 4559–4571. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Newland, S.A.; Gibbs, S.E.; Tourlomousis, P.; Fernandes dos Santos, P.; Patel, M.N.; Hall, S.W.; Walczak, H.; Schramm, G.; Haas, H.; et al. The Schistosoma mansoni T2 ribonuclease omega-1 modulates inflammasome-dependent IL-1β secretion in macrophages. Int. J. Parasitol. 2015, 45, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Grainger, J.R.; Smith, K.A.; Hewitson, J.P.; McSorley, H.J.; Harcus, Y.; Filbey, K.J.; Finney, C.A.M.; Greenwood, E.J.D.; Knox, D.P.; Wilson, M.S.; et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-β pathway. J. Exp. Med. 2010, 207, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Popa, G.L.; Popa, M.I. Recent Advances in Understanding the Inflammatory Response in Malaria: A Review of the Dual Role of Cytokines. J. Immunol. Res. 2021, 2021, 10–12. [Google Scholar] [CrossRef]
- Długosz, E.; Basałaj, K.; Zawistowska-Deniziak, A. Cytokine production and signalling in human THP-1 macrophages is dependent on Toxocara canis glycans. Parasitol. Res. 2019, 118, 2925–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Długosz, E.; Wasyl, K.; Klockiewicz, M.; Wiśniewski, M. Toxocara canis mucins among other excretory-secretory antigens induce in vitro secretion of cytokines by mouse splenocytes. Parasitol. Res. 2015, 114, 3365–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, Y.L.L.; Neto, J.R.C.; Costa, A.W.F.; Silva, M.V.T.; Silva, M.V.; Celes, M.R.N.; Oliveira, M.A.P.; Joosten, L.A.B.; Ribeiro-Dias, F.; Gomes, R.S.; et al. Interleukin-32γ in the Control of Acute Experimental Chagas Disease. J. Immunol. Res. 2022, 2022, 7070301. [Google Scholar] [CrossRef] [PubMed]
- Somoza, M.; Bertelli, A.; Pratto, C.A.; Verdun, R.E.; Campetella, O.; Mucci, J. Trypanosoma cruzi Induces B Cells That Regulate the CD4+ T Cell Response. Front. Cell. Infect. Microbiol. 2022, 11, 789373. [Google Scholar] [CrossRef]
- Maizels, R.M.; Smits, H.H.; McSorley, H.J. Modulation of Host Immunity by Helminths: The Expanding Repertoire of Parasite Effector Molecules. Immunity 2018, 49, 801–818. [Google Scholar] [CrossRef] [Green Version]
- Zakeri, A.; Hansen, E.P.; Andersen, S.D.; Williams, A.R.; Nejsum, P. Immunomodulation by helminths: Intracellular pathways and extracellular vesicles. Front. Immunol. 2018, 9, 2349. [Google Scholar] [CrossRef]
- Zakeri, A. Helminth-induced apoptosis: A silent strategy for immunosuppression. Parasitology 2017, 144, 1663–1676. [Google Scholar] [CrossRef]
- Motran, C.C.; Ambrosio, L.F.; Volpini, X.; Celias, D.P.; Cervi, L. Dendritic cells and parasites: From recognition and activation to immune response instruction. Semin. Immunopathol. 2017, 39, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Popple, S.J.; Burrows, K.; Mortha, A.; Osborne, L.C. Remote regulation of type 2 immunity by intestinal parasites. Semin. Immunol. 2021, 53, 101530. [Google Scholar] [CrossRef] [PubMed]
- Drurey, C.; Maizels, R.M. Helminth extracellular vesicles: Interactions with the host immune system. Mol. Immunol. 2021, 137, 124–133. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, Q.; Zhang, T.; Fan, J. C7ORF41 Regulates Inflammation by Inhibiting NF-κB Signaling Pathway. Biomed Res. Int. 2021, 2021, 7413605. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-κB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef] [PubMed]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-κB: At the Borders of Autoimmunity and Inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar] [CrossRef]
- Lu, T.; Stark, G.R. NF-κB: Regulation by Methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, C.; Bucci, I.; Napolitano, G. The role of the transcription factor Nuclear Factor-kappa B in thyroid autoimmunity and cancer. Front. Endocrinol. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Aggarwal, B.B. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia 2002, 16, 1053–1068. [Google Scholar] [CrossRef] [Green Version]
- Visekruna, A.; Volkov, A.; Steinhoff, U. A key role for NF-κb transcription factor c-rel in T-lymphocyte-differentiation and effector functions. Clin. Dev. Immunol. 2012, 2012, 239368. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sudom, A.; Min, X.; Cao, Z.; Gao, X.; Ayres, M.; Lee, F.; Cao, P.; Johnstone, S.; Plotnikova, O.; et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 2012, 287, 27326–27334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurumi, A.; Zhao, C.; Li, W.X. Canonical and non-canonical JAK/STAT transcriptional targets may be involved in distinct and overlapping cellular processes. BMC Genom. 2017, 18, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.R.; Ramakrishnan, P. Regulation of Nuclear Factor-kappaB Function by O-GlcNAcylation in Inflammation and Cancer. Front. Cell Dev. Biol. 2021, 9, 751761. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.; Carmody, R. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Orsini, E.M.; Perelas, A.; Southern, B.D.; Grove, L.M.; Olman, M.A.; Scheraga, R.G. Stretching the Function of Innate Immune Cells. Front. Immunol. 2021, 12, 767319. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Q.; Liu, H.; Zhang, X. Interplay between Non-Canonical NF-κB Signaling and Hepatitis B Virus Infection. Front. Immunol. 2021, 12, 730684. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnico, I.; Lanzillotta, A.; Benarese, M.; Alghisi, M.; Baiguera, C.; Battistin, L.; Spano, P.F.; Pizzi, M. Chapter 24 NF-KappaB Dimers in the Regulation of Neuronal Survival, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 85, ISBN 9780123748935. [Google Scholar]
- Bhatt, D.; Ghosh, S. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante, A.A.; Pacheco, M.A. Malaria Molecular Epidemiology: An Evolutionary Genetics Perspective. Microbiol. Spectr. 2019, 7, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2021; ISBN 978-92-4-004049-6. electronic version.
- Punsawad, C.; Maneerat, Y.; Chaisri, U.; Nantavisai, K.; Viriyavejakul, P. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar. J. 2013, 12, 260. [Google Scholar] [CrossRef] [Green Version]
- Thiam, A.; Sanka, M.; Diallo, R.N.; Torres, M.; Mbengue, B.; Nunez, N.F.; Thiam, F.; Diop, G.; Victorero, G.; Nguyen, C.; et al. Gene expression profiling in blood from cerebral malaria patients and mild malaria patients living in Senegal. BMC Med. Genom. 2019, 12, 148. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Sha, W.; Shulaev, V.; Stins, M.F.; Sullivan, D.J. Plasmodium falciparum-infected erythrocytes induce NF-κB regulated inflammatory pathways in human cerebral endothelium. Blood 2009, 114, 4243–4252. [Google Scholar] [CrossRef] [Green Version]
- Rénia, L.; Howland, S.W.; Claser, C.; Gruner, A.C.; Suwanarusk, R.; Teo, T.H.; Russell, B.; Lisa, N.P. Cerebral malaria Mysteries at the blood-brain barrier. Virulence 2012, 3, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.K.; Sullivan, D.J.; Stins, M.F. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-κB. Infect. Immun. 2006, 74, 3262–3270. [Google Scholar] [CrossRef] [Green Version]
- Toda, H.; Diaz-Varela, M.; Segui-Barber, J.; Roobsoong, W.; Baro, B.; Garcia-Silva, S.; Galiano, A.; Gualdrón-López, M.; Almeida, A.C.G.; Brito, M.A.M.; et al. Plasma-derived extracellular vesicles from Plasmodium vivax patients signal spleen fibroblasts via NF-kB facilitating parasite cytoadherence. Nat. Commun. 2020, 11, 2761. [Google Scholar] [CrossRef]
- Viriyavejakul, P.; Khachonsaksumet, V.; Punsawad, C. Liver changes in severe Plasmodium falciparum malaria: Histopathology, apoptosis and nuclear factor kappa B expression. Malar. J. 2014, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Prato, M.; Gallo, V.; Giribaldi, G.; Aldieri, E.; Arese, P. Role of the NF-κB transcription pathway in the haemozoin- and 15-HETE-mediated activation of matrix metalloproteinase-9 in human adherent monocytes. Cell. Microbiol. 2010, 12, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Punsawad, C.; Krudsood, S.; Maneerat, Y.; Chaisri, U.; Tangpukdee, N.; Pongponratn, E.; Nantavisai, K.; Udomsangpetch, R.; Viriyavejakul, P. Activation of nuclear factor kappa B in peripheral blood mononuclear cells from malaria patients. Malar. J. 2012, 11, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.S.; Tam, W.; Sen, R.; Pereira, M.E.A. Cell-specific activation of nuclear factor-κB by the parasite Trypanosoma cruzi promotes resistance to intracellular infection. Mol. Biol. Cell 2000, 11, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Z.; Plazolles, N.; Baltz, T.; Coustou, V. Identification of Trans-Sialidases as a Common Mediator of Endothelial Cell Activation by African Trypanosomes. PLoS Pathog. 2013, 9, e1003710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suman, S.; Rachakonda, G.; Mandape, S.N.; Sakhare, S.S.; Villalta, F.; Pratap, S.; Lima, M.F.; Nde, P.N. Phospho-proteomic analysis of primary human colon epithelial cells during the early Trypanosoma cruzi infection phase. PLoS Negl. Trop. Dis. 2018, 12, e0006792. [Google Scholar] [CrossRef]
