

Analysis of Hop Stunt Viroid Diversity in Grapevine (Vitis vinifera L.) in Slovakia: Coexistence of Two Particular Genetic Groups
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Grapevine Samples
2.2. High-Throughput Sequencing and Analysis
2.3. RT-PCR Detection, Sanger Sequencing, and Phylogenetic Analysis
3. Results and Discussion
3.1. Identification of HSVd in the Grapevine Virome
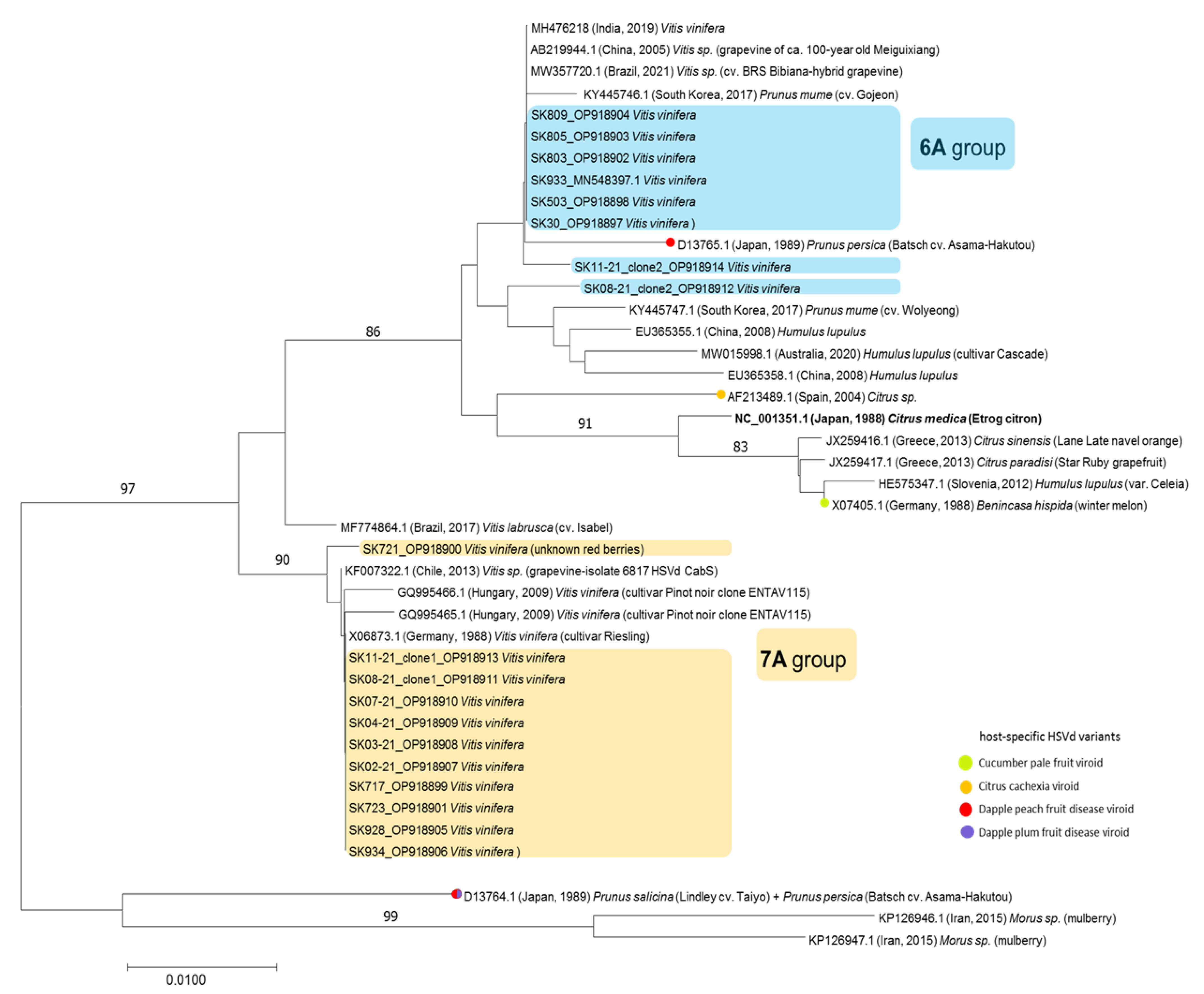
3.2. Identification of Two Molecularly Different HSVd Groups
3.3. Mixed Infection of HSVd Variants
3.4. Group-Defining Mutations vs. RNA Secondary Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flores, R.; Daròs, J.-A.; Hernández, C.; Navarro, B.; Di Serio, F. Viroids. eLS 2020, 1, 192–203. [Google Scholar] [CrossRef]
- Diener, T.O. Potato spindle tuber ‘‘virus’’. IV. A replicating, low-molecular weight RNA. Virology 1971, 45, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Diener, T.O. Discovering viroids—A personal perspective. Nat. Rev. Microbiol. 2003, 1, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, K.; Adkar-Purushothama, C.R.; Tassios, E.; Samiotaki, M.; Andronis, C.; Lisón, P.; Nikolaou, C.; Perreault, J.-P.; Kalantidis, K. Revisiting the Non-Coding Nature of Pospiviroids. Cells 2022, 11, 265. [Google Scholar] [CrossRef]
- Hammond, R.W.; Owens, R.A. Viroids: New and Continuing Risks for Horticultural and Agricultural Crops; APS: St. Paul. MN, USA, 2006. [Google Scholar] [CrossRef]
- Silletti, M.R.; Navarro, B.; Bozzano, G.; Trisciuzzi, V.N.; Di Serio, F. The PSTVd viroid, a threat to tomatoes and potatoes. Informatore Agrario 2009, 65, 89–90. [Google Scholar]
- Hadidi, A.; Vidalakis, G.; Sano, T. Chapter 2—Economic Significance of Fruit Tree and Grapevine Viroids. In Viroids and Satellites; Hadidi, A., Flores, R., Randles, J.W., Palukaitis, P., Eds.; Academic Press-Elsevier: Cambridge, MA, USA, 2017; pp. 15–25. ISBN 978-0-12-801498-1. [Google Scholar]
- Hammond, R.W. Economic significance of viroids in vegetable and field crops. In Viroids and Satellites; Hadidi, A., Flores, R., Randles, J.W., Palukaitis, P., Eds.; Academic Press-Elsevier: Cambridge, MA, USA, 2017; pp. 5–13. ISBN 978-0-12-801498-1. [Google Scholar]
- Mackie, A.E.; Barbetti, M.J.; Rodoni, B.; McKirdy, S.J.; Jones, R.A.C. Effects of a Potato Spindle Tuber Viroid Tomato Strain on the Symptoms, Biomass, and Yields of Classical Indicator and Currently Grown Potato and Tomato Cultivars. Plant Dis. 2019, 103, 3009–3017. [Google Scholar] [CrossRef]
