
Mechanism of Immune Evasion in Mosquito-Borne Diseases
by
 and
and
Swagato Bhattacharjee
1,
Debanjan Ghosh
2,
Rounak Saha
3,
Rima Sarkar
1,
Saurav Kumar
1,
Manoj Khokhar
4 and
Rajan Kumar Pandey
5,*
1
DBT Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram 695014, India
2
Department of Biotechnology, Pondicherry University, Puducherry 605014, India
3
Department of Biochemistry and Molecular Biology, Pondicherry University, Puducherry 605014, India
4
Department of Biochemistry, AIIMS, Jodhpur 342005, India
5
Department of Medical Biochemistry and Biophysics, Karolinska Institute, 171 77 Solna, Sweden
*
Author to whom correspondence should be addressed.
Pathogens 2023, 12(5), 635; https://doi.org/10.3390/pathogens12050635
Submission received: 28 January 2023
/
Revised: 20 April 2023
/
Accepted: 21 April 2023
/
Published: 23 April 2023
(This article belongs to the Special Issue Advances in Mosquito-Borne Pathogens and Diseases)
Abstract
:In recent decades, mosquito-borne illnesses have emerged as a major health burden in many tropical regions. These diseases, such as malaria, dengue fever, chikungunya, yellow fever, Zika virus infection, Rift Valley fever, Japanese encephalitis, and West Nile virus infection, are transmitted through the bite of infected mosquitoes. These pathogens have been shown to interfere with the host’s immune system through adaptive and innate immune mechanisms, as well as the human circulatory system. Crucial immune checkpoints such as antigen presentation, T cell activation, differentiation, and proinflammatory response play a vital role in the host cell’s response to pathogenic infection. Furthermore, these immune evasions have the potential to stimulate the human immune system, resulting in other associated non-communicable diseases. This review aims to advance our understanding of mosquito-borne diseases and the immune evasion mechanisms by associated pathogens. Moreover, it highlights the adverse outcomes of mosquito-borne disease.
Keywords:
mosquito-borne diseases; malaria; dengue; Zika; chikungunya; West Nile fever; Rift Valley fever; virus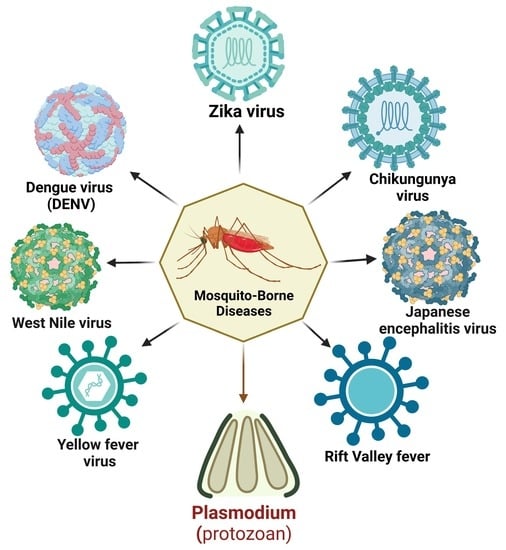
1. Introduction
Vector-borne diseases are a major public health concern worldwide, accounting for a substantial proportion of the global disease burden. As per a World Health Organization (WHO) report, nearly 700,000 deaths are caused annually by vector-borne contagious diseases [1]. Mosquitoes are a potential vector for disease transmission and can disproportionately impact impoverished communities, particularly children. Regardless of acquiring centuries of control strategies, mosquito-borne diseases are burgeoning. These conditions are responsible for enormous mortality and morbidity worldwide [2] (Figure 1).
Among mosquito-borne diseases, malaria is a potentially fatal disease caused by Plasmodium parasites and transmitted by hitherto infected female anopheles mosquito bites. When the vector mosquito feeds on an infected person’s blood, Plasmodium parasites get ingested along with the blood meal. Subsequent bites to a healthy person may transmit the parasite, which leads to malaria. Its symptoms begin with the first liver schizont rupture and merozoite release in the peripheral circulation [3]. Five species of the malarial parasite have been reported, Plasmodium falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi, leading to clinical symptoms in humans. P. falciparum is the most lethal species associated with the majority of malaria-related deaths worldwide. Apart from its ability to impair neuro-cognitive functions, this species is known to develop resistance to anti-malarial drugs. P. vivax, the most widely distributed species, is responsible for a significant proportion of malaria cases worldwide. Other Plasmodium species, namely P. ovale, differ in the latency period and resemble P. vivax clinically [3]. P. malariae is a relatively rare species but has distinct clinical outcomes. Compared to other malaria types, the number of merozoites produced with each schizont rupture is lower; thus, the parasitemias are lower in these patients [4]. Because of the longer parasite life cycle, patients experience fever every 72 h during an infection. P. knowlesi, despite its limited distribution, shows a higher severity rate than other common strains [5].
Mosquitoes can also harbor viral pathogens and cycle them between the human population. Among mosquito-borne viral infections, dengue is the most common disease caused by dengue virus 1–4 (DENV 1–4). DENVs are most commonly spread through the bite of an infected female Aedes sp. mosquito. A person infected with a particular dengue virus serotype can sometimes induce short-term cross-reactivity with other serotypes as well. Although the majority of dengue virus infections are asymptomatic or only cause mild disease, severe disease can occur and is characterized by plasma leakage, a pathophysiologic process in which the protein-rich fluid component of blood leaks into the surrounding tissue, resulting in extravascular fluid accumulation and shock, coagulopathy, and end-organ impairment [6].