- Huang, H.; Calderon, T.M.; Berman, J.W.; Braunstein, V.L.; Weiss, L.M.; Wittner, M.; Tanowitz, H.B. Infection of endothelial cells with Trypanosoma cruzi activates NF-κB and induces vascular adhesion molecule expression. Infect. Immun. 1999, 67, 5434–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.-C.; Peixoto, J.R.; de Arruda, L.B.; Campos, M.A.; Gazzinelli, R.T.; Golenbock, D.T.; Akira, S.; Previato, J.O.; Mendonça-Previato, L.; Nobrega, A.; et al. Expression of Functional TLR4 Confers Proinflammatory Responsiveness to Trypanosoma cruzi Glycoinositolphospholipids and Higher Resistance to Infection with T. cruzi. J. Immunol. 2004, 173, 5688–5696. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.M.; Oliveira, A.C.; Bellio, M. The immune response to Trypanosoma cruzi: Role of toll-like receptors and perspectives for vaccine development. J. Parasitol. Res. 2012, 2012, 507874. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, T.; Pacheco-Fernández, T.; Vázquez-Mendoza, A.; Nieto-Yañez, O.; Juárez-Avelar, I.; Reyes, J.L.; Terrazas, L.I.; Rodriguez-Sosa, M. MGL1 Receptor Plays a Key Role in the Control of T. cruzi Infection by Increasing Macrophage Activation through Modulation of ERK1/2, c-Jun, NF-κB and NLRP3 Pathways. Cells 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Doyle, P.S.; Zhou, Y.M.; Hsieh, I.; Greenbaum, D.C.; McKerrow, J.H.; Engel, J.C. The trypanosoma cruzi protease cruzain mediates immune evasion. PLoS Pathog. 2011, 7, e1002139. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Pan, W.; Tabel, H. Experimental African trypanosomiasis: IFN-γ mediates early mortality. Eur. J. Immunol. 2003, 33, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Driessler, F.; Venstrom, K.; Sabat, R.; Asadullah, K.; Schottelius, A.J. Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: A role for p50. Clin. Exp. Immunol. 2004, 135, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosschaerts, T.; Morias, Y.; Stijlemans, B.; Hérin, M.; Porta, C.; Sica, A.; Mantovani, A.; De Baetselier, P.; Beschin, A. IL-10 limits production of pathogenic TNF by M1 myeloid cells through induction of nuclear NF-κB p50 member in Trypanosoma congolense infection-resistant C57BL/6 mice. Eur. J. Immunol. 2011, 41, 3270–3280. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Gupta, S.; Davidson, M.; Garg, N.J. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J. Biol. Chem. 2010, 285, 11596–11606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.F.; Capettini, L.S.A.; da Silva, J.F.P.; Sales-Junior, P.; Cruz, J.S.; Cortes, S.F.; Lemos, V.S. Mechanisms of vascular dysfunction in acute phase of Trypanosoma cruzi infection in mice. Vascul. Pharmacol. 2016, 82, 73–81. [Google Scholar] [CrossRef]
- Dubey, J.P. The history of Toxoplasma gondii—The first 100 years. J. Eukaryot. Microbiol. 2008, 55, 467–475. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Atenea 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Chang, H.R.; Grau, G.E.; Pechère, J.C. Role of TNF and IL-1 in infections with Toxoplasma gondii. Immunology 1990, 69, 33–37. [Google Scholar]
- Yap, G.; Pesin, M.; Sher, A. Cutting Edge: IL-12 Is Required for the Maintenance of IFN-γ Production in T Cells Mediating Chronic Resistance to the Intracellular Pathogen, Toxoplasma gondii. J. Immunol. 2000, 165, 628–631. [Google Scholar] [CrossRef] [Green Version]
- Denkers, E.Y.; Gazzinelli, R.T. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 1998, 11, 569–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gov, L.; Karimzadeh, A.; Ueno, N.; Lodoen, M.B. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio 2013, 4, e00255-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkers, E.Y.; Kim, L.; Butcher, B.A. In the belly of the beast: Subversion of macrophage proinflammatory signalling cascades during Toxoplasma gondii infection. Cell. Microbiol. 2003, 5, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gazzinelli, R.T.; Wysocka, M.; Hieny, T.; Scharton -Kersten, T.; Cheever, A.; Kuhn, R.; Muller, W.; Trincheri, G.; Sher, A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 1996, 157, 798–805. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Van Rooijen, N.; et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 2008, 130, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.; Mercado, E.L.; Adelaja, A.; Ho, J.Q.; Cheng, Q.J.; Ghosh, G.; Hoffmann, A. An NFκB activity calculator to delineate signaling crosstalk: Type I and II interferons enhance NFκB via distinct mechanisms. Front. Immunol. 2019, 10, 1425. [Google Scholar] [CrossRef] [Green Version]
- Caamaño, J.; Alexander, J.; Craig, L.; Bravo, R.; Hunter, C.A. The NF-kappa B family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J. Immunol. 1999, 163, 4453–44561. [Google Scholar]
- Shapira, S.; Speirs, K.; Gerstein, A.; Caamano, J.; Hunter, C.A. Suppression of NF-κB activation by infection with Toxoplasma gondii. J. Infect. Dis. 2002, 185, S66–S72. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhai, T.; Yu, Y.; Li, X.; Gong, P.; Zhang, X.; Li, G.; Li, J. Delayed IL-12 production by macrophages during Toxoplasma gondii infection is regulated by miR-187. Parasitol. Res. 2020, 119, 1023–1033. [Google Scholar] [CrossRef]
- Butcher, B.A.; Kim, L.; Johnson, P.F.; Denkers, E.Y. Toxoplasma gondii Tachyzoites Inhibit Proinflammatory Cytokine Induction in Infected Macrophages by Preventing Nuclear Translocation of the Transcription Factor NF-κB. J. Immunol. 2001, 167, 2193–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, B.A.; Denkers, E.Y. Mechanism of entry determines the ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect. Immun. 2002, 70, 5216–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandori, W.J.; Lima, T.S.; Mallya, S.; Kao, T.H.; Gov, L.; Lodoen, M.B. Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin Dindependent release of IL-1β during infection of primary human monocytes. PLoS Pathog. 2019, 15, e1007923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, T.S.; Gov, L.; Lodoen, M.B. Evasion of Human Neutrophil-Mediated Host Defense during Toxoplasma gondii Infection. MBio 2018, 13, e02027-17. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.H.; Kim, S.G.; Shin, J.H.; Ham, D.W.; Shin, E.H. Toxoplasma gra16 inhibits nf-κb activation through pp2a-b55 upregulation in non-small-cell lung carcinoma cells. Int. J. Mol. Sci. 2020, 21, 6642. [Google Scholar] [CrossRef]
- Zhang, A.M.; Shen, Q.; Li, M.; Xu, X.C.; Chen, H.; Cai, Y.H.; Luo, Q.L.; Chu, D.Y.; Yu, L.; Du, J.; et al. Comparative studies of macrophage-biased responses in mice to infection with Toxoplasma gondii ToxoDB #9 strains of different virulence isolated from China. Parasites Vectors 2013, 6, 308. [Google Scholar] [CrossRef] [Green Version]
- Shapira, S.; Harb, O.S.; Margarit, J.; Matrajt, M.; Han, J.; Hoffmann, A.; Freedman, B.; May, M.J.; Roos, D.S.; Hunter, C.A. Initiation and termination of NF-κB signaling by the intracellular protozoan parasite Toxoplasma gondii. J. Cell Sci. 2005, 118, 3501–3508. [Google Scholar] [CrossRef] [Green Version]
- Molestina, R.E.; Payne, T.M.; Coppens, I.; Sinai, A.P. Activation of NF-ΚB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IΚB to the parasitophorous vacuole membrane. J. Cell Sci. 2003, 116, 4359–4371. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; An, R.; Chen, L.; Shen, Y.; Chen, Y.; Cheng, L.; Jiang, Z.; Zhang, A.; Yu, L.; Chu, D.; et al. Toxoplasma gondii virulence factor rop18 inhibits the host nf-kb pathway by promoting p65 degradation. J. Biol. Chem. 2014, 289, 12578–12592. [Google Scholar] [CrossRef] [Green Version]
