1. Introduction
Proteus mirabilis (PM) is a Gram-negative bacterium first described by Gustav Hauser in 1885. He named this genus of Proteobacteria after the Greek god Proteus due to the bacteria’s changeability in form as it alternates between an immobile and a mobile, so-called “swarming” condition [
1].
PM possesses the enzyme urease that splits urea in urine into carbon dioxide (CO
2) and ammonia (NH
3), which results in a rise in urinary pH [
2]. Consequently, salts precipitate as a result of reduced solubility, resulting in the crystallization and subsequent crystal formation of (MgNH
3PO
4) and apatite (CaPO
4) and eventual catheter encrustation and struvite stone formation [
3]. Biofilm formation and catheter encrustation, both hallmarks of
PM-induced catheter-associated urinary tract infections (CAUTIs), are financially and physically burdensome outcomes of urinary catheter use [
4]. The first step in
PM pathogenicity in the urinary tract is colonization, as the microorganism can attach to epithelial cells with various fimbriae and adhesins and potentially internalize within these cells [
5]. Various virulence factors of
PM have been described: fimbriae are outer-membrane pili that extend from the bacterial surface and usually facilitate adherence to surfaces, of which the chaperone-usher fimbriae are linear, unbranching, outer-membrane pili produced by Gram-negative bacteria that mediate their form of secretion and assembly [
6,
7].
PM HI4320 encodes 17 chaperone-usher fimbrial operons, and these fimbriae are regarded as key factors in adhesion and bacterial aggregation for
PM in vivo [
8,
9]. To date, not much is known regarding the role of specific cell surface components in the adhesion to and invasion of uroepithelial cells and the overall pathogenicity of
PM [
10]. To begin addressing this gap in knowledge, we made use of transposon and Targetron mutants of genes involved in the synthesis of
PM cell surface components, generated through previous studies, to identify
PM HI4320 fitness factors, to assess their role in PM pathogenicity with specific emphasis on key adhesion and invasion processes [
11]. These include transposon mutagenesis of
ucaJ,
fim14C,
PMI3191,
arnA,
pbpC, and
waaE.
Urothelial cell adhesin (UCA) frimbriae are adhesion proteins that have been observed to play a critical role in the attachment to the urothelium and are related to the K99 fimbriae of enterotoxigenic
E. coli [
12]. MrpJ is a transcriptional regulator encoded at the 3′ end of the mrp fimbrial operon in
PM. Overexpression of MrpJ decreases flagellum-mediated motility in
PM in order to regulate the opposing processes of motility and adhesion. UcaJ is one of 14 paralogues of MrpJ found in
PM and functions as an inhibitor of motility when attachment is indicated [
13].
Fim14C (PMI2998) is the major pilin subunit of a putative chaperone usher fimbrial operon, and
fim8F (PMI1464) is part of another putative chaperone usher fimbrial operon [
14,
15].
PMI3191 encodes for a glycosyltransferase that has been shown to contribute to
PM lipopolysaccharide (LPS) [
16].
ArnA (PMI1045) is the name of a gene that encodes for the bifunctional polymyxin resistance protein ArnA that physiologically catalyzes the oxidative decarboxylation of UDP-glucuronic acid to UDP-4-keto-arabinose, which is important in LPS formation [
8]. This protein is also a significant contributor to bacterial resistance to antimicrobial peptides, as well as resistance to antibiotics, including polymyxin [
17]. The penicillin-insensitive transglycosylase gene
pbpC (PMI1850) encodes for a protein that is involved in the pathway peptidoglycan biosynthesis with regard to cell wall formation. The penicillin-binding protein 1C (pbpC) is one of several in
PM HI4320 and represents a conserved feature across Gram-negative bacteria [
18,
19]. The gene
waaE encodes for an LPS core biosynthesis glycosyl transferase, an enzyme required for LPS biosynthesis. This outer membrane component of Gram-negative bacteria is critical for antimicrobial peptide resistance, as well as swarming motility [
20,
21].