Chikungunya virus (CHIKV), a Togaviridae with a single-stranded, positive-sense RNA genome, is transmitted mainly by the mosquito vector Aedes aegypti and, to some extent, A. albopictus, mainly in regions of Paraguay, Bolivia, Argentina, and Thailand [7]. Patients infected with CHIKV generally encounter intense asthenia, myalgia, headache, and arthralgia. Unlike other acute infections, CHIKV infection is dedicated to attacking the skeletal muscles, joints, and myotendinous insertions [8].
Yellow fever virus (YFV), a flavivirus causing yellow fever, has been considered one of the deadliest infectious diseases. Endemic to the tropics and sub-tropics, this virus is transmitted via Haemogogus janthinomys, H. leucocelaenus, Sabethe, and Aedes mosquitoes. Naturally, YFV circulated between mosquitoes and non-human primates in a sylvatic cycle. Following urbanization, YFV has entered the urban cycle, infecting humans and resulting in human-to-human circulation [9]. A. africanus maintains a sylvatic cycle in the rainforest, whereas A. bromelia was found to serve as a vector mediating urban cycles. The subsequent return of sick individuals to non-endemic, more densely populated places can set off an urban cycle perpetuated by A. aegypti mosquitoes [10]. Individuals typically experience an incubation period of 3 to 6 days after being bitten by an infected mosquito, followed by flu-like symptoms before a remission period of 1 to 2 days. Following remission, some patients (20–60%) progress to a more toxic phase of the disease, characterized by hemorrhagic fever, jaundice, thrombocytopenia, and liver and renal failure [11].
Zika virus (ZIKV) is an arbovirus that usually causes asymptomatic infections in the human host, but several neurological impairments have been reported in several cases. ZIKV is transmitted in the human population mainly through bites of anthropophilic mosquitoes such as A. aegypti, A. albopictus, and A. hensilii. Its urban cycle uses humans as reservoirs and continuously multiplies in them. Apart from this cycle, a sylvatic transmission cycle operates between non-human primates and arboreal canopy-dwelling mosquitoes. ZIKV can persist in mosquito eggs, leading to transovarial transmission, where the virus infects ovarian tissues, and transegg transmission, where the virus infects its host during fertilization [12]. Besides the direct human-to-human transmission, ZIKV has also been found to travel to fetuses from infected mothers (in utero).
Other mosquito-transmitted viruses, including the Rift Valley fever virus (RVFV) and Japanese encephalitis virus (JEV), follow the epizootic transmission cycle, where the virus amplifies in domesticated animals before infecting humans. Rift Valley fever is an emerging zoonotic viral disease caused by the RVFV of the Bunyaviridae family. Cattle, sheep, goats, and camels are especially vulnerable to RVF and serve as viral amplification hotspots. Infection of domestic animals is initiated by female Aedes mosquitoes (the primary mosquito vectors) with a disseminated salivary gland with an RVFV infection during probing or blood-feeding. After the primary infection, Culex and Mansonia sp. can channel RVFV between viremic domestic animals and humans [13]. JEV, the causative agent of human encephalitis, is primarily transmitted by Culex tritaeniorhynchus and C. annulirostris. C. pipiens and A. japonicus could be considered potentially important vectors in the case of JEV introduction in new regions [14].
West Nile virus (WNV), another emerging neurotropic flavivirus, is predominantly found to cycle between mosquitoes and birds. Culex pipens and C. quinqifasciatus serve as WNV vectors in major parts of Asia, Africa, and America, whereas C. australicus and C. globcoxitus have been found to be predominant in Australia. While feeding on infected viraemic birds, female Culex spp. mosquitoes pick up WNV. The virus replicates in the mosquito’s midgut epithelial cells and spreads to the salivary glands and other organs via hemolymph [15]. The pathogenesis of WNV includes an initial infectivity period followed by viral amplification and, finally, neuro-invasion in the central nervous system [16].
From the mosquito perspective, A. aegypti is distributed worldwide and found in most tropical countries. A. albopictus, on the other hand, is an opportunistic daytime and outdoor feeder that prefers humans and can be found feeding and resting indoors. Vertical transmission (VT) of viral pathogens is common in mosquito hosts. Similarly, viruses can also spread among mosquito populations through venereal transmission, which involves viral transfer during mosquito mating. Males cannot contract the virus through a blood meal, but they can contract it through venereal transmission from an infected female partner. Transmission by different species depends on some specific alterations in their non-structural proteins. Aedes aegypti was found to be more prone to ZIKV infection owing to a mutation in its non-structural protein 1 (NS1), whereas A. triseriatus and A. taeniorhynchus were not susceptible [17,18]. Other Aedes species, such as A. furcifer, were found to transmit DENV to humans, whereas Aedes luteocephalus transmit DENV and YFV in non-human primates [19]. In this context, several cases of co-infection were reported where patients were positive for both CHIKV and DENV [20]. In the genus Culex, C. quinquefasciatus, the most abundant species in tropical Africa, serves as a vector for the transmission of CHIKV and Japanese encephalitis virus (JEV). Meanwhile, C. antennatus is speculated to play an essential role as an epizootic vector of WNV [21]. The Anopheles genera support the transmission of Plasmodium sp. and certain nematodes but do not support arboviral transmission [22]. This review explains the complex mechanisms utilized by mosquito-borne pathogens to evade host immune systems. Malaria, Dengue fever, chikungunya, and other illnesses present substantial health challenges in tropical regions. Additionally, this review investigates the effects of these evasion strategies on disease progression and other outcomes. Through analysis of current scientific discoveries, we aim to enhance the understanding of the intricate interplay between these pathogens and the host’s immune responses, ultimately contributing to developing effective strategies to tackle these diseases and improve global health.