- Leng, J.; Butcher, B.A.; Egan, C.E.; Abi Abdallah, D.S.; Denkers, E.Y. Toxoplasma gondii Prevents Chromatin Remodeling Initiated by TLR-Triggered Macrophage Activation. J. Immunol. 2009, 182, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Bąska, P.; Norbury, L.J.; Zawistowska-Deniziak, A.; Wiśniewski, M.; Januszkiewicz, K. Excretory/secretory products from two Fasciola hepatica isolates induce different transcriptional changes and IL-10 release in LPS-activated bovine “BOMA” macrophages. Parasitol. Res. 2017, 116, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Bąska, P.; Wiśniewski, M.; Krzyżowska, M.; Długosz, E.; Zygner, W.; Górski, P.; Wedrychowicz, H. Molecular cloning and characterisation of in vitro immune response against astacin-like metalloprotease Ace-MTP-2 from Ancylostoma ceylanicum. Exp. Parasitol. 2013, 133, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Mammari, N.; Halabi, M.A.; Yaacoub, S.; Chlala, H.; Dardé, M.L.; Courtioux, B. Toxoplasma gondii Modulates the Host Cell Responses: An Overview of Apoptosis Pathways. Biomed Res. Int. 2019, 2019, 6152489. [Google Scholar] [CrossRef] [Green Version]
- Nash, P.B.; Purner, M.B.; Leon, R.P.; Clarke, P.; Duke, R.C.; Curiel, T.J. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J. Immunol. 1998, 160, 1824–1830. [Google Scholar]
- Payne, T.M.; Molestina, R.E.; Sinai, A.P. Inhibition of caspase activation and a requirement for NF=ΚB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J. Cell Sci. 2003, 116, 4345–4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, D.J.; Godbout, M.; Contreras, I.; Forget, G.; Olivier, M. A novel form of NF-κB is induced by Leishmania infection: Involvement in macrophage gene expression. Eur. J. Immunol. 2008, 38, 1071–1081. [Google Scholar] [CrossRef]
- Rossi, M.; Fasel, N. How to master the host immune system? Leishmania parasites have the solutions! Int. Immunol. 2018, 30, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Ready, P.D. Leishmaniasis emergence in Europe. Eurosurveillance 2010, 15, 29–39. [Google Scholar] [CrossRef]
- Gupta, G.; Oghumu, S.; Satoskar, A.R. Mechanisms of Immune Evasion in Leishmaniasis. Adv. Appl. Microbiol. 2013, 82, 155–184. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hoffmann, K.F.; Mendez, S.; Kamhawi, S.; Udey, M.C.; Wynn, T.A.; Sacks, D.L. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 2001, 194, 1497–1506. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.L.; Wille, U.; Villegas, E.N.; Hunter, C.A.; Farrell, J.P. IL-10 mediates susceptibility to Leishmania donovani infection. Eur. J. Immunol. 2001, 31, 2848–2856. [Google Scholar] [CrossRef]
- Grigoriadis, G.; Zhan, Y.; Grumont, R.J.; Metcalf, D.; Handman, E.; Cheers, C.; Gerondakis, S. The Rel subunit of NF-κB-like transcription factors is a positive and negative regulator of macrophage gene expression: Distinct roles for Rel in different macrophage populations. EMBO J. 1996, 15, 7099–7107. [Google Scholar] [CrossRef] [PubMed]
- Guizani-Tabbane, L.; Ben-Aissa, K.; Belghith, M.; Sassi, A.; Dellagi, K. Leishmania major Amastigotes Induce p50/c-Rel NF-κB Transcription Factor in Human Macrophages: Involvement in Cytokine Synthesis. Infect. Immun. 2004, 72, 2582–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricardo-Carter, C.; Favila, M.; Polando, R.E.; Cotton, R.N.; Bogard Horner, K.; Condon, D.; Ballhorn, W.; Whitcomb, J.P.; Yadav, M.; Geister, R.L.; et al. Leishmania major inhibits IL-12 in macrophages by signalling through CR3 (CD11b/CD18) and down-regulation of ETS-mediated transcription. Parasite Immunol. 2013, 35, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecoeur, H.; Rosazza, T.; Kokou, K.; Varet, H.; Coppée, J.Y.; Lari, A.; Commère, P.H.; Weil, R.; Meng, G.; Milon, G.; et al. Leishmania amazonensis Subverts the Transcription Factor Landscape in Dendritic Cells to Avoid Inflammasome Activation and Stall Maturation. Front. Immunol. 2020, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, P.M.; de Menezes-Neto, A.; Borges, V.M.; Descoteaux, A.; Torrecilhas, A.C.; Xander, P.; Revach, O.Y.; Regev-Rudzki, N.; Soares, R.P. Immunomodulatory Properties of Leishmania Extracellular Vesicles during Host-Parasite Interaction: Differential Activation of TLRs and NF-κB Translocation by Dermotropic and Viscerotropic Species. Front. Cell. Infect. Microbiol. 2020, 10, 380. [Google Scholar] [CrossRef]
- Ranatunga, M.; Rai, R.; Richardson, S.C.W.; Dyer, P.; Harbige, L.; Deacon, A.; Pecorino, L.; Getti, G.T.M. Leishmania aethiopica cell-to-cell spreading involves caspase-3, AkT, and NF-κB but not PKC-δ activation and involves uptake of LAMP-1-positive bodies containing parasites. FEBS J. 2020, 287, 1777–1797. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, A.; Das, S.; Kumar, A.; Abhishek, K.; Verma, S.; Mandal, A.; Singh, R.K.; Das, P. Leishmania donovani activates hypoxia inducible factor-1α and miR-210 for survival in macrophages by downregulation of NF-κB mediated pro-inflammatory immune respons. Front. Microbiol. 2018, 9, 385. [Google Scholar] [CrossRef]
- Neves, B.M.; Silvestre, R.; Resende, M.; Ouaissi, A.; Cunha, J.; Tavares, J.; Loureiro, I.; Santarém, N.; Silva, A.M.; Lopes, M.C.; et al. Activation of phosphatidylinositol 3-kinase/akt and impairment of nuclear factor-κB: Molecular mechanisms behind the arrested maturation/activation state of leishmania infantum-infected dendritic cells. Am. J. Pathol. 2010, 177, 2898–2911. [Google Scholar] [CrossRef]
- Cleenewerk, L.; Garssen, J.; Hogenkamp, A. Clinical Use of Schistosoma mansoni Antigens as Novel Immunotherapies for Autoimmune Disorders. Front. Immunol. 2020, 11, 1821. [Google Scholar] [CrossRef]
- Tang, C.-L.; Zou, J.-N.; Zhang, R.-H.; Liu, Z.-M.; Mao, C.-L. Helminths protect against type 1 diabetes: Effects and mechanisms. Parasitol. Res. 2019, 118, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Terrazas, C.A.; Alcántara-Hernández, M.; Bonifaz, L.; Terrazas, L.I.; Satoskar, A.R. Helminth-excreted/secreted products are recognized by multiple receptors on DCs to block the TLR response and bias Th2 polarization in a cRAF dependent pathway. FASEB J. 2013, 27, 4547–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Chauhan, A.; Sukumaran, P.; Sharma, J.; Singh, B.B.; Mishra, B.B. Inhibition of store-operated calcium entry in microglia by helminth factors: Implications for immune suppression in neurocysticercosis. J. Neuroinflamm. 2014, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Uddin, J.; Gonzalez, A.E.; Gilman, R.H.; Garcia, H.H.; Verastegui, M.; Moore, L.J.; Evans, C.A.W.; Read, R.C.; Friedland, J.S. Neurocysticercal antigens stimulate chemokine secretion from human monocytes via an NF-κB-dependent pathway. Microbes Infect. 2006, 8, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.; Shahin, A. Induction of interferon-γ by Taenia crassiceps glycans and Lewis sugars in naive BALB/c spleen and peritoneal exudate cells. Mol. Immunol. 2007, 44, 1623–1630. [Google Scholar] [CrossRef]
- Artis, D.; Shapira, S.; Mason, N.; Speirs, K.M.; Goldschmidt, M.; Caamaño, J.; Liou, H.-C.; Hunter, C.A.; Scott, P. Differential Requirement for NF-κB Family Members in Control of Helminth Infection and Intestinal Inflammation. J. Immunol. 2002, 169, 4481–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Zhang, X.; Edwards, J.P.; Mosser, D.M. NF-κB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006, 281, 26041–26050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSchoolmeester, M.L.; Manku, H.; Else, K.J. The innate immune responses of colonic epithelial cells to Trichuris muris are similar in mouse strains that develop a type 1 or type 2 adaptive immune response. Infect. Immun. 2006, 74, 6280–6286. [Google Scholar] [CrossRef] [Green Version]