- Takeda, R.; Petrov, A.I.; Leontis, N.B.; Ding, B. A Three-Dimensional RNA Motif in Potato spindle tuber viroid Mediates Trafficking from Palisade Mesophyll to Spongy Mesophyll in Nicotiana benthamiana. Plant Cell 2011, 23, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Archual, A.J.; Amin, A.A.; Ding, B. A Genomic Map of Viroid RNA Motifs Critical for Replication and Systemic Trafficking. Plant Cell 2008, 20, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Flores, R.; Gas, M.-E.; Molina-Serrano, D.; Nohales, M.-Á.; Carbonell, A.; Gago, S.; De la Peña, M.; Daròs, J.-A. Viroid Replication: Rolling-Circles, Enzymes and Ribozymes. Viruses 2009, 1, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qu, J.; Ji, S.; Wallace, A.J.; Wu, J.; Li, Y.; Gopalan, V.; Ding, B. A Land Plant-Specific Transcription Factor Directly Enhances Transcription of a Pathogenic Noncoding RNA Template by DNA-Dependent RNA Polymerase II. Plant Cell 2016, 28, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Mudiyanselage, S.D.D.; Qu, J.; Tian, N.; Jiang, J.; Wang, Y. Potato Spindle Tuber Viroid RNA-Templated Transcription: Factors and Regulation. Viruses 2018, 10, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.; Daròs, J.A.; Hernández, C. Avsunviroidae family: Viroids containing hammerhead ribozymes. Adv. Virus Res. 2000, 55, 271–323. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kagami, Y.; Kurokawa, M.; Nishimura, S.; Ukawa, S.; Kubo, S. Studies on hop stunt disease in Japan. Rep. Res. Lab. Kirin Brew. 1973, 16, 49–62. [Google Scholar]
- Sasaki, M.; Shikata, E. On Some Properties of Hop Stunt Disease Agent, a Viroid. Proc. Jpn. Acad. Ser. B 1977, 53, 109–112. [Google Scholar] [CrossRef] [Green Version]
- van Dorst, H.J.M.; Peters, D. Some biological observations on pale fruit, a viroid-incited disease of cucumber. Neth. J. Plant Pathol. 1974, 80, 85–96. [Google Scholar] [CrossRef]
- Sano, T.; Uyeda, I.; Shikata, E.; Ohno, T.; Okada, Y. Nucleotide sequence of cucumber pale fruit viroid: Homology to hop stunt viroid. Nucleic Acids Res. 1984, 12, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Semancik, J.S.; Roistacher, C.N.; Rivera-Bustamante, R.; Duran-Vila, N. Citrus Cachexia Viroid, a New Viroid of Citrus: Relationship to Viroids of the Exocortis Disease Complex. J. Gen. Virol. 1988, 69, 3059–3068. [Google Scholar] [CrossRef]
- Reanwarakorn, K.; Semancik, J.S. Correlation of Hop Stunt Viroid Variants to Cachexia and Xyloporosis Disease of Citrus. Phytopathology 1999, 89, 568–574. [Google Scholar] [CrossRef] [Green Version]
- Hataya, T.; Tsushima, T.; Sano, T. Hop stunt viroid. In Viroids and Satellites; Hadidi, A., Flores, R., Randles, J., Palukaitis, P., Eds.; Academic Press-Elsevier: Cambridge, MA, USA, 2017; pp. 199–210. ISBN 978-0-12-801498-1. [Google Scholar]
- Vamenani, R.; Rahimian, H.; Alavi, S.M.; Parizi, A.P.; Razzaz, T.M. Genetic diversity of hops stunt viroid from symptomatic and asymptomatic citrus trees in Iran. J. Phytopathol. 2019, 167, 484–489. [Google Scholar] [CrossRef]
- Sano, T.; Hataya, T.; Terai, Y.; Shikata, E. Hop stunt viroid strains from dapple fruit disease of plum and peach in Japan. J. Gen. Virol. 1989, 70, 1311–1319. [Google Scholar] [CrossRef]