2. Immune Evasion in Mosquitoes
Mosquito midgut tissues are the first to be infected [23] and therefore have been the primary area of focus for studying the immune responses of the vector. Researchers aim to identify the molecules and pathways involved in the infection process of mosquito-borne pathogens. The major innate immune pathways involved in vector immune responses are the toll pathway, IMD pathway, JAK-STAT pathway, RNAi pathway (mi-RNA and pi-RNA pathways), and the interferon-mediated antiviral response. Mosquito-borne pathogens can shut down all these defense mechanisms by downregulating the responsible genes for these pathways [24,25]. According to a transcriptome study, the toll pathway plays a role in restricting the mosquito’s capacity for infection. Toll, Rel1A, and Spätzle (Spz) are the genes responsible for Toll activation, whereas cactus is the negative regulator.
In the mosquito, the anti-plasmodial response in Anopheles is enacted by the circulating thioester-containing protein 1 (TEP 1) responsible for humoral immunity at the ookinete stage. Apart from this, Enterobacter, Pseudomonas, Asaia, and Panteoa induce the secretion of antimicrobial peptides (AMPs) against the invading Plasmodium [26]. The innate immune system of Anopheles controls pathogen invasion by regulating three signaling cascades: the immune deficiency, the Toll, and the JAK-STAT pathways [27]. Immune evasion by Plasmodium includes the activation of the Pfs47 gene, which allows the parasite to inhibit several caspases responsible for the Jun-N-terminal-kinase-mediated activation of mid-gut apoptosis [28]. Furthermore, Pfs47 inhibits midgut nitration responses, which are required to activate the complement-like system [29].
Arboviruses can inhibit the antibacterial and antiparasitic activity of the IMD pathway in Aedes aegypti, leading to enhanced viral replication [30]. Functional studies have revealed that genes involved in the JAK-STAT pathway are essential to the vector’s immune system. This pathway is upregulated as an early response to arbovirus infection [31]. However, DENV and other mosquito-borne pathogens have evolved mechanisms to downregulate the JAK-STAT immune response [24]. Interfering RNA is one of the most successful antiviral defense mechanisms in mosquitoes [32]. Different viral proteins/factors can inhibit the vector’s RNAi processing pathways to prevent the degradation of their genomic material. The cytokine-like element named Vago establishes coordination between an IFN-like antiviral immunity pathway and the canonical innate immunity pathway (JAK-STAT) in culex mosquitoes [33].
3. Immune Evasion in the Vertebrate Host
3.1. Immune Response at the Skin Barrier
Immunity at the skin level is a crucial first line of defense against pathogen invasion. This is especially true for vector-borne diseases such as malaria, dengue, chikungunya, yellow fever, Zika virus infection, Rift Valley fever, Japanese encephalitis, and West Nile virus infection. Post-pathogen entry via mosquito bite into host skin, the immune response at the skin level plays a vital role in preventing the establishment of the infection [34]. This response involves activating local immune cells, such as dendritic cells, macrophages, and natural killer cells, which recognize and eliminate pathogens. The skin also contains antimicrobial peptides and proteins that can kill or neutralize invading pathogens [34]. The skin’s immune response is complex and can be modulated by various factors, including age, sex, and previous exposure to the pathogen. Understanding the skin’s immune response to these vector-borne pathogens is critical for developing effective preventive and therapeutic strategies. In the case of malaria, the sporozoites are blocked at the dermis and gradually removed by phagocytic cells. SPECT-1 (sporozoite microneme protein essential for cell traversal) and SPECT-2 (perforin-like protein 1) proteins are used by these sporozoites to avoid dermal blockade and phagocytic ingestion by immune cells [35]. Numerous mosquito-borne viruses, such as DENV and WNV, actively replicate in migratory Langerhans cells (LCs), thereby indicating that this ability might be a desirable method inside vertebrate hosts [36]. Different receptors have been found or suggested as entry points for DENV, which enters the cell through receptor-mediated endocytosis. It has been demonstrated that DENV can be bound in vitro by heparin sulfate, proteoglycan, and glycosaminoglycans, which are frequently expressed on different mammalian cell types [37].
3.2. Immune Evasion Strategies by Plasmodium Parasites
Malaria is a major public health concern and is most prevalent in tropical Africa [38]. The primary survival strategy adopted by Plasmodium species inside the human host is immune evasion. This involves a common mechanism wherein parasitic proteins interact with specific receptors on the erythrocyte’s surface. This mechanism is shared by all Plasmodium species [39]. Malaria symptoms and clinical manifestations typically become apparent during the asexual reproduction of the parasite within the red blood cells (RBCs) of the vertebrate host [40]. The parasite has adapted to modify RBCs’ morphology and surface features to evade the host’s immune system. Other mechanisms to evade the host’s immune system involve the suppression of Kupffer cells and dendritic cells (DCs) [41] (Figure 2).
Among the five Plasmodium species, P. falciparum is the most severe, causing high morbidity and mortality, and is found predominantly in sub-Saharan Africa, South-East Asia, and Eastern Mediterranean countries. P. vivax is the second most widespread species, causing moderate illness, and is distributed throughout Ethiopia, Sudan, Guatemala, Brazil, Colombia, Indonesia, Myanmar, India, Pakistan, Papua New Guinea, and Afghanistan. P. ovale, a less severe species, causes low to moderate malaria cases and is mainly endemic to tropical Western Africa. P. malariae, which causes mild malaria, is distributed across sub-Saharan Africa and the southwest Pacific. Lastly, P. knowlesi, a zoonotic species, causes moderate- to high-severity malaria and is primarily found in Southeast Asian regions of Malaysian Borneo, extending to peninsular Malaysia [42].