- Kron, M.A.; Metwali, A.; Vodanovic-Jankovic, S.; Elliott, D. Nematode asparaginyl-tRNA synthetase resolves intestinal inflammation in mice with T-cell transfer colitis. Clin. Vaccine Immunol. 2013, 20, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Mukherjee, S.; Maiti, T.K.; Bhattacharya, S.; Babu, S.P.S. A novel ligand of Toll-like receptor 4 from the sheath of Wuchereria bancrofti Microfilaria induces proinflammatory response in macrophages. J. Infect. Dis. 2017, 215, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Guan, F.; Jiang, W.; Bai, Y.; Hou, X.; Jiang, C.; Zhang, C.; Jacques, M.L.; Liu, W.; Lei, J. Purinergic P2X7 receptor mediates the elimination of Trichinella spiralis by activating NF-κB/NLRP3/IL-1b pathway in macrophages. Infect. Immun. 2021, 89, e00683-20. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yu, J.; Zhang, Z.; Zhai, P.; Zhang, Y.; Meng, S.; Yu, Y.; Li, X.; Song, M. Immunomodulatory effects of Trichinella spiralis excretory-secretory antigens on macrophages. Exp. Parasitol. 2019, 196, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wu, X.; Wang, X.; Guan, Z.; Gao, F.; Yu, J.; Yu, L.; Tang, B.; Liu, X.; Song, Y.; et al. Regulation of cytokine expression in murine macrophages stimulated by excretory/secretory products from Trichinella spiralis in vitro. Mol. Cell. Biochem. 2012, 360, 79–88. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, P.; Ganga, L.; Satoeya, N.; Mishra, S.; Vishwakarma, A.L.; Srivastava, M. Infective larvae of Brugia malayi induce polarization of host macrophages that helps in immune evasion. Front. Immunol. 2018, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, S.; Anuradha, R.; Pavan Kumar, N.P.; George, P.J.; Kumaraswami, V.; Nutman, T.B. Toll-like receptor- and filarial antigen-mediated, mitogen-activated protein kinase- and NF-κB-dependent regulation of angiogenic growth factors in filarial lymphatic pathology. Infect. Immun. 2012, 80, 2509–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruszewska-Cheruiyot, M.; Donskow-Łysoniewska, K.; Piechna, K.; Krawczak, K.; Doligalska, M. L4 stage Heligmosomoides polygyrus prevents the maturation of dendritic JAWS II cells. Exp. Parasitol. 2019, 196, 12–21. [Google Scholar] [CrossRef]
- Doligalska, M.; Brodaczewska, K.; Donskow-Lysoniewska, K. The antiapoptotic activity of Heligmosomoides polygyrus antigen fractions. Parasite Immunol. 2012, 34, 589–603. [Google Scholar] [CrossRef]
- Semnani, R.T.; Liu, A.Y.; Sabzevari, H.; Kubofcik, J.; Zhou, J.; Gilden, J.K.; Nutman, T.B. Brugia malayi Microfilariae Induce Cell Death in Human Dendritic Cells, Inhibit Their Ability to Make IL-12 and IL-10, and Reduce Their Capacity to Activate CD4+ T Cells. J. Immunol. 2003, 171, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Babu, S.; Bhat, S.Q.; Kumar, N.P.; Lipira, A.B.; Kumar, S.; Karthik, C.; Kumaraswami, V.; Nutman, T.B. Filarial lymphedema is characterized by antigen-specific Th1 and Th17 proinflammatory responses and a lack of regulatory T cells. PLoS Negl. Trop. Dis. 2009, 3, e420. [Google Scholar] [CrossRef] [Green Version]
- Fordjour, F.A.; Asiedu, E.; Larbi, A.; Kwarteng, A. The role of nuclear factor kappa B (NF-κB) in filarial pathology. J. Cell Commun. Signal. 2021, 15, 185–193. [Google Scholar] [CrossRef]
- Aurora, A.B.; Biyashev, D.; Mirochnik, Y.; Zaichuk, T.A.; Sánchez-Martinez, C.; Renault, M.A.; Losordo, D.; Volpert, O.V. NF-κB balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood 2010, 116, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, J.P.; Gao, L. Proinflammatory Cytokines Increase Vascular Endothelial Growth Factor Expression in Alveolar Epithelial Cells. Mediat. Inflamm. 2015, 2015, 387842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jothi, D.J.; Dhanraj, M.; Solaiappan, S.; Sivanesan, S.; Kron, M.; Dhanasekaran, A. Brugia malayi asparaginyl—Trna synthetase stimulates endothelial cell proliferation, vasodilation and angiogenesis. PLoS ONE 2016, 11, e0146132. [Google Scholar] [CrossRef] [Green Version]
- Castro, V.N.; Rodrigues, J.L.; Cardoso, D.T.; Resende, S.D.; Magalhães, F.C.; Souza, D.C.; Requeijo, M.H.; Negrão-Corrêa, D.; Geiger, S.M. Systemic Cytokine and Chemokine Profiles in Individuals With Schistosoma mansoni Infection and Low Parasite Burden. Front. Immunol. 2018, 9, 2975. [Google Scholar] [CrossRef]
- Brunet, L.R.; Finkelman, F.D.; Cheever, A.W.; Kopf, M.A.; Pearce, E.J. IL-4 protects against TNF-alpha-mediated cachexia and death during acute schistosomiasis. J. Immunol. 1997, 159, 777–785. [Google Scholar]
- Braz, M.M.; Ramalho, F.S.; Cardoso, R.L.; Zucoloto, S.; Costa, R.S.; Ramalho, L.N.Z. Slight activation of nuclear factor kappa-B is associated with increased hepatic stellate cell apoptosis in human schistosomal fibrosis. Acta Trop. 2010, 113, 66–71. [Google Scholar] [CrossRef]
- Chen, T.T.W.; Cheng, P.C.; Chang, K.C.; Cao, J.P.; Feng, J.L.; Chen, C.C.; Lam, H.Y.P.; Peng, S.Y. Activation of the NLRP3 and AIM2 inflammasomes in a mouse model of Schistosoma mansoni infection. J. Helminthol. 2019, 94, e72. [Google Scholar] [CrossRef]
- Liu, M.; Wu, Q.; Chen, P.; Büchele, B.; Bian, M.; Dong, S.; Huang, D.; Ren, C.; Zhang, Y.; Hou, X.; et al. A Boswellic acid-containing extract ameliorates schistosomiasis liver granuloma and fibrosis through regulating NF-κB signaling in mice. PLoS ONE 2014, 9, e100129. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Jin, F.; Du, Y.; Yang, K.; Yao, L.; Mei, Z.; Huang, W. Genistein improves schistosomiasis liver granuloma and fibrosis via dampening NF-kB signaling in mice. Parasitol. Res. 2017, 116, 1165–1174. [Google Scholar] [CrossRef]
- Hamilton, C.M.; Dowling, D.J.; Loscher, C.E.; Morphew, R.M.; Brophy, P.M.; O’Neill, S.M. The Fasciola hepatica tegumental antigen suppresses dendritic cell maturation and function. Infect. Immun. 2009, 77, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Bąska, P.; Zawistowska-Deniziak, A.; Żdziarska, A.M.; Wasyl, K.; Wiśniewski, M.; Cywińska, A.; Klockiewicz, M.; Januszkiewicz, K.; Wędrychowicz, H. Fasciola hepatica—The pilot study of in vitro assessing immune response against native and recombinant antigens of the fluke. Acta Parasitol. 2013, 58, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bąska, P.; Zawistowska-Deniziak, A.; Norbury, L.J.; Wiśniewski, M.; Januszkiewicz, K. Fasciola hepatica isolates induce different immune responses in unmaturated bovine macrophages. J. Vet. Res. 2019, 63, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roig, J.; Saiz, M.L.; Galiano, A.; Trelis, M.; Cantalapiedra, F.; Monteagudo, C.; Giner, E.; Giner, R.M.; Recio, M.C.; Bernal, D.; et al. Extracellular vesicles from the helminth Fasciola hepatica Prevent DSS-induced acute ulcerative colitis in a T-lymphocyte independent mode. Front. Microbiol. 2018, 9, 1036. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.J.; Hamilton, C.M.; Donnelly, S.; La Course, J.; Brophy, P.M.; Dalton, J.; O’Neill, S.M. Major secretory antigens of the Helminth Fasciola hepatica activate a suppressive dendritic cell phenotype that attenuates Th17 cells but fails to activate Th2 immune responses. Infect. Immun. 2010, 78, 793–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo, V.; Valdés Fernandez, B.N.; Rodríguez-Valentín, M.; Ruiz-Jiménez, C.; Ramos-Benítez, M.J.; Méndez, L.B.; Espino, A.M. Fasciola hepatica GST downregulates NF-κB pathway effectors and inflammatory cytokines while promoting survival in a mouse septic shock model. Sci. Rep. 2019, 9, 2275. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Caban-Hernandez, K.; Figueroa-Santiago, O.; Espino, A.M. Fasciola hepatica fatty acid binding protein inhibits TLR4 activation and suppresses the inflammatory cytokines induced by LPS in vitro and in vivo. Physiol. Behav. 2015, 194, 3924–3936. [Google Scholar] [CrossRef] [Green Version]
- Ashour, D.S.; Shohieb, Z.S.; Sarhan, N.I. Upregulation of Toll-like receptor 2 and nuclear factor-kappa B expression in experimental colonic schistosomiasis. J. Adv. Res. 2015, 6, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Celias, D.P.; Corvo, I.; Silvane, L.; Tort, J.F.; Chiapello, L.S.; Fresno, M.; Arranz, A.; Motrán, C.C.; Cervi, L. Cathepsin L3 from fasciola hepatica induces NLRP3 inflammasome alternative activation in murine dendritic cells. Front. Immunol. 2019, 10, 552. [Google Scholar] [CrossRef]
- Chen, T.T.W.; Wu, L.S.H.; Hsu, P.W.C.; Pang, C.Y.; Lee, K.M.; Cheng, P.C.; Peng, S.Y. Mitochondrial dynamics in the mouse liver infected by Schistosoma mansoni. Acta Trop. 2015, 148, 13–23. [Google Scholar] [CrossRef]
Figure 1.