- Astruc, N.; Marcos, J.F.; Macquaire, G.; Candresse, T.; Pallás, V. Studies on the diagnosis of hop stunt viroid in fruit trees: Identification of new hosts and application of a nucleic acid extraction procedure based on non-organic solvents. Eur. J. Plant Pathol. 1996, 102, 837–846. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.W.; Zhu, D.Z.; Zong, X.J.; Wei, H.R.; Chen, X.; Hammond, R.W.; Liu, Q.Z. First Report of Hop stunt viroid From Sweet Cherry With Dapple Fruit Symptoms in China. Plant Dis. 2017, 101, 394. [Google Scholar] [CrossRef]
- Marquez-Molins, J.; Gomez, G.; Pallas, V. Hop stunt viroid: A polyphagous pathogenic RNA that has shed light on viroid-host interactions. Mol. Plant Pathol. 2021, 22, 153–162. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Wu, M.-L.; Shen, T.-L.; Yeh, H.-H.; Hung, T.-H. Multiplex detection, distribution, and genetic diversity of Hop stunt viroid and Citrus exocortis viroid infecting citrus in Taiwan. Virology J. 2015, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Di Serio, F.; Ambrós, S.; Sano, T.; Flores, R.; Navarro, B. Viroid Diseases in Pome and Stone Fruit Trees and Koch’s Postulates: A Critical Assessment. Viruses 2018, 10, 612. [Google Scholar] [CrossRef] [Green Version]
- Maddahian, M.; Massumi, H.; Heydarnejad, J.; Hosseinipour, A.; Khezri, A.; Sano, T. Biological and molecular characterization of hop stunt viroid variants from pistachio trees in Iran. J. Phytopathol. 2019, 167, 163–173. [Google Scholar] [CrossRef]
- Sano, T.; Mimura, R.; Ohshima, K. Phylogenetic analysis of hop and grapevine isolates of hop stunt viroid supports a grapevine origin for hop stunt disease. Virus Genes 2001, 22, 53–59. [Google Scholar] [CrossRef]
- Kawaguchi-Ito, Y.; Li, S.-F.; Tagawa, M.; Araki, H.; Goshono, M.; Yamamoto, S.; Tanaka, M.; Narita, M.; Tanaka, K.; Liu, S.-X.; et al. Cultivated Grapevines Represent a Symptomless Reservoir for the Transmission of Hop Stunt Viroid to Hop Crops: 15 Years of Evolutionary Analysis. PLoS ONE 2009, 4, e8386. [Google Scholar] [CrossRef]
- Vozárová, Z.; Sihelská, N.; Predajňa, L.; Šoltys, K.; Glasa, M. First report of Grapevine yellow speckle viroid-1 infecting grapevine in Slovakia. J. Plant Pathol. 2016, 98, 697. [Google Scholar] [CrossRef]
- Glasa, M.; Predajňa, L.; Šoltys, K.; Sabanadzovic, S.; Olmos, A. Detection and molecular characterisation of Grapevine Syrah virus-1 isolates from Central Europe. Virus Genes 2015, 51, 112–121. [Google Scholar] [CrossRef]
- Glasa, M.; Predajňa, L.; Sihelská, N.; Šoltys, K.; Ruiz-García, A.B.; Olmos, A.; Wetzel, T.; Sabanadzovic, S. Grapevine virus T is relatively widespread in Slovakia and Czech Republic and genetically diverse. Virus Genes 2018, 54, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Glasa, M.; Predajňa, L.; Komínek, P.; Nagyová, A.; Candresse, T.; Olmos, A. Molecular characterization of divergent grapevine Pinot gris virus isolates and their detection in Slovak and Czech grapevines. Arch. Virol. 2014, 159, 2103–2107. [Google Scholar] [CrossRef] [PubMed]