There is a significant difference in disease severity between P. falciparum and P. vivax. The former causes severe malaria, characterized by a high parasitemia level and the sequestration of infected red blood cells (RBCs) in the microvasculature of different organs, including the brain, lungs, and placenta [43]. In contrast, the latter has a lower parasitemia level and does not sequester in organs, leading to a less severe disease. However, P. vivax uniquely forms hypnozoites in the liver, which is a dormant stage that causes relapses [44,45].
Studies suggest that the immune response to P. falciparum and P. vivax infections differs. P. falciparum infections result in a strong proinflammatory response, essential for controlling the infection but also causing tissue damage and contributing to severe malaria pathogenesis. P. vivax infections, on the other hand, elicit a less intense immune response, which may be related to the low parasitemia levels observed during infections [46]. Additionally, it can modulate the host’s immune response to promote its survival, for example, by reducing proinflammatory cytokine levels [47]. In an earlier study, blood was taken for inspection to compare the immune response against P. vivax- and P. falciparum-infected patients. This whole blood sample was taken before and after infection once the count exceeded 10,000 parasites/mL. CD38 is a surface glycoprotein marker that facilitates signal transduction and cell adhesion and regulates Ca2+ levels that are upregulated on lymphocytes after activation [48]. When comparing the frequencies of CD38+ T cells, it was noted that P. falciparum infection was associated with higher numbers of CD38+ CD4+ T cells, whereas P. vivax infection was associated with higher numbers of CD38+ CD8+ T cells. However, no discernible differences in the frequency of CD38+ B cells were observed, irrespective of the presence of P. falciparum or P. vivax [49]. Little is known regarding the immune response to the dormant hypnozoite stage of P. vivax. However, recent studies suggest the immune response to hypnozoites is different from that of the blood-stage parasite, and the host’s immune system may not be able to recognize or eliminate dormant hypnozoites effectively [50]. P. falciparum and P. vivax differ significantly in their ability to cause disease, sequestrate in organs, and form dormant stages. These differences also affect the immune response to infections [51] (Table 1).
3.2.1. Immune Evasion Strategies at the Liver Stage
Malaria’s pre-erythrocytic and blood stages are initiated when a female Anopheles mosquito carrying the malaria parasite feeds on human blood [40,55]. Post mosquito bites, sporozoites are discharged into the victim’s dermis and enter into the bloodstream, where they must surpass Kupffer cells (KC) and endothelial cells (EC) to invade the hepatocytes and start an infection [55,56]. Sporozoites can also pass through the gaps between EC and KC by modulating cytokinin activity, upregulating anti-inflammatory Th2 cytokines, and downregulating inflammatory Th1 cytokines [57,58]. Additionally, sporozoite-produced circumsporozoite protein (CSP) can interact with KC surface proteins, namely LRP-1 (low-density lipoprotein receptor-related protein) and proteoglycan, and inhibit ROS production by producing intracellular cAMP/EPAC [58]. Notably, the sporozoites induce KC apoptosis, while the CSP reduces KC-specific expression of MHC-1, decreasing its antigen presentation capability [59]. Thus, the sporozoites can effectively minimize KC functions and make their entry into liver hepatocytes [39].
3.2.2. Immune Evasion in the Hepatocyte Stage
The sporozoites successfully enter the hepatocytes after infiltrating the sinusoidal layer and downregulate the mTOR pathway, thus affecting the proteins needed for cell division, proliferation, and autophagy. The sporozoites always travel through several hepatocytes until they reach a final hepatocyte, where they develop into a merozoite [60]. Hepatocyte growth factor (HGF) is secreted by the transmigrated hepatocytes, which bind the c-MET receptor and makes the hepatocyte vulnerable to infection by rearranging the host cell’s actin cytoskeleton [61]. By upregulating the PI-3 kinase/MAPK and ATK pathway, this interaction confers resistance to apoptosis in the hepatocyte [62]. Sporozoite-released CSP binds to high-sulfate HSPGs (heparan sulfate proteoglycans) on hepatocytes and undergoes cleavage, exposing the TSR (thrombospondin repeat) domain. The binding of the TSR domain to HSPG on hepatocytes facilitates the invasion of the hepatocytes by sporozoites [63,64]. The sporozoite is encased in a parasitophorous vacuole to protect them from host lysosomal degradation. The cleaved CSP makes its way into the cytoplasm of the hepatocytes using its PEXEL domain [65]. Cleaved CSP inhibits host cell protein synthesis, benefiting sporozoite survival [66]. It also suppresses the NF-κB signaling pathway [65] and significantly lowers p53 expression in the infected hepatocytes, allowing parasite survival [67].
3.2.3. Immune Evasion Strategies at the Pre-Erythrocytic Stage
The invasive form of the parasite, the merozoites [68], leads to the asymptomatic phase of the disease, referred to as the pre-erythrocytic stage. Merozoites save themselves from resident KC and DC by covering themselves in merosomes, which are derived from the host hepatocytes [69]. Following maturity, the merosomes (vesicles containing merozoites) bud off the hepatocyte and are released into the bloodstream [69]. This initiates the erythrocytic stage of the infection. Subsequently, initiating the erythrocytic stage of infection can take several days after the exit of merosomes from the liver [39].
3.2.4. Immune Evasion Strategies at the Erythrocytic Stage
Following the release of merozoites into the bloodstream, they invade the host RBCs and begin their maturation phase within the RBCs. The merozoite matures into a trophozoite within a membrane-bound parasitophorous vacuole inside the host RBC [41], which further undergoes schizogony to produce the multinucleated schizont stage containing daughter merozoites [70]. These infected RBCs (iRBCs) release their merozoites into the bloodstream upon maturation, which continues to infect new RBCs. This mechanism leads to the development of the malarial parasite’s asexual stage inside of host RBCs [41].