Schematic model of NF-κB activation through canonical and non-canonical pathways. Canonical activation involves TGF-β activated kinase-1 (TAK1) which phosphorylates Inhibitory Kappa B Kinase β (IKKβ) complexed with IKKα and IKKγ (NEMO). This leads to phosphorylation of the α Inhibitor of κB (IκBα), its detachment from the p56/p50 dimer, ubiquitination, and proteasomal degradation. Released p65/p50 dimer migrates to the nucleus and binds to DNA sequences leading to transcription of appropriate genes. During the noncanonical pathway, NF-κB-inducing kinase (NIK) phosphorylates the IKKα dimer which phosphorylates p100 leading to its disruption and release of the RelB/p52 dimer. The dimer migrates to the nucleus and regulates the transcription of particular genes.
Figure 1.
Schematic model of NF-κB activation through canonical and non-canonical pathways. Canonical activation involves TGF-β activated kinase-1 (TAK1) which phosphorylates Inhibitory Kappa B Kinase β (IKKβ) complexed with IKKα and IKKγ (NEMO). This leads to phosphorylation of the α Inhibitor of κB (IκBα), its detachment from the p56/p50 dimer, ubiquitination, and proteasomal degradation. Released p65/p50 dimer migrates to the nucleus and binds to DNA sequences leading to transcription of appropriate genes. During the noncanonical pathway, NF-κB-inducing kinase (NIK) phosphorylates the IKKα dimer which phosphorylates p100 leading to its disruption and release of the RelB/p52 dimer. The dimer migrates to the nucleus and regulates the transcription of particular genes.

Figure 2.
The impact of Plasmodium spp., Trypanosoma cruzi, Toxoplasma gondii, and Leishmania spp. on NF-κB activity and outcomes. (A) Plasmodium spp. increase NF-κB activity in specific cell populations which is associated with pathology in the brain, inducing cerebral malaria symptoms (apoptosis in brain endothelial cells and intravascular leukocytes), and may facilitate hidden parasite populations in the spleen. Plasmodium spp. also trigger an inflammatory response in monocytes, but patients with decreased NF-κB activity in PBMCs show more severe malaria symptoms. (B) Reduced NF-κB activity facilitates infection of T. cruzi. Enhanced NF-κB activity in heart tissue during T. cruzi infections leads to heart failure. (C) RelB-deprived mice do not survive T. gondii infection. T. gondii deactivate NF-κB signaling, reducing the immune response in macrophages and neutrophils. (D) Leishmania spp. reduce NF-κB activity in infected macrophages and DC, facilitating parasite survival.
Figure 2.
The impact of Plasmodium spp., Trypanosoma cruzi, Toxoplasma gondii, and Leishmania spp. on NF-κB activity and outcomes. (A) Plasmodium spp. increase NF-κB activity in specific cell populations which is associated with pathology in the brain, inducing cerebral malaria symptoms (apoptosis in brain endothelial cells and intravascular leukocytes), and may facilitate hidden parasite populations in the spleen. Plasmodium spp. also trigger an inflammatory response in monocytes, but patients with decreased NF-κB activity in PBMCs show more severe malaria symptoms. (B) Reduced NF-κB activity facilitates infection of T. cruzi. Enhanced NF-κB activity in heart tissue during T. cruzi infections leads to heart failure. (C) RelB-deprived mice do not survive T. gondii infection. T. gondii deactivate NF-κB signaling, reducing the immune response in macrophages and neutrophils. (D) Leishmania spp. reduce NF-κB activity in infected macrophages and DC, facilitating parasite survival.

Figure 3.