- Glasa, M.; Predajňa, L.; Šoltys, K.; Sihelská, N.; Nagyová, A.; Wetzel, T.; Sabanadzovic, S. Analysis of Grapevine rupestris stem pitting-associated virus (GRSPaV) in Slovakia reveals differences in intra-host population diversity and naturally occurring recombination events. Plant Pathol. J. 2017, 33, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasa, M.; Predajňa, L.; Sihelská, N.; Šoltys, K.; Ruiz-García, A.B. Analysis of Virome by High-Throughput Sequencing Revealed Multiple Infection and Intra-Virus Diversity in a Single Grapevine Plant. Acta Hortic. Regiotect. 2020, 23, 35–39. [Google Scholar] [CrossRef]
- Vončina, D.; Almeida, R.P.P. Screening of some Croatian autochthonous grapevine varieties reveals a multitude of viruses, including novel ones. Arch. Virol. 2018, 163, 2239–2243. [Google Scholar] [CrossRef] [Green Version]
- Meshi, T.; Ishikawa, M.; Watanabe, Y.; Yamaya, J.; Okada, Y.; Sano, T.; Shikata, E. The sequence necessary for the infectivity of hop stunt viroid cDNA clones. Mol. Gen. Genet. 1985, 200, 199–206. [Google Scholar] [CrossRef]
- López-Carrasco, A.; Flores, R. Dissecting the secondary structure of the circular RNA of a nuclear viroid in vivo: A “naked” rod-like conformation similar but not identical to that observed in vitro. RNA Biol. 2017, 14, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Mühlbach, H.-P.; Sänger, H.L. Viroid replication is inhibited by α-amanitin. Nature 1979, 278, 185–188. [Google Scholar] [CrossRef]
- Rackwitz, H.R.; Rohde, W.; Sänger, H.L. DNA-dependent RNA polymerase II of plant origin transcribes viroid RNA into full-length copies. Nature 1981, 291, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Schindler, I.M.; Mühlbach, H.P. Involvement of nuclear DNA-dependent RNA polymerases in potato spindle tuber viroid replication: A reevaluation. Plant Sci. 1992, 84, 221–229. [Google Scholar] [CrossRef]
- Jiang, J.; Smith, H.N.; Ren, D.; Mudiyanselage, S.D.D.; Dawe, A.L.; Wang, L.; Wang, Y. Potato Spindle Tuber Viroid Modulates Its Replication through a Direct Interaction with a Splicing Regulator. J. Virol. 2018, 92, e01004-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Li, S.; Hou, W.; Fan, Z.; Xiao, H.; Lu, M.; Sano, T.; Zhang, Z. Global Transcriptomic Changes Induced by Infection of Cucumber (Cucumis sativus L.) with Mild and Severe Variants of Hop Stunt Viroid. Front. Microbiol. 2017, 8, 2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassenegger, M.; Spieker, R.L.; Thalmeir, S.; Gast, F.U.; Riedel, L.; Sänger, H.L. A single nucleotide substitution converts potato spindle tuber viroid (PSTVd) from a noninfectious to an infectious RNA for nicotiana tabacum. Virology 1996, 226, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.; Gago, S.; Duran-Vila, N. A single nucleotide change in Hop stunt viroid modulates citrus cachexia symptoms. Virus Res. 2008, 138, 130–134. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Sample | Host | Locality | Year of Sampling | GenBank Accession Number |