3.2.5. Antigenic Variation on the Surface of iRBCs
The iRBCs evade the host’s immune response by expressing various surface antigens encoded by multigene families known as variant surface antigens (VSA) [71,72,73,74,75]. Three VSAs have been identified in P. falciparum, namely P. falciparum erythrocyte membrane protein 1 (PfEMP1) coded by the var genes, repetitive interspersed repeats (RIFIN) coded by the rif genes, and sub-telomeric variant open reading frame (STEVOR) coded by the stevor genes [76].
There are approximately 60 var genes that express PfEMP1 [77] on RBC surfaces [78]. Approximately 150 rif genes per genome are clustered at the sub-telomeric region of the chromosome [77,79]. RIFINs are classified into two sub-families, namely A-type RIFIN and B-type RIFIN. The former are trafficked to the iRBC membrane with the help of the PEXEL motif, while the latter is present inside the parasite [80,81]. Lately, RIFINS were also found in merozoites with A-type RIFIN, localizing the apical tip and B-type RIFIN present in the cytoplasm of merozoites [82]. RIFINS have evolved host immune evasion mechanisms, such as rosetting, which will be discussed in the next section. The STEVOR protein family is similar to RIFIN and comprises 28 copies of the stevor gene in the P. falciparum 3D7 reference strain [77]. STEVOR also contains a PEXEL motif that targets the proteins involved in export to the surface of the iRBC membrane [83].
3.2.6. Cytoadherence and Sequestration as an Immune Evasion Strategy
The spleen clears out abnormal and old RBCs and kills bloodborne pathogens, which places the circulating iRBCs in grave danger [41]. The Plasmodium-infected RBC bind to human endothelial cells and then evade the spleen via a process known as cytoadherence. This causes the iRBCs to sequester in the microvasculature of various organs [41]. The var gene-encoded PfEMP1 mediates the sequestration of iRBCs [84]. The antigenic variability and cytoadhesive properties of iRBCs are contributed by the Duffy binding ligand (DBLs) and cysteine-rich interdomain region (CIDRs) domains of the PfEMP1 proteins [85,86,87]. The acid terminal segment (ATS) domain assists in projecting PfEMP1 onto the surface of RBCs [88]. PfEMP1 also binds to numerous host cell receptors, such as intercellular adhesion molecule 1 (ICAM1), CD36, chondroitin sulfate A (CSA), endothelial protein C, heparin sulfate, IgM, α-2 macroglobulin, thrombospondin, and complement receptor 1 (CR1), and promoting the process of cytoadherence and sequestration [89,90,91,92,93,94,95,96]. Sequestration and cytoadherence allow the iRBCs to adhere to endothelial cells, which protects them from being cleared by the spleen.
3.2.7. Rosetting as an Immune Evasion Strategy
Rosetting is the process where iRBCs adhere to healthy RBCs and form a rosette-like structure. It helps the iRBCs to elude the host’s immune system and shields the newly released merozoites against host-invasion-inhibitory antibodies [97]. It also provides a suitable microenvironment for the emerging merozoites to invade the healthy RBCs [98]. Recent literature has demonstrated that STEVOR, RIFIN, and PfEMP1 can induce rosetting by binding to glycophorin C and blood group A antigen, respectively [99]. This shows that rosette formation is mediated by three different strategies the parasite has evolved, indicating a crucial role in parasite survival.
Different variants of PfEMP1 bind to different RBC receptors, such as CR1, heparin sulfate, and α2 macroglobulin, and mediate the process of rosetting [94,100,101,102,103]. It has been shown that PfEMP1 binds to both α2 macroglobulin and IgM [104]. The binding of IgM increases the avidity of iRBCs, thus mediating rosetting [105]. Recent studies also show that cytoadhesion and rosetting can be mediated simultaneously by a single PfEMP1 molecule from the P. falciparum rosetting strain [96].
A study by Goel et al. demonstrated that one member of the A-type RIFIN family could form large rosettes by adhering to the blood group A antigen on RBCs [106]. RIFINs can also form smaller rosettes by binding to the glycophorin A receptor on the surface of O blood group RBCs [106]. In addition, RIFIN-mediated rosettes seem to shield PfEMP1 from antibody identification, suggesting their function as protective antigens and in immune evasion [106,107]. Recent research has demonstrated that STEVOR can independently promote rosetting without PfEMP1 by binding to glycophorin C on healthy RBCs [108,109]. The study conducted by Niang et al. also proved that merozoites are protected from invasion-inhibitory antibodies with the aid of STEVOR-mediated rosettes [99,108]. Rosetting suggests that Plasmodium species has used this mechanism for its survival and efficient transmission in the host.