Collectively, the different genes being targeted are involved in the production of cell surface components with different functionalities, providing a broader overview of bacterial surface structures critical for PM adhesion and invasion of bladder epithelial cells and the overall infectious process. The identification of these key components represents a critical first step towards the development of targeted therapies to block key steps in the pathogenicity of PM and prevent associated morbidity and mortality.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
The studies were carried out using wildtype
PM HI4320, a clinical isolate from the urine of a female nursing home patient with a long-term indwelling catheter (≥30 days), [
22,
23] along with 6 isogenic mutants of this same strain (PMI0532, PMI1045, PMI2998, PMI3191, PMI1850, PMI1464) (
Table 1) generated as part of a previous study [
24]. Briefly, the affected gene was disrupted by the insertion of a transposon carrying the kanamycin resistance cassette. A seventh isogenic mutant in
waaE (PMI3136) was generated for this study via the TargeTron method: a kanamycin resistance cassette was inserted into the gene of interest, following the Sigma TargeTron group II intron protocol, as previously described [
24]. In addition, two complemented strains (
cPMI3191 and
cwaaE) were generated. Briefly, complementation vectors were constructed by amplifying the indicated gene along with approximately 500 bp of flanking sequence and ligating the amplified sequences into a linearized pGEN-MCS vector. Successful complementation was verified by selection on ampicillin plates and by PCR.
For bacterial culture, bacterial strains were separately cultured from freezer stocks (−80 °C) in fresh Luria broth (LB) media (10 g/L tryptone, 5 g/L yeast extract, 0.5 g/L NaCl) ± kanamycin or ampicillin. Strains were incubated in a shaking incubator at 225 RPM and 37 °C overnight. The following day, all cultures were sub-cultured in fresh media (LB, DMEM, or RPMI depending on the experiment) and left for a subsequent overnight subculture under the same conditions prior to use for experiments.
2.2. Catheter Adhesion Studies
To assess the potential for
PM WT and mutants to interact with the catheter surface and form biofilm over time, an adhesion assay using 24G polyurethane (PU) catheters to be used for eventual in vivo analyses was performed in artificial urine.
PM wildtype and the 9 mutant and complemented strains were separately cultured from freezer stocks in fresh LB media. Strains were incubated at 37 °C overnight. The following day, all cultures were sub-cultured in fresh LB media and left overnight in the same conditions. Bacterial stock solutions of approximately 1 × 10
8 CFU/mL were prepared in artificial urine. The artificial urine was prepared as described by Brooks and Keevil [
25].
Sterile catheter pieces were placed inside sterile Eppendorf tubes containing artificial urine and one of the 10 PM strains, and tubes were placed on a rotator and placed inside an incubator for either 4 or 24 h at 37 °C. At each timepoint, catheters were removed and washed gently in sterile PBS to remove any loosely adhered bacteria. Catheters from each strain were transferred to tubes of fresh PBS, sonicated, vortexed, serially diluted, and plated on LB agar for CFU counts. Agar plates were incubated overnight at 37 °C until bacterial colonies were visible, and distinguishable CFU counts between 2 to 50 colonies were documented.
2.3. Adhesion and Invasion Assays
For adhesion and invasion assays, three different human cell lines were used, including the kidney epithelial cell lines A498 and HEK293 and the human bladder epithelial cells line T24. Cells were cultured from liquid nitrogen freezer stocks. A498 and HEK293 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco™, ThermoFisher, Whaltam, MA, USA) and T24 cells with Roswell Park Memorial Institute (RPMI; Gibco™, ThermoFisher, Whaltam, MA, USA) 1640 media, all supplemented with 10% fetal bovine serum (FBS). Once cells had stabilized in their growing patterns and were 80% confluent, they were detached using Trypsin/EDTA(Gibco™, ThermoFisher, Whaltam, MA, USA). Cells were counted using an automatic cell counter and 1 × 104–1 × 105 cells were seeded in each well of a 24-well tissue culture plate. Cells were incubated at 37 °C and 5% CO2 and allowed to grow until 80% confluent. On the day of the experiments, two of the wells were trypsinized to quantify the number of cells per well and the average cell number used to determine the number of bacteria to be added to each well. Cell media were removed, and all cells were gently washed thrice with sterile PBS. Next, each of the bacterial strains to be tested was added at a multiplicity of infection (MOI) of 1:100. The plates were gently centrifuged at 500× g for 5 min to bring bacteria in close proximity to the epithelial cells without forcing them to directly interact. Plates were then incubated at 37 °C and 5% CO2 for either 2 h or 24 h.
To assess bacterial adhesion, bacterial cell media were removed after 2 h of incubation, and the monolayers were washed thrice gently with sterile PBS to remove any loosely adherent bacteria. Cells were then lysed using 0.1% Triton-X solution for 15 min at room temperature. The resultant cell lysates were then serially diluted in sterile PBS and plated on LB agar plates followed by overnight incubation at 37 °C and CFU counting the following day. Counts between 2 and 50 visible colonies were recorded and used for bacterial quantification.