The impact of helminths on NF-κB activity and outcomes. (A) B. malayi infection results in decreased NF-κB activity and induces M2 and eventually Mreg macrophages, while patients with lymphatic pathology show increased angiogenesis associated with NF-κB activation. H. polygyrus induces semi-maturation of DCs and induces Th2 and regulatory events through modulation of NF-κB activity. Products released by T. spiralis affect NF-κB activity in LPS-activated macrophages, significantly reducing proinflammatory cytokine production. (B) T. solium larval antigens activate the NF-κB pathway in monocytes inducing chemokine release. M. corti antigens inhibit LPS-induced inflammatory phenotypes in microglia cells via NF-κB modulation. (C) F. hepatica tegumental antigens temporarily prevent LPS-induced NF-κB in DC, suppressing maturation. S. mansoni induces NF-κB activation in human hepatic stellate cells which is associated with liver fibrosis; a similar situation occurs in S. Japonicum-infected mice.
Figure 3.
The impact of helminths on NF-κB activity and outcomes. (A) B. malayi infection results in decreased NF-κB activity and induces M2 and eventually Mreg macrophages, while patients with lymphatic pathology show increased angiogenesis associated with NF-κB activation. H. polygyrus induces semi-maturation of DCs and induces Th2 and regulatory events through modulation of NF-κB activity. Products released by T. spiralis affect NF-κB activity in LPS-activated macrophages, significantly reducing proinflammatory cytokine production. (B) T. solium larval antigens activate the NF-κB pathway in monocytes inducing chemokine release. M. corti antigens inhibit LPS-induced inflammatory phenotypes in microglia cells via NF-κB modulation. (C) F. hepatica tegumental antigens temporarily prevent LPS-induced NF-κB in DC, suppressing maturation. S. mansoni induces NF-κB activation in human hepatic stellate cells which is associated with liver fibrosis; a similar situation occurs in S. Japonicum-infected mice.


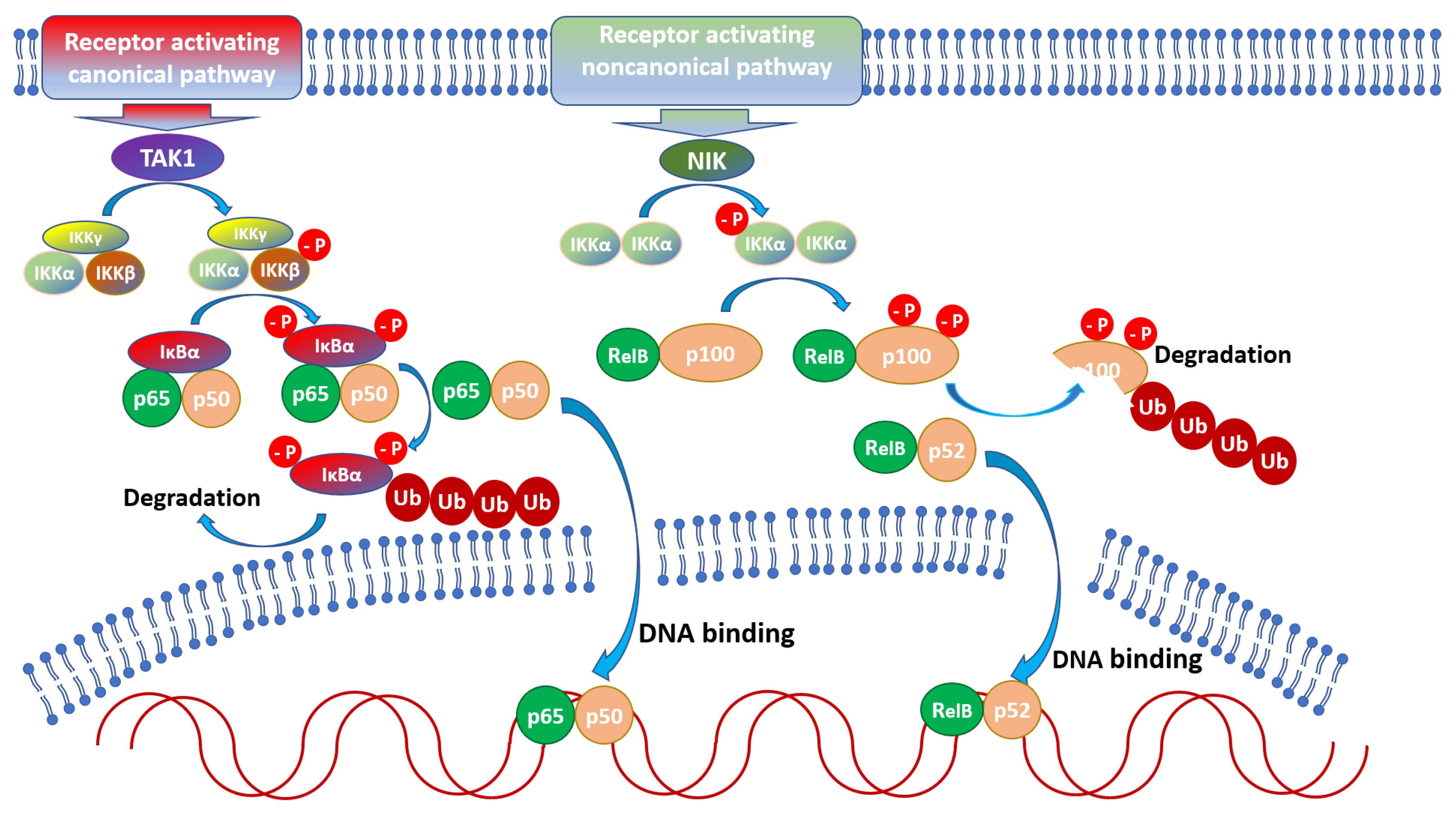{kind=link}
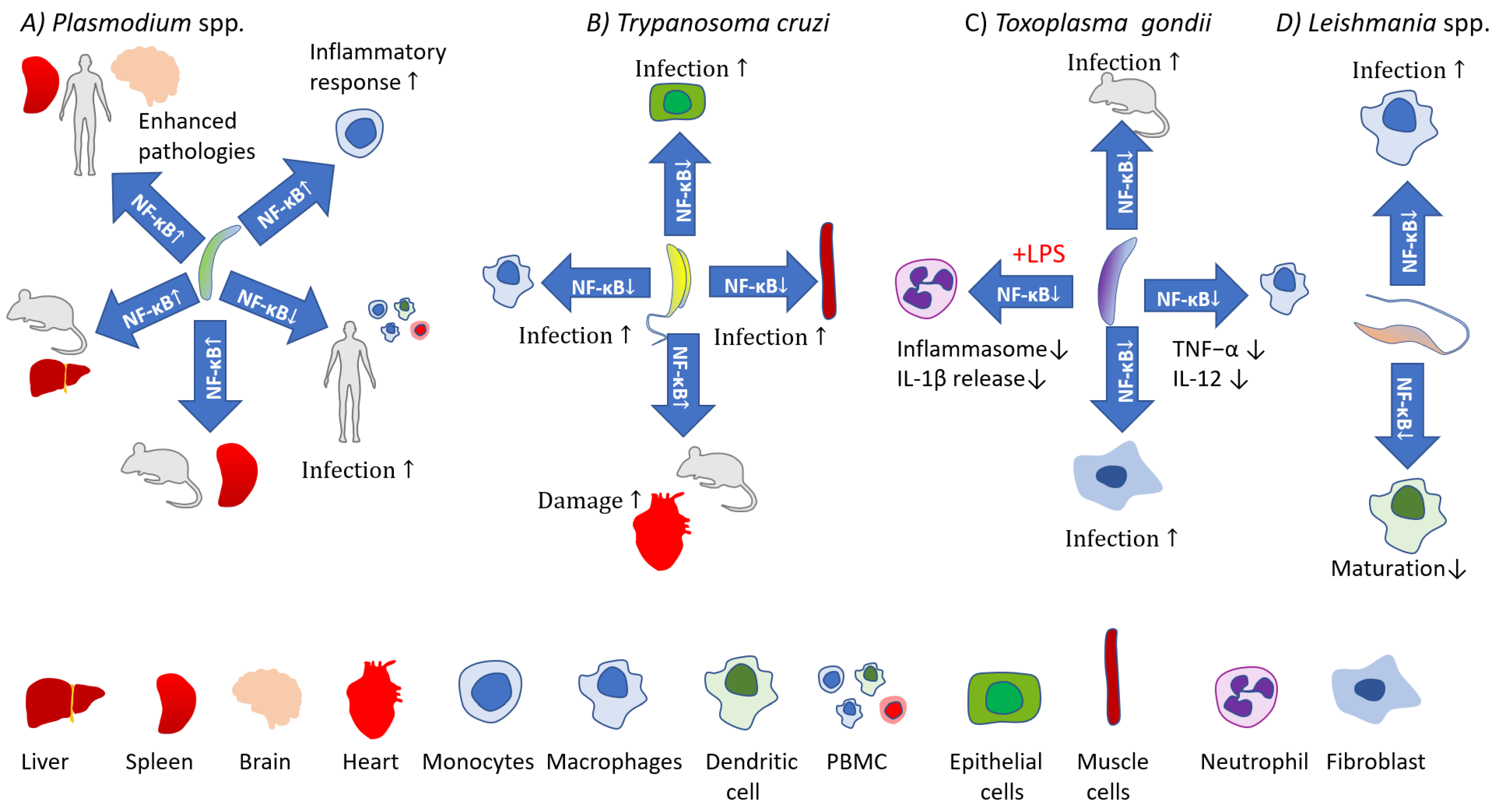{kind=link}
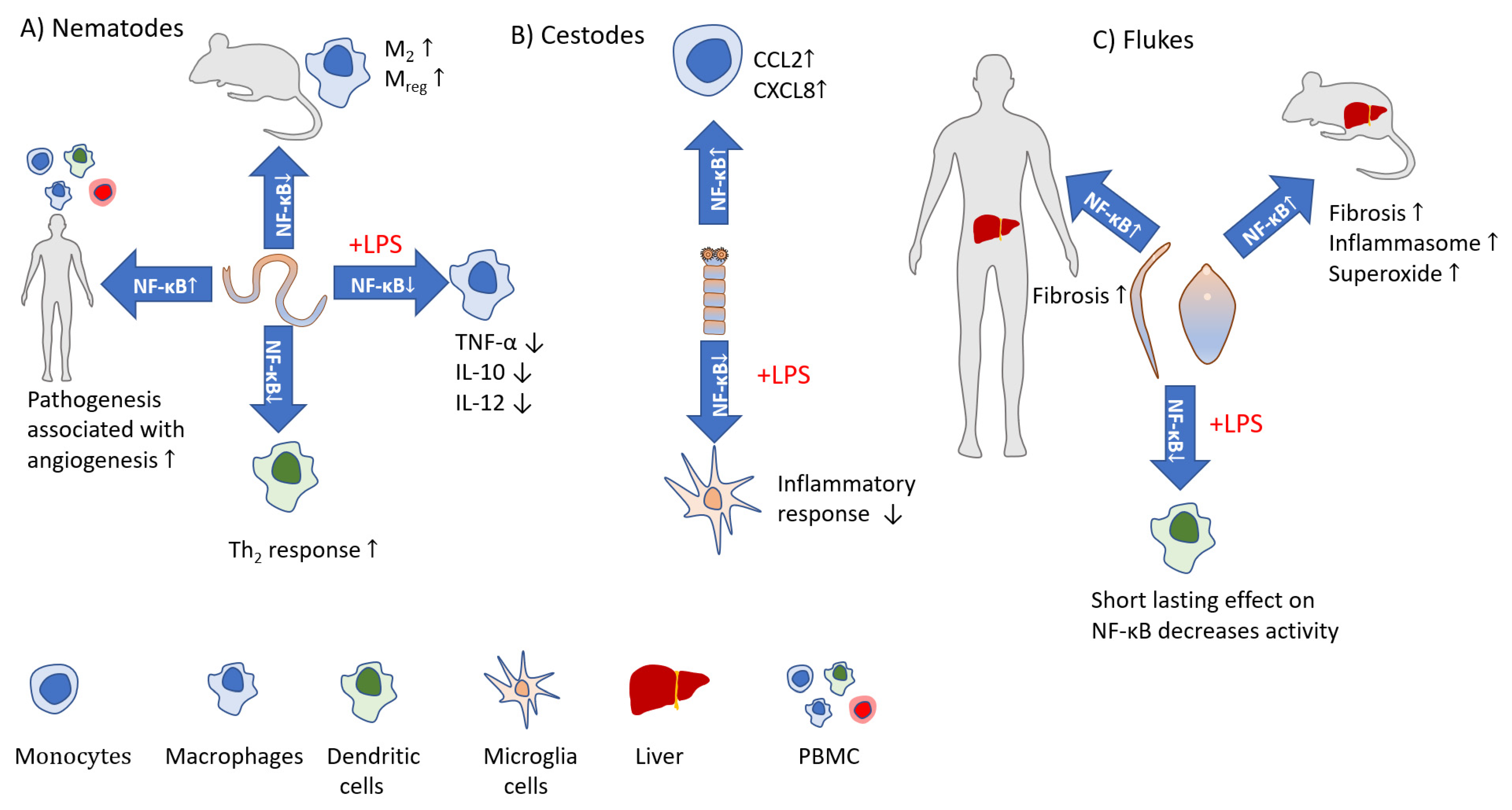{kind=link}
Table 1.
Plasmodium spp. impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| P. falciparum [53] |
| Human monocytes | Translocation of p65, p50 to nucleus of monocytes; degradation of IκBα. | Enhanced activity of MMP-9, enhanced production of proinflammatory cytokines TNF and IL-1β. |
| P. falciparum P. vivax [54] |
| Peripheral blood mononuclear cells | Elevated phospho-NF-κB p65 levels (P. vivax and uncomplicated P. falciparum infection). Reduced phospho-NF-κB p65 levels in complicated P. falciparum, but elevated post-treatment. | Possible association of lower NF-κB features in patients with complicated P. falciparum malaria may be induced by increased IL-10 level during the course of the infection. |
| P. falciparum [46] |
| Brain tissue | Increase in expression of nuclear NF-κB p65 in the brain (neurons, glial cells, ECs, and intravascular leukocytes). | NF-κB p65 levels correlate with histopathological changes (apoptosis) in brain. NF-κB p65 modulates apoptosis in the brain endothelial cells and intravascular leukocytes (but not glial cells, neurons) of fatal cerebral malaria patients. |
| P. vivax [51] |
| Human spleen fibroblasts | Translocation of NF-κB to the nucleus. | Upregulation of ICAM-1 surface expression, facilitating adhesion of infected reticulocytes. |
| P. falciparum [48] |
| Human brain microvascular endothelial cells | Upregulation of NF-κB activation cascade, upregulation of NF-κB subunits (p100, p105, cREL, RELB) and upregulation of NF-κB inhibitory proteins (IκBα, IκBε). Increased p65 translocation to nucleus. | Increased proinflammatory cytokine release (CCL20 and TNF-α). |
| P. falciparum [52] |
| Liver tissue | p65 expression in B cells and Kupffer cells correlates with severity of the disease. | Apoptosis of Kupffer cells and portal tract lymphocytes is related to NF-κB activation. |
| P. falciparum [50] |
| Human brain microvascular endothelial cells | Induction of nuclear translocation of NF-κB (p65). | Increase in ICAM-1 surface expression. |
Table 2.