3.2.8. Malaria and Its Association with Other Disease Outcomes
Cancer: Although cancer and malaria have been historically studied separately, recent evidence suggests the importance of investigating their potential biological interactions in light of their evolutionary history and epidemiology. According to epidemiological data, there appears to be an inverse relationship between cancer and several mosquito-borne diseases, whereby cancer cases rise as the chances of contracting mosquito-borne infections decrease [110]. Parasites may alter the balance between immunosuppression and immunity against a tumor by modifying the availability and presentation of cross-reactive antigens, influencing the induction of pre-existing immunity, and modifying the components of the tumor microenvironment [111]. Malaria pathogenesis may selectively induce certain immunological events, such as the production of inflammatory cytokines, that can potentially lead to cancer [112]. p53, the master regulator of oncogenesis, is well known for its role in cell cycle regulation and apoptosis. Kaushansky et al. observed a reduction in p53 levels during malarial parasite infection and demonstrated that this decrease in p53 levels led to an increase in hepatic infection in mice as their model organism. Conversely, they found that boosting p53 levels significantly reduced hepatocyte infection [67]. The decrease in p53 activity observed during malarial parasite infection could potentially contribute to developing hepatic cancer. In addition, P. vivax, in its sporozoite stage, utilizes the Duffy antigen receptor for chemokines (DARC) receptor to invade red blood cells. Interestingly, the same receptor has been found to sequester chemokines that are essential for tumor metastasis and angiogenesis in a breast cancer model [113]. In addition, DARC has been found to interact with the tumor suppressor protein KAI1 (CD82), which plays a crucial role in inhibiting tumor cell proliferation and promoting senescence. This interaction was supported by direct evidence obtained through DARC knockout mice, which demonstrated that the metastasis-arresting property of CD82 was severely compromised [114]. Malarial parasites attach to the placenta using variant surface antigens during pregnancy. Interestingly, these same antigens have been found to interact with chondroitin sulfate A (CSA), a glycosaminoglycan that enhances aggressiveness and metastasis [115,116]. Epidemiologically, malarial infection has been associated with Burkitt’s lymphoma (eBL) [117]. Post-infection, Robbiani et al. reported the rapid clonal proliferation of B cells, which can lead to genomic instability and the alteration of B cells to non-Hodgkin’s lymphoma through the action of an activation-induced deaminase, an enzyme responsible for DNA mutations and double-stranded breaks [118]. In a rodent model displaying immunity against the malarial parasite, Burkitt’s lymphoma was observed after repeated Plasmodium infections, suggesting that the immune response against malaria could act as a switch leading to eBL in the presence of Epstein–Barr virus (EBV) [119].
Cardiovascular diseases: Evidence suggests Plasmodium infections, specifically those caused by P. falciparum, may increase the risk of cardiovascular disease and heart attacks. This is thought to occur through the chronic inflammation and endothelial dysfunction that can result from repeated or chronic malaria infections [120,121]. Brainin et al. conducted a cohort study in Denmark and found that individuals with Plasmodium infections had an increased risk of heart attack [122]. Cardiovascular complications in malaria are mainly attributed to the altered cytoadhesive properties of P. falciparum-infected erythrocytes. These parasites use platelet bridges to connect infected red blood cells (RBCs) and ultimately form aggregates through autoagglutination, leading to microvascular obstruction, ischemia, and tissue damage. This mechanism of cytoadhesion and sequestration of infected RBCs in the microvasculature has been implicated in the pathogenesis of severe malaria syndromes, including cerebral malaria and acute respiratory distress syndrome (ARDS), which are characterized by widespread endothelial dysfunction and multiorgan failure [122,123]. Furthermore, these agglutinated aggregates accumulate in myocardial capillaries, obstructing blood flow and contributing to the progression of cardiovascular disease. Additionally, Wennicke et al. found that plasmodial glycosylphosphatidylinositol (GPI), which is involved in the pathogenesis, can lead to apoptosis in cardiomyocytes [124]. Plasmodial GPI has been shown to induce the production of pro-inflammatory cytokines such as TNF-α and IL-6, which can cause excessive nitric oxide (NO) production. This NO overproduction can lead to apoptosis and inflammatory cardiomyopathy, characterized by contractile dysfunction and myocardial energy depletion. Therefore, it is suggested that the pathogenesis of malaria-related cardiovascular complications involves the dysregulation of cytokine and NO production through GPI-mediated mechanisms [125,126]. P. falciparum-infected erythrocytes can lead to various symptoms, such as ECG changes, myocarditis, tachycardia, pericardial effusion, and acute heart failure in patients with malarial infections [127,128]. Repeated infection by the Plasmodium parasite in endemic areas is reported to enhance angiotensin II and sphingosine-I phosphate (S1P), leading to hypertension and endothelial dysfunction [129,130]. A prospective study conducted by Kingston et al., with a sample size of 45 patients with either severe or acute P. falciparum infection, was found to be significantly associated with reduced cardiac index reserve and hypovolemia, as assessed by transthoracic echocardiography and invasive hemodynamic monitoring. These findings suggest that malaria-induced hemodynamic alterations can contribute to the development of cardiovascular complications in affected patients [131]. Plasmodium infections are associated with an increased risk of cardiovascular disease and heart attacks. The pathogenesis of malaria-related cardiovascular complications involves the dysregulation of cytokine and NO production through GPI-mediated mechanisms, as well as hemodynamic alterations.