To assess bacterial invasion, the media were removed and cells were washed thrice with sterile PBS at the 2 h and 24 h timepoints. Cells were then incubated in fresh media containing 1% Penicillin-Streptomycin for an additional 4 h at 37 °C and 5% CO2 to kill adherent bacteria. Next, cells were washed with PBS and lysed with 0.1% Triton-X at room temperature for 15 min. Internalized bacteria were quantified via CFU counts as described above.
2.4. LPS Gel Electrophoresis
To verify the complementation of genes affecting LPS synthesis, we extracted bacterial LPS via hot aqueous-phenol extraction followed by separation by SDS-PAGE (15%) (
Supplementary Figure S1).
PM WT and mutants were grown in 5 mL LB and incubated at 37 °C overnight. The culture was diluted 1:10 with LB, and a 1.5 ml suspension was created to an OD
600 of 0.5. The bacteria were centrifuged at 10,600×
g for 10 min, the supernatant was removed, and the LPS extraction was started immediately. Bacterial LPS was extracted via hot aqueous-phenol extraction according to Davies et al. [
26]. LPS was separated by SDS-PAGE run on a 15% SDS-polyacrylamide gel.
2.5. In Vivo CAUTI Model
2.5.1. Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Animal Care Committee of The University of British Columbia (Protocol A17-0297).
2.5.2. Catheter Modification
Under aseptic conditions, 24G angiocatheters (Terumo Surflash® Polyurethane I.V. Catheter 24 Gauge x 3/4”, Cat. No. SRFF2419, Terumo, Vaughn, Canada) were temporarily removed from the needles, and a 4 mm piece was cut off using a sterile blade. The angiocatheter and small piece were then re-assembled back onto the needle tip. Prior to being used for the in vivo experiments, all catheters were gas-sterilized with ethylene oxide to further ensure sterility and avoid any contamination.
2.5.3. Mouse Experiments
Mice used in these experiments were male C57Bl/6 mice (Harlan
®) at 12 weeks of age. Mice underwent inhalational anesthesia with 3% isoflurane. Under ultrasound guidance, (Vevo 770
® High-Resolution Imaging System, VisualSonics, Bothell, WA, USA) a 4 mm piece of a modified 24G catheter was implanted into the mouse bladder [
27]. The day following catheter implantation, all mice underwent anesthesia as described above, and a bacterial solution containing 5 × 10
5 CFU/mL in 50 μL PBS of one of the strains to be tested was percutaneously injected into the mouse bladder lumen using a 30G needle. Following bacterial injection, all mice were maintained under anesthesia at 1% isoflurane for an additional hour to promote bacterial attachment to the catheter and epithelium and to avoid premature micturition. On day 3, animals were anesthetized as above and urine was collected using a 30G needle under ultrasound guidance for subsequent quantification of planktonic bacteria via CFU counts, as described above. Subsequently, all mice were euthanized on day 4. All catheters were removed from the mouse bladders and aseptically transferred to tubes containing sterile PBS. After gentle washing with PBS, catheter samples were sonicated at 50/60 Hz for 10 min in an ultrasonic water bath (No. 21811-820, VWR
®) followed by vortexing for 30 s at high speed to promote detachment of bacteria from the catheter. The PBS-bacterial solutions were then serially diluted and plated on LB agar plates for CFU counts, as outlined above.
2.6. Statistical Analysis
Significance was assessed using one-way analysis of variance (ANOVA), a nonparametric Kruskal–Wallis test, and the Wilcoxon signed-rank test. p-values were two-tailed at a 95% confidence interval. The analysis was performed with GraphPad Prism (version 8) software (GraphPad Software, San Diego, CA, USA).
4. Discussion
Proteus mirabilis is a notoriously strong biofilm former and is therefore a frequent cause of urinary tract infections in individuals with indwelling foreign bodies within the urinary tract, such as bladder catheters, ureteral stents or nephrostomy tubes, and often leads to the encrustation of devices and/or infection stones [
28]. To date, not much is known regarding the role of specific bacterial surface components in
PM pathogenicity, especially the adherence to indwelling devices leading to biofilm formation and subsequent adhesion and invasion of uroepithelial cells. To address this gap in knowledge, the present work investigates the role of different bacterial components in mediating these key processes involved in
PM pathogenicity using both in vitro and in vivo settings.