Trypanosoma spp. impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| T. cruzi [57] |
| Primary human colon epithelial cells | Increase in p65 and IKKα/β phosphorylation, increase in phosphorylation of NF-κB upstream regulators (TAK1 and IRAK4). | Modulated expression of NF-κB signaling molecules, proposed to promote pro-inflammatory signaling pathways. |
| T. cruzi [61] |
| Mouse macrophages | Increased phosporylation of NF-κB p65 (partially dependent on MGL1 receptor). | IL-10, TNF-α, and nitric oxide (NO) production. |
| T. cruzi [68] |
| Thoracic aortic rings from infected C57BL/6 mice | Increase in expression of NF-κB p65. | Increased expression of COX-2 and thromboxane synthase in aortas leading to vascular contraction. |
| T. congolense,T. vivax, T. b. gambiense, T. b. brucei [56] |
| Bovine aortic endothelial cells, in vivo experiments 13 different human and murine endothelial cell lines | Translocation of NF-κB to nucleus, IκBα phosphorylation, NF-κB activation via classical pathway (mediated by parasite trans-sialidase). | Endothelial cell activation, and subsequent pro-inflammatory response–production of cytokines IL-1β and IL-6, nitric oxide and expression of adhesion molecules. |
| T. cruzi [67] |
| Human cardiomyocytes (AC16) | Increased nuclear translocation of p65, enhanced expression of NF-κB dependent genes. | Enhanced mRNA expression of TNF-α and IL-β. |
| T. cruzi [55] |
| Mink lung epithelial cells (Mv1lu), murine endothelial line (SVEC4–10), primary cultures of human fibroblasts, rat myoblasts (L6E9 and H9c2), primary human vascular smooth muscle cells, bovine aortal muscle cells | Nuclear translocation of p65 in epithelial cells, endothelial cells, and fibroblasts, enhanced expression of NF-κB dependent genes. | NF-κB activation by parasites limits infection levels whereas experimental blocking of NF-κB signaling increases parasite burden. |
| T. cruzi [58] |
| Human umbilical vein endothelial cells (HUVEC) | Nuclear translocation of active NF-κB (p65 and p50). | Induction of vascular cell adhesion molecule 1 (VCAM-1) and E-selectin and the upregulation of intercellular adhesion molecule 1 (ICAM-1). |
| T. cruzi [62] |
| J774 macrophages | Proteolytic cleavage of NF-κB p65 by cruzain. | Impairment of macrophage activation pathways (reduced IL-12, increased L-arginase). |
Table 3.
Toxoplasma gondi impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| T. gondii [82] |
| Mouse primary peritoneal macrophages | Phosphorylation of p65 through downregulation of miR-187. | Delayed production of IL-12. |
| T. gondii [87] |
| Human Non-Small Cell Lung Cancer H1299 cells deficient in p53 | GRA16 prevents NF-κB activation; decreased total and nuclear levels of p65, decreased IKKβ level, decreased phosphorylation of IkBα. | Decrease in cell survival, induce cell apoptosis. |
| T. gondii [86] |
| Human neutrophils | Reduction in LPS-induced IκBα degradation and p65 phosphorylation. | Reduction in release of LPS induced IL-1β. |
| T. gondii [88] |
| In vivo: mice. In vitro: bone marrow derived macrophages, RAW 264.7, adherent peritoneal macrophages, HELA cells | Virulent strain resulted in less p65 translocation to the nucleus and IκBα phosphorylation compared to avirulent strain. | Avirulent strains induced increased TNF-α and IL-12 release compared to virulent strains. Avirulent and virulent strain polarized macrophages towards M1 and M2 phenotypes, respectively. |
| T. gondii [89] |
| Human foreskin fibroblasts, NIH 3T3 fibroblasts, MEF, HeLa, and COS cell lines, as well as primary cultures of mouse and human macrophages | Lack of nuclear p65/RelA translocation despite IκB degradation. No increase in expression of NF-κB dependent genes upon infection. | The results show that T. gondi may use undefined mechanisms to interfere with NF-κB signaling. |
| T. gondii [84] |
| Mouse bone marrow-derived macrophages | Inhibition of LPS-induced NF-κB translocation. | Blocked production of proinflammatory TNF-α and delayed (24 h) IL-12 in response to LPS. |
| T. gondii [81] |
| In vivo: mice. In vitro: mouse bone marrow derived macrophages | In vivo: activation of NF-κB, higher expression of p65, p50 from 24 h. In vitro: no p65 and cRel translocation to nucleus. | In vitro: reduced capacity to increase transcription of IL-12, IL-18, and iNOS in response to LPS and IFN-γ. |
| T. gondii [90] |
| 3T3 mouse embryonic fibroblasts, 3T3 p65−/− fibroblasts | nuclear translocation of p50 and p65. Higher affinity to DNA for p50, p52, p65, and RelB. Phosphorylation but no IκB degradation. | Upregulation of antiapoptotic responses. |
| T. gondii [91] |
| Human foreskin fibroblast, RAW264.7 (mouse macrophage cell line), U937 (human macrophage cell line) | ROP18+ strain induced p65 phosphorylation at Ser468 and promotes its degradation. | ROP18+: reduced LPS-induced IL-6, IL-12, and TNF-α; M2-biased phenotypes. ROP18-: enhanced LPS-induced IL-6, IL-12, and TNF-α; M1-biased activation. This strain has a relative inability to inhibit the NF-κB pathway. |
| T. gondii [92] |
| Mouse bone marrow-derived macrophages | No change in LPS induced p65 accumulation in nucleus as well as NF-kB binding to DNA. Significant diminished ability of p65 to bind to TNF-α promoter. | Describes T. gondi’s ability to interfere with TNF-α transcription. |
| T. gondii [74] |
| Primary human monocytes and THP-1 cells | Type II: increased p65 accumulation in nucleus. | Type II: expression of IL-1β. |
| T. gondii [85] |
| Primary human peripheral blood monocytes | Increase in phosphorylation of p65. | Expression of IL-1β. |
| T. gondii [80] |
| Wild type and RelB−/− C57B6 mice. | Infection induces NF-κB DNA binding activities of p65 and RelB containing complexes in the spleen. | RelB−/− mice show high mortality in response to the infection with negligible levels of IFN-γ and diminished NK cell activity. |
| T. gondii [83] |
| In vivo: Mice (injected intra peritoneally with tachyzoites). In vitro: mouse peritoneal macrophages macrophage cell lines (RAW 264.7 and THP-1) | In vivo: no NF-κB translocation in macrophages or neutrophils within 4h of infection. In vitro: rapid IκB phosphorylation and degradation but NF-κB p50-p65 heterodimers did not translocate to the nucleus. | In vitro: little or no production of IL-12 and TNF-α; LPS triggering unable to promote IL-12 and TNF-α production. |
Table 4.
Leishmania spp. impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| L. major [105] |
| Promonocytic human cell line U937 and fresh human peripheral blood monocytes | Inhibition of DNA binding activity of p50/p65 heterodimer. Induction of p50/p50 and p50/c-Rel heterodimer. | Increase in IL-10 and TNF-α secretion. |
| L. amazonensis [107] |
| murine immature bone marrow-derived DCs | Expression of p65 and RelB. Down regulation of genes encoding for activators (IL-1 receptors, TIRAP, MYD88, TIFA, EIF2AK2, USP7), upregulation of genes encoding inhibitors of the NF-κB pathway (OPTN, TNFAIP3, TAX1BP1, PTPN1, USP10). | Alternative NF-κB pathway is favored, likely promoting MHC I-restricted antigen presentation. Amastigote infection does not change maturation status whereas antibody opsonized amastigotes induce semi-mature DC phenotype. |
| L. braziliensis, L. infantum (WT and modified), L. amazonensis [108] |
| Murine peritoneal Macrophages, THP-1 macrophages | Only L. amazonensis EV were able to induce p65 translocation. | Only L. amazonensis EV induced NO, TNF-α, IL-6, and IL-10 via TLR4 and TLR2. |
| L. aethiopica [109] |
| Human THP-1 cells | Reduction of total p65 protein, lower expression of total Iκ-Bα protein, decrease in IkBα phosphorylation (Ser 32/36), no p65 cleavage to p35. | NF-κB signal pathway downregulation proposed to promote induction of apoptosis to facilitate spreading. |
| L. donavani [110] |
| Mouse macrophages and mouse macrophages with inhibited miR-210 expression | During the infection p50 and p65 expression relatively unchanged in cytoplasm and nucleus. During the infection and miR-210 inhibition increased p50/p65 translocation to nucleus. | Infection: marginally elevated TNF-α, IL-12, and IL-10. miR-210 inhibition during infection: TNF-α, IL-12 more elevated, IL-10 less elevated, increased O2- and Nox. |
| L. major [106] |
| BM derived mouse macrophages | Infection did not inhibit NF-κB activation: no impact on p65 translocation, no impact on LPS-induced IκBα degradation, no activation of NF-κB dependent gene expression. | Infection inhibits LPS-induced IL-12 (not NF-κB related) |
| L. infantum [111] |
| Mouse bone marrow-derived DCs | Cleavage of p65 to p35, IκB-α phosphorylation and degradation unchanged. | Slightly increased expression of IL-12, unchanged IL-10, IL-6, TNF-α expression. Reduced LPS-induced IL-12 and IL-6, increased IL-10, TNF-α unchanged. |
| L. donovani, L. major, L. mexicana, L. (Viannia) braziliensis,L. tarentolae [98] |
| B10R macrophage, primary bone marrow-derived macrophages, mouse and human macrophage cell lines Raw264.7, J774 and THP-1 | Excluding non-pathogenic L. tarentolae: p65 cleaved into p35, p35 translocation to nucleus, nuclear p35/p50. | Induction of chemokine gene expression (MIP-2/CXCL2, MCP-1/CCL2, MIP-1a/CCL3, MIP-1b/CCL4). |
Table 5.