Neurological disorder: P. falciparum-mediated malaria is known to cause cerebral malaria, a severe disease that can result in neurological complications such as seizures, loss of consciousness, and coma [132]. The pathogenesis of cerebral malaria involves the sequestration of infected erythrocytes in the brain microvasculature and the resulting immune response, leading to endothelial damage, vascular leakage, and hypoxia. These pathological changes can ultimately lead to neurological dysfunction and contribute to the high mortality rate associated with cerebral malaria [133]. Gene expression profiling studies have identified shared pathways and molecular signatures between cerebral malaria and neurodegenerative diseases, suggesting potential common mechanisms. The overexpression of the SNCA gene, which encodes alpha-synuclein, was observed in children with cerebral malaria. Alpha-synuclein aggregation is known to cause Parkinson’s disease, a neurodegenerative disorder that results in brain dysfunction. This suggests a potential link between the molecular pathogenesis of cerebral malaria and Parkinson’s disease [134]. A 53-year-old male diagnosed with cerebral malaria after traveling from Uganda later exhibited symptoms of neurodegenerative disease. The patient initially experienced prolonged seizures but later presented with ischaemic lesions associated with Parkinsonism. This case highlights the potential long-term neurological complications of malaria infections and the need for the continued monitoring of patients even after treatment for the acute infection. The mechanisms underlying the link between cerebral malaria and Parkinsonism remain unclear. Still, some studies have suggested that the aggregation of alpha-synuclein, a protein involved in Parkinson’s disease, may also play a role in the pathogenesis of cerebral malaria. Further research is needed to elucidate the precise mechanisms involved and to identify potential therapeutic targets for these debilitating conditions [135]. The overexpression of the PTEN-induced kinase I (PINKI) gene, which is involved in the clearance of dysfunctional mitochondria, was observed in subjects with cerebral malaria. This gene has also been implicated in the development of neurodegeneration and neuroinflammation, which are correlated with mitochondrial dysfunction and reported in several diseases, such as Alzheimer’s disease and multiple sclerosis. The findings suggest that cerebral malaria could induce mitochondrial dysfunction, which may contribute to neurodegenerative complications [136]. The HSPA1A gene encodes heat shock protein 70, which acts as a molecular chaperone and prevents protein aggregation by preventing misfolding. Overexpression of the HSPA1A gene has been observed in children with cerebral malaria and patients with epilepsy. The upregulation of HSPA1A mRNA expression is thought to result from inflammation and stress caused by brain injury. These findings suggest a potential link between the protective role of HSPA1A in preventing protein aggregation and the pathogenesis of neurological disorders associated with malaria and epilepsy [137]. Post-malaria neurologic syndrome (PMNS) is a rare neurological syndrome that occurs after a symptom-free period of acute malaria and is often misdiagnosed or unreported. Two hypotheses have been postulated to explain the development of PMNS: the ischemic hypothesis and the cytokine storm hypothesis. The ischemic hypothesis suggests that PMNS might result from the cytoadherence of P. falciparum and the blockage of brain microvasculature, leading to ischemic damage in the brain [138].
On the other hand, the cytokine storm hypothesis suggests that inflammatory cytokines such as IL-2 and IL-6 could damage the central nervous system by causing neuroinflammation and oxidative stress [139]. Malaria infection has been found to reduce the production of L-arginine and nitric oxide [140]. Nitric oxide is an important molecule that regulates endothelial cell function, including suppressing adhesion receptors. Low levels of nitric oxide in the system may contribute to the increased cytoadherence of Plasmodium-infected erythrocytes [141]. Although PMNS and autoimmune encephalitis converge in the symptom-free interval and after treatment with corticosteroids, no autoantibodies have been reported against malaria [141]. Neurocognitive decline, seizures, peripheral neuropathies, and tremors are mostly reported as clinical outcomes of PMNS [142]. Cerebral malaria, a severe and life-threatening complication of malaria, is commonly observed in sub-Saharan Africa and is often linked with epileptic seizures [143]. Studies have reported that some pediatric survivors of cerebral malaria have developed seizures and epilepsy within two years of the initial infection, highlighting the long-term neurological consequences of the disease [144].
Renal disorders: As per the literature, malaria has been associated with a rapid deterioration of the kidney. Plasmodium infection can lead to renal disorders due to the cytoadherence of infected red blood cells in the kidney vasculature, resulting in tissue damage and reduced kidney function [145]. Additionally, the immune response to the infection can cause inflammation and damage to the kidney. Certain antimalarial drugs can also cause kidney damage as a side effect. Therefore, it is important to monitor kidney function in patients with Plasmodium infection, especially those with pre-existing kidney conditions [146,147]. A retrospective study found that 5% of malaria patients with induced acute kidney injury (AKI) developed chronic kidney failure [148]. Patients with severe malaria are known to produce higher levels of proinflammatory cytokines [149]. A study conducted by Sinnai et al. on C57BL/6 mice infected with the murine malaria parasite P. berghei showed elevated levels of proinflammatory cytokines, establishing the potential roles of cytokines in kidney-related disorders [150]. Apart from this, endothelial activation induced the sequestration of RBCs, leading to impaired renal flow, and increased leukocyte infiltration may reflect the pathogenesis in AKI [151]. Autoantibody formation leads to immune-complex-mediated complement activation and may cause renal injury in malaria patients [152]. Monitoring kidney function is crucial, as severe malaria may increase proinflammatory cytokines and endothelial activation, contributing to the pathogenesis of acute kidney injury.
3.3. Dengue Virus and Associated Immune Evasion Strategies
3.3.1. Viral Sensing by the Host’s Immune System
DENV infection is a growing global burden, affecting countries regardless of economic status. DENV exists in four serotypes (DENV 1-4), all of which have the potential to cause severe conditions. These four serotypes facilitate different clinical manifestations ranging from mild symptomatic to asymptomatic. Severe forms are more likely to manifest in those who have been exposed to a secondary DENV infection of a different serotype. Additionally, there is a higher risk of developing dengue shock syndrome (DSS) and antibody-dependent enhancement (ADE) post-secondary infection by heterogenous dengue serotypes. ADE occurs when non-neutralizing antibodies from a previous infection bind to a different serotype, facilitating viral entry into immune cells and leading to increased viral replication and disease severity. This means that the immune response to a second infection with a different serotype can be stronger and more damaging than the response to the first infection [153].
The successful transmission of the dengue virus commonly activates several immune responses within the host’s body. This includes the recognition of pathogen-associated molecular patterns (PAMPs) by the pattern recognition receptors (PRRs) of our innate immune system [154]. Toll-like receptor-3 (TLR3), TLR-7, retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated protein 5 (MDA5) are among the pattern recognition receptors (PRRs) that induce an antiviral state by activating cytokine and chemokine production [155]. The RIG-I-like receptor pathway (RLR) primarily detects viral nucleic acid through the cytosolic sensors RIG-1/DDX58, MDA5, and Laboratory of Genetics and Physiology 2 (LGP2 or DHX58). Upon activation, RIG-I and MDA5 interact with mitochondrial antiviral-signalling protein (MAVS) through the caspase activation and recruitment domain, which activates IκB kinase ε (IKKε), TANK-binding kinase-1 (TBK1), and phosphorylates IFN regulatory factors (IRF3, IRF7), leading to the production of type I interferon, mainly IFN-β [156,157]. Additionally, viral RNA is recognized by TLR3 and converges into MAVS, stimulating IRF3, IKKε, and IFN-β. The outer membrane of the mitochondria plays a vital role in initiating and amplifying the innate immune system [158].