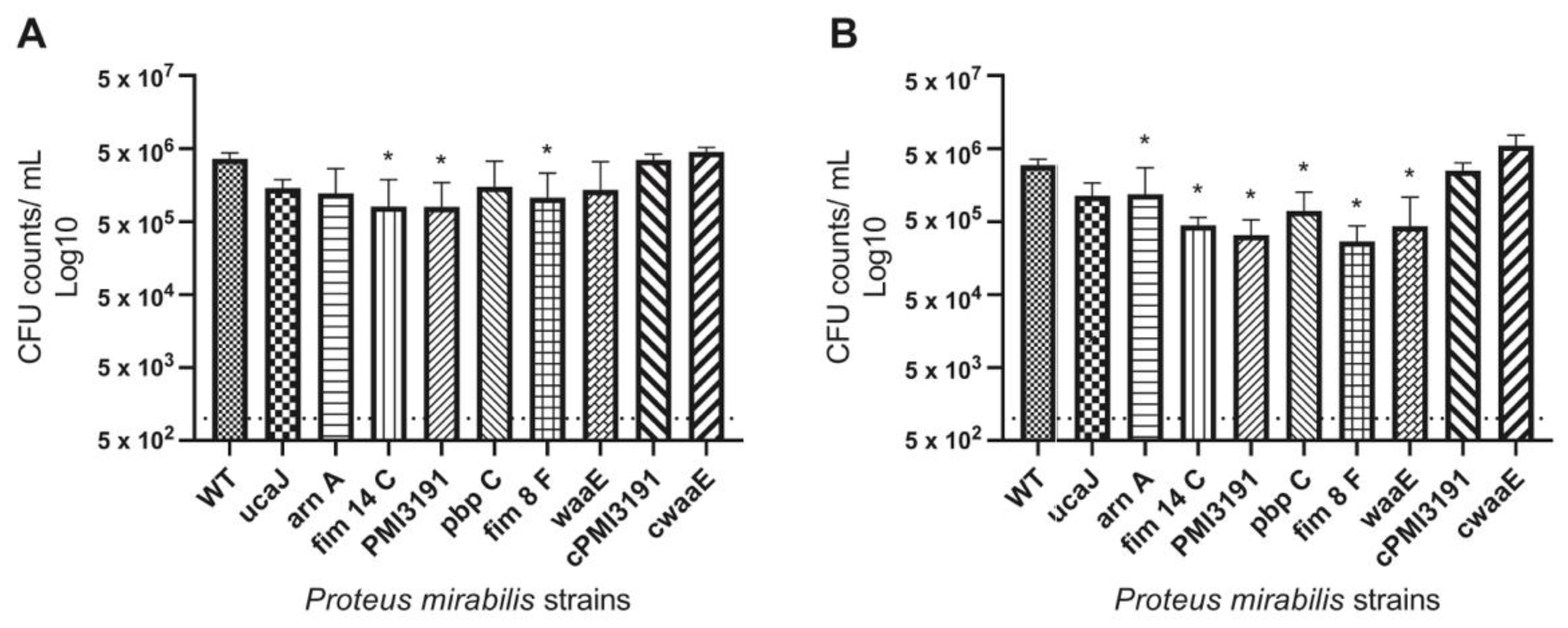
All mutants were found to have decreased adhesion to PU catheters. Considering that these experiments were performed in artificial urine, which has limited proteins, adhesion in this setting is mainly driven by charge and hydrophobicity characteristics rather than the direct interaction of adhesins with proteins deposited on the material surface [
29]. Interestingly, the level of adhesion to the catheter surface was similar for WT and mutant strains at both the 4 h and 24 h timepoints. While one would expect greater adhesion levels of a given strain at a later timepoint, this may not be the case in the present study due to the charge-driven adhesion taking place in this environment, plateauing within the first few hours and not changing significantly as the bacteria transitioned from initial adhesion to colonization in the first 24 h. It is unlikely that the observed pattern is due to the loss of viability in planktonic bacteria at the later timepoint due to a higher pH, as
P. mirabilis has been shown to prefer a higher pH to undergo adhesion processes. Furthermore, bacteria within a biofilm remain alive for at least several days in a higher-pH environment [
30]. A similar pattern of adhesion to the same type of catheter was also observed in vivo, with the exception of the
PMI3191 and
waaE mutants, which adhered to a lesser extent than the other mutants. Interestingly, lower numbers were also recovered from the urines of animals infected with these two mutants despite similar inoculation numbers, suggesting that lower adhesion may be the result of fewer bacteria being retained in the urinary tracts of these animals. That being said, reduced adhesiveness and bacterial numbers of these mutants in urine did not translate to the bladder tissue, as both adhesion and invasion levels were similar to the remaining mutants. This may suggest that despite lower numbers being retained in the urine, these mutants were more efficient at adhering to the bladder tissue. The fact that complementation of
PMI3191 and
waaE resulted in adhesion and invasion levels similar to the WT strain suggests that changes in these key pathogenic steps were driven by changes in LPS structure, as these genes express LPS biosynthesis genes, and at least knocking out
waaE resulted in significant changes in LPS banding patterns that were resolved by complementation.