|---|---|---|---|---|
| SK30 * | V. vinifera, cv. Veltliner | Pezinok | 2012 | OP918897 |
| SK503 * | V. vinifera, cv. Dornfelder | Šenkvice | 2015 | OP918898 |
| SK933 * | V. vinifera, unknown white berries | Pezinok | 2017 | MN548397 |
| SK717 | V. vinifera, unknown white berries | Pezinok | 2016 | OP918899 |
| SK721 | V. vinifera, unknown red berries | Pezinok | 2016 | OP918900 |
| SK723 | V. vinifera, cv. Muller-Thurgau | Pezinok | 2016 | OP918901 |
| SK803 | V. vinifera, cv. Palava | Pezinok | 2017 | OP918902 |
| SK805 | V. vinifera, cv. Veltliner | Pezinok | 2017 | OP918903 |
| SK809 | V. vinifera, cv. Welshriesling | Pezinok | 2017 | OP918904 |
| SK928 | V. vinifera, cv. Silvaner | Limbach | 2017 | OP918905 |
| SK934 | V. vinifera, cv. Dornfelder | Cífer | 2017 | OP918906 |
| SK02-21 | V. vinifera, unknown red berries | Pezinok | 2021 | OP918907 |
| SK03-21 | V. vinifera, unknown red berries | Pezinok | 2021 | OP918908 |
| SK04-21 | V. vinifera, cv. Traminer | Pezinok | 2021 | OP918909 |
| SK07-21 | V. vinifera, unknown white berries | Pezinok | 2021 | OP918910 |
| SK08-21 | V. vinifera, cv. Veltliner | Pezinok | 2021 | OP918911 (clone 1) OP918912 (clone 2) |
| SK11-21 | V. vinifera, cv. Muller-Thurgau | Pezinok | 2021 | OP918913 (clone 1) OP918914 (clone 2) |
| Nucleotide Position * | 6A Group Nucleotide/Number of Sequences/% | 7A Group Nucleotide/Number of Sequences/% |
|---|---|---|
| 26 | C/151/62.66 | A/90/37.34 |
| 32 | A/156/64.20 | U/85/35.00 ** |
| 44/45 | Δ/158/65.56 | A/83/34.44 |
| 46/47 | A/162/66.94 | U/80/33.06 |
| 47/48 | A/185/77.40 | U/53/22.18 *** |
| 256/257 | U/170/70.83 | G/70/29.17 |
| 257/258 | G/166/69.17 | U/74/30.83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaxin, P.; Predajňa, L.; Achs, A.; Šubr, Z.; Mrkvová, M.; Glasa, M. Analysis of Hop Stunt Viroid Diversity in Grapevine (Vitis vinifera L.) in Slovakia: Coexistence of Two Particular Genetic Groups. Pathogens 2023, 12, 205. https://doi.org/10.3390/pathogens12020205
Alaxin P, Predajňa L, Achs A, Šubr Z, Mrkvová M, Glasa M. Analysis of Hop Stunt Viroid Diversity in Grapevine (Vitis vinifera L.) in Slovakia: Coexistence of Two Particular Genetic Groups. Pathogens. 2023; 12(2):205. https://doi.org/10.3390/pathogens12020205
Chicago/Turabian StyleAlaxin, Peter, Lukáš Predajňa, Adam Achs, Zdeno Šubr, Michaela Mrkvová, and Miroslav Glasa. 2023. "Analysis of Hop Stunt Viroid Diversity in Grapevine (Vitis vinifera L.) in Slovakia: Coexistence of Two Particular Genetic Groups" Pathogens 12, no. 2: 205. https://doi.org/10.3390/pathogens12020205