Cestodes impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| Taenia crassiceps [114] |
| Mouse BMDCc | Tc-ES did not induce p65 phosphorylation (Ser536), but attenuated LPS-induced p65 phosphorylation (Ser536). | Inhibition of DC maturation, cytokine production, and the ability of LPS-treated DCs to prime Th1 responses. |
| Taenia solium [116] |
| Human primary monocytes | Increased degradation of IkB-α but not IkBβ; increased DNA binding by p65, p50, and cRel. | Induced CCL2, CXCL8, and CCL3 secretion. |
| Mesocestoides corti [115] |
| Mouse microglia cells | Decrease in LPS-induced p65 phosphorylation and acetylation. | Inhibition of LPS-induced secretion of IL-6 and TNF-α cytokines. |
| Taenia crassiceps [117] |
| Mouse spleen cells | p65 level increased, p50 level was not affected. | Release of IFN-γ from spleen cells stimulated with N glycans upon stimulation with Taenia glycans; Lewis X sugars upregulated p65 expression and had no impact on p50 expression. |
Table 6.
Nematodes impact on NF-κB activity and outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| Brugia malayi [121] |
| Colitic mice | Changes in NF-κB signaling pathway. | Mitigation of colitis symptoms. |
| Wuchereria bancrofti [122] |
| Mouse macrophages (RAW 264.7) HEK293-TLR4 cells | Increased NF-κB phosphorylation Enhanced expression of NF-κB dependent gens. | Wb-MfP induces proinflammatory response through TLR4. |
| Trichuris muris [120] |
| Colon epithelial cell line (CMT93) | Dynamic increase in p65 phosphorylation. Increase at 4 h post-exposure, back to baseline level by 24 h. | Increased release of proinflammatory factors IFN-γ, TNF, and CCL2. |
| Trichuris muris [118] |
| B6 mice and NF-κB1 KO, NF-κB2 KO, c-Rel KO mice | Enhanced activity of NF-κB upon infection in B6 mice. | c-Rel KO mice: expelled worms and lymph node cells released low level of IFN-γ. NF-κB1 KO and NF-κB2 KO mice: unable to terminate infection, NF-κB1 KO mice lymph node cells released high levels of IFN-γ, and mice developed intestinal immunopathology. |
| Trichinella spiralis [123] |
| Human macrophage cell line (U937) | Downregulation of NF-κB following P2X7R blockade. | Inhibits NLRP3 inflammasome activation and decreases macrophage capacity to kill T. spiralis larvae. |
| Trichinella spiralis [124] |
| Mouse macrophages (RAW264.7) | Reduced LPS-induced p65 expression levels. | Increased regulatory IL-10, reduced LPS-induced proinflammatory (TNF-α and IL-12) and regulatory cytokines (IL-10). |
| Trichinella spiralis [125] |
| Macrophage cell line (J774A.1) | Reduced LPS-induced p65 nuclear translocation. | Increased levels of anti-inflammatory cytokines (IL-10, TGF-β) and arginase 1, and (ML) iNOS. Reduced LPS-induced expression of proinflammatory (TNF-α, IL-1β, IL-6, IL-12) cytokines and (Ad3+Ad5+NBL) iNOS. |
| Brugia malayi [126] |
| BALB/c mice | Expression downregulation for p65 and p50/105 in M2 macrophages and dramatic decrease in p65 and p50/105 in Mreg. | Alternatively activated (M2) and regulatory (Mreg) macrophages appeared 3 and 7 dpi, respectively. |
| Brugia malayi [127] |
| TLR ligands | Use of NF-κB inhibitor. | Diminished production of Angiopoetin-I and VEGF-A in response to TLR2 ligand in the presence of NF-κB inhibitor. |
| Heligmosomoides polygyrus [128] |
| Immature DC line (JAWS II) | Decreased translocation of p50 NF-κB into the nucleus. | Relative lack of DC activation: TNF-α, TGF-β, IL-6, MCP-1 unchanged, decreased expression of regulatory cytokine (IL-10) and proinflammatory factor (IL-12p70), increased expression of IL-4 (Th2 type). |
| Heligmosomoides polygyrus [129] |
| MLN cells from naive and infected mice | Various changes in p65 and p50 levels in cytoplasm and nucleus nucleus. | Inhibition of apoptosis. |
Table 7.
Flukes impact on NF-κB activity and the main outcomes.
| Parasite | Infection/Life Stage/Antigen | Host/Location/Model | Effect on NF-κB | Outcome |
|---|---|---|---|---|
| Schistosoma mansoni [138] |
| Liver biopsy | Increased p65 presence in hepatic stellate cells’ (HSC) cytoplasm and enhanced translocation to nucleus in infected patients. | Increase in apoptotic HSC number in schistosome-induced fibrosis. |
| Schistosoma mansoni [139] |
| Mice | Increase of p65 expression in liver. | May promote inflammasome activation. |
| S. mansoni [149] |
| Mice | Increased p65 (Ser 276) phosphorylation in colonic tissue. | NF-κB shows significant role during the course of schistosomiasis. |
| Fasciola hepatica [145] |
| Colitic mice | Diminished p65 expression in response to Fh-EV in colitic mice. | Protective effect of Fh-EV: amelioration of pathological colitis symptoms and reduced the amount of pro-inflammatory cytokines. |
| Fasciola hepatica [142] |
| Mouse bone marrow-derived DCs | Suppressed LPS–induced expression of p65. | Fh-ES suppressed LPS–induced IL-12p70, IL-10 and TNF-α release; modulated LPS-stimulated DC cytokine production and cell surface marker expression. |
| Fasciola hepatica [147] |
| Mouse bone-marrow derived macrophages; Mice with induced septic shock, THP-1 cells | Blocked TLRs from inducing NF-κB activation. | Reduced expression of LPS induced mRNA encoding IL-1β and TNF-α and release of IL-1β, IL-12p70, IL-6, IFN-γ, TNF-α and IL-2. |
| Fasciola hepatica [148] |
| Mouse bone-marrow derived macrophages; Mice with induced septic shock, THP-1 cells, HEK293 cells | Blocked LPS-induced NF-κB activation. | Reduced mRNA expression encoding IL-1β, IL-12p35, IL-12p40, TNF-α, IL-6, NOS-2 Reduced release of GM-CSF, IL-12p70, IL-3, IL-9, IL-10, IL-15, TNF-α and IFN-γ. |
| Fasciola hepatica [146] |
| Mouse DC and HEK293 cells | rFh-GST-si, (but not rFh-CL1) increased p65 phosphorylation and NF-κB activation. | Induction of IL-6, IL-12p40, and MIP-2, but also prevented IL-17 and IL-23 release. |
| Fasciola hepatica [150] |
| Mouse bone marrow-derived DCs | No change in p65 expression and IκB-α. | Increased release of IL-1β and IL-18 by NLRP3 dependent mechanism. |
| Schistosoma mansoni [151] |
| Liver cells | Increase in p65 expression and translocation to nucleus. | Possible role of p65 in inhibiting apoptosis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bąska, P.; Norbury, L.J. The Role of Nuclear Factor Kappa B (NF-κB) in the Immune Response against Parasites. Pathogens 2022, 11, 310. https://doi.org/10.3390/pathogens11030310
AMA Style
Bąska P, Norbury LJ. The Role of Nuclear Factor Kappa B (NF-κB) in the Immune Response against Parasites. Pathogens. 2022; 11(3):310. https://doi.org/10.3390/pathogens11030310
Chicago/Turabian StyleBąska, Piotr, and Luke J. Norbury. 2022. "The Role of Nuclear Factor Kappa B (NF-κB) in the Immune Response against Parasites" Pathogens 11, no. 3: 310. https://doi.org/10.3390/pathogens11030310
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.