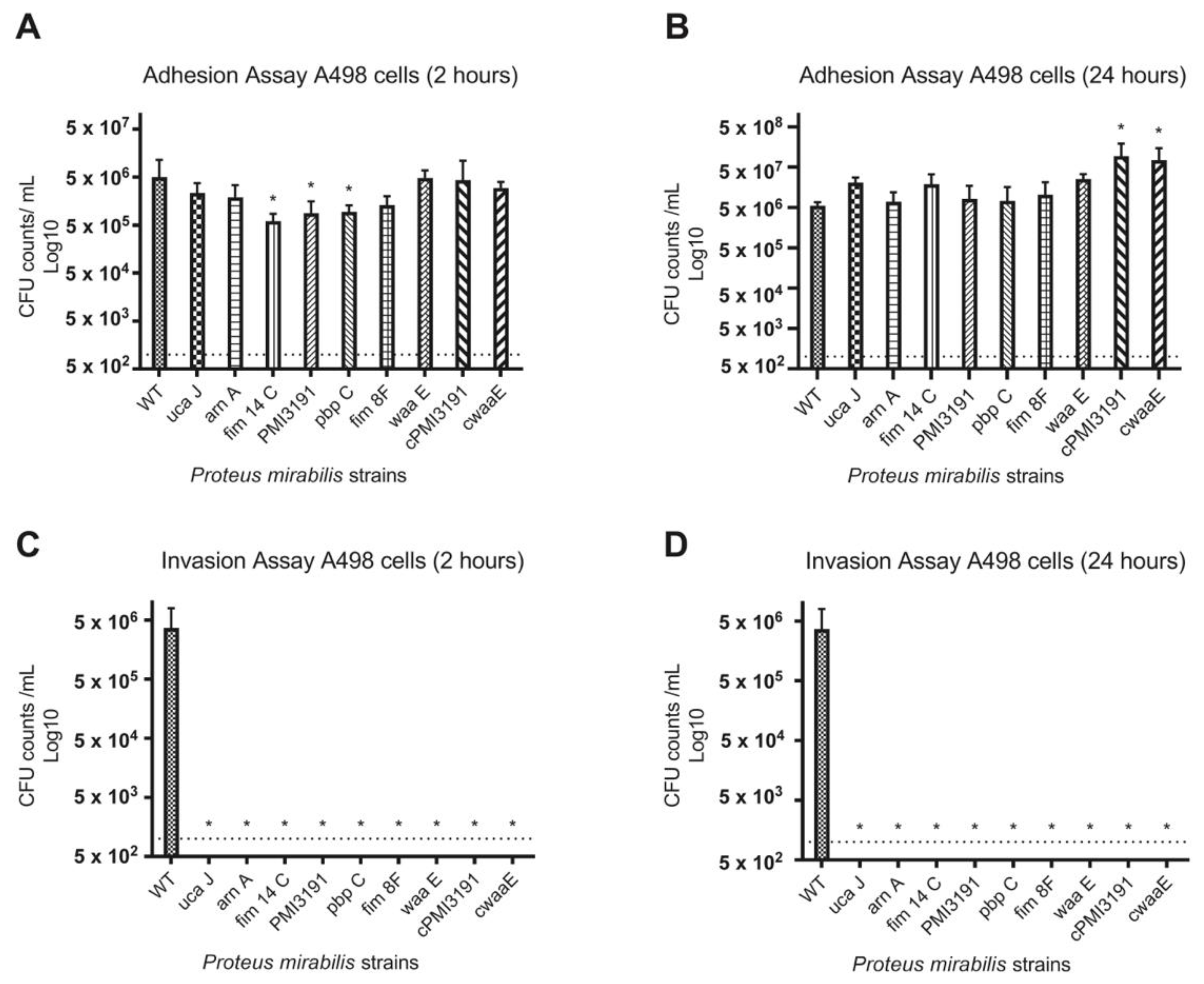
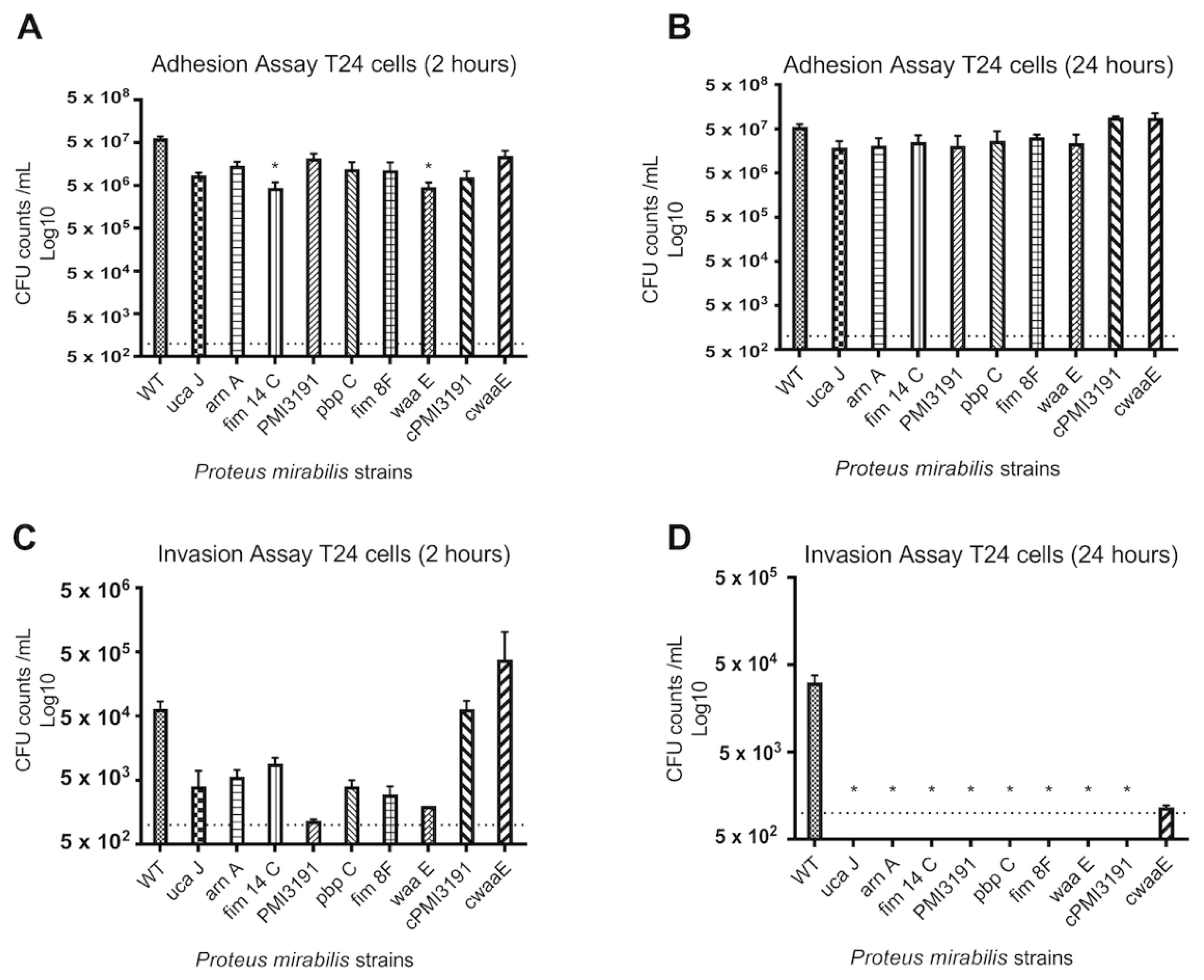
None of the mutants were found invading the bladder tissue when the animals were sacrificed 48 h post-infection. To better understand the adhesion and invasion patterns of WT and mutant strains, these key processes were assessed in three different cell lines representing the bladder and kidney. Overall, we found adhesion and invasion patterns to differ between the different cell types. While adhesion was somewhat affected for some mutants at the 2 h timepoint for all cell types, no differences were observed at 24 h post-infection. In contrast, none of the mutants were found to invade A498 cells at either timepoint, while some initial invasion was evident for HEK293 cells that were lost by 24 h. A similar pattern was also observed for T24 bladder cells, which suggests that a lack of invasion in the mice may be due to an inability of the mutants to invade bladder cells. In terms of the role for LPS structure in these processes, complementation of
PMI3191 and
waaE resulted in adhesion levels that were greater than the corresponding mutant and, in some cases, similar to WT. The fact that complementation of
PMI3191 and
waaE resulted in the rescue of the invasion phenotype for both HEK293 and T24 cells suggests that the LPS structure plays a role in mediating the invasion of uroepithelial cells by
PM. Changes in LPS glycosylation such as those expected from the loss of a glycosyltransferase (i.e., PMI3191) have previously been shown to affect the invasiveness of
Shigella flexneri, with strains with increased glycosylation showing enhanced invasion [
31]. Similarly, alterations of the LPS inner core were shown to significantly affect the invasiveness of uropathogenic
E. coli, which may explain the loss of invasion of the
waaE mutant [
32]. That said, this same role was not observed in A498 cells, as complementation of
PM3191 and
waaE did not rescue the invasiveness in this cell type, suggesting that a role for
PM LPS in invasion may be cell-dependent. This certainly requires further investigation.
Fimbriae have been the subject of continued investigation in
PM virulence, and their crucial role in infection pathogenicity by aiding adhesion and aggregation in vivo has been demonstrated [
9]. Fimbriae-deficient strains have shown significantly decreased infectivity when compared to the wildtype, with decreased kidney and bladder colonization and adherence [
33]. These findings are in accordance with our results, as
ucaJ, fim8F, and
fim14c mutants showed substantially lower CFU counts in the adhesion assays at both timepoints in the majority of experiments.
UcaJ is a uroepithelial cell adhesin (UCA) mutant, and UCA fimbriae have been identified to play a critical role in the attachment of
PM to epithelial cells of the urinary tract [
12]. Pellegrino et al. observed that an
ucaA mutant demonstrated diminished adhesion to the urothelium, compared to wildtype [
34]. Their results suggested that the urothelial cell adhesin of
PM may be a crucial factor in kidney colonization, as well as the onset and propagation of CAUTI.
ArnA is a bifunctional polymyxin resistance protein that is required for the modification of lipid A in LPS, and this modification leads to bacterial resistance to cationic antimicrobial peptides of the host immune system, as well as resistance to certain antibiotics, such as polymyxin [
17]. These proteins have seen to be conserved across many different Gram-negative bacteria, including
E. coli and
Pseudomonas aeruginosa [
17]. Another study looking at the disruption of the
arnA gene in a
PM CAUTI mouse model specifically indicated that it leads to a decrease in bacterial fitness in the urinary tract, as well as the spleen, which may be indicative of bacteremia [
11]. CFU counts of
arnA mutant bacteria that adhered to kidney epithelial cells were comparable to those of WT, but there were substantially lower CFU counts in the bladder cells when compared to WT, indicating that
arnA plays a role in the interaction of
PM with the uroepithelium.
Penicillin-binding proteins (PBPs) are high-molecular-weight proteins that are attributed to crucial steps in peptidoglycan synthesis of Gram-negative bacteria [
18].
PM has been identified to possess PBPs, and the
pbpC mutant strain used in this study lacks enzymatic abilities associated with peptidoglycan synthesis [
19]. Interestingly the
pbpC mutant showed the same patterns in the adhesion and invasion assays as mutants in fimbriae, suggesting that changes in peptidoglycan structure play a role in these important processes.
Collectively, the present work provides a broad overview of bacterial surface structures critical for different critical steps in the pathogenicity and the overall infectious process of PM, including the adhesion to indwelling medical devices, as well as the adhesion and invasion of urinary tissue cells and the tissue itself. These data suggest a role for all surface components studied in these key processes. Considering that no mutant resulted in the complete loss of tissue adhesion and invasion indicates that these processes likely involve the dynamic interplay between multiple components. Future experiments should focus on utilizing PM strains with mutations on multiple key components to determine whether targeting more than one results in the loss of infection. To that end, the data presented herein represent a critical first step towards the development of targeted therapies to block key steps in the pathogenicity of PM and prevent associated morbidity and mortality.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}