During DENV infection, the STING/cGAS pathway is activated by cyclic GMP-AMP synthase (cGAS), a secondary messenger molecule, which stimulates TBK1, phosphorylates IRF3, and facilitates the production of IFN-I [159]. This pathway recognizes DNA molecules and is also essential in developing innate immune responses against viral RNA. DENV infection causes mitochondrial damage, releasing mitochondrial DNA (mtDNA) into the cytosol and activating the cGAS-STING pathway [160,161,162]. Recent studies show that mtDNA also activates TLR9, which recognizes nonmethylated CpG islands in dendritic cells [163]. Additionally, the inflammasome secretes interleukin (IL)-1β, inducing the release of mtDNA and activating the cGAS/STING pathway to produce IFN [164]. Thus, DENV infections trigger these two major pathways, RIG-I/MAVS and cGAS/STING, which need further study to understand their crosstalk (Figure 3).
3.3.2. Immune Evasion Strategies of Dengue Virus Strains
The immune response to dengue virus infection is complex and can have different effects on the four virus serotypes [165]. When a person becomes infected with one of the serotypes, their immune system mounts a response to fight the infection [166]. This response includes the production of antibodies specific to the infecting serotype, which can provide lifelong immunity to that particular serotype. However, this immunity is only partial and temporary for the other three serotypes [167].
This increased viral replication can lead to a more severe form of the disease, such as dengue hemorrhagic fever (DHF) or dengue shock syndrome [168]. DHF is characterized by bleeding, plasma leakage, and organ dysfunction, while DSS involves dangerously low blood pressure and can be fatal [169]. The risk of developing severe dengue is generally higher during secondary infections with a different serotype. However, the specific interactions between the immune system and the different serotypes are still not fully understood [170].
3.3.3. Countermeasures to Hijack RLR Signaling
DENV RNA molecules have intrinsic factors that help evade and hijack the host’s innate immune system. The sensing interference mainly depends on two specific alterations: partial degradation by host nucleases and 2′-O-methylation. 2′-O-methylation imitates cellular mRNA, enabling it to evade the host’s immune barrier. Additionally, DENV RNA interferes with the activation of RIG-I by preventing the formation of a signaling complex, ultimately suppressing the production of type I interferon. Understanding these evasion mechanisms is crucial for developing effective antiviral strategies [171,172]. When DENV RNA is partially degraded by host nucleases, it generates a sub-genomic flavivirus RNA (sfRNA). The DENV sfRNAs are known to inhibit the deubiquitylation of TRIM25 by ubiquitin-specific protease 15 (USP15). TRIM25 is an important factor in RIG-I signaling, and its inhibition ultimately suppresses the production of type I interferon, allowing the virus to evade the host’s immune system. These findings suggest that sfRNAs play a crucial role in the pathogenesis of DENV infections [173]. The inhibition of TRIM25 by DENV sfRNAs hinders the polyubiquitylation of RIG-I and prevents its dimerization with the CARD domains, which is a crucial step in the activation of the RIG-I signaling pathway. This ultimately leads to the suppression of the interferon signaling process, allowing the virus to evade the host’s immune system. These findings highlight the significance of the interaction between viral RNA and host factors in the pathogenesis of DENV infections [174].
The NS3 of the DENV has a phosphomimetic RxEP motif that inhibits the translocation of RIG-I to the mitochondria by restraining 14-3-3ε [175]. The 14-3-3ε protein is a primary requirement for the association of RIG-I/TRIM25 and eventually interacting with MAVS in mitochondria [176]. A morphological change in the mitochondria has also been reported to modulate antiviral signaling [177]. The strategy of mitochondrial morphodynamics used by the DENV is not unique since similar strategies are used by other viruses, including severe acute respiratory syndrome-related coronavirus (SARS–CoV) and hepatitis virus [178]. Non-structural proteins of DENV, including NS2A, NS2B, and NS4B, block the phosphorylation of IRF3 by inhibiting the kinase activity of IKBKE and TBK1 [179].
3.3.4. Countermeasures to Hijack the cGAS/STING Pathway and IFN Signaling
After DENV infection, IFN induction is triggered by activating the cGAS/STING pathway by releasing mtDNA in the cytoplasm [159]. The NS2B protein of DENV identifies cGAS for lysosomal degradation. On the other hand, the protease activity of the NS2B-NS3 complex cleaves STING, thereby inhibiting the production of type I IFN at an optimal level [162,180]. During DENV infection, the JAK-STAT signaling cascade is downregulated by NS4B, specifically by hijacking STAT1 activation by an unknown mechanism yet to unfold [181]. It is hypothesized that it prevents STAT1 activation through dephosphorylation or by degrading the activated STAT1. In addition, the NS5 protein attaches to STAT2, thereby decreasing its expression level and ultimately preventing the normal function of the JAK-STAT pathway [182].
3.3.5. Role of microRNA in DENV Pathogenesis
MicroRNA (miRNA) plays a critical role during viral infection as viruses use these miRNAs to hijack the host’s immune system and facilitate viral replication. A microarray analysis study was carried out using a blood sample of a patient with DENV infection, expressing 348 miRNAs after DENV infection [183]. Another patient’s serum infected with DENV-1 was analyzed, and it was found that 12 miRNAs were downregulated and 41 miRNAs were upregulated [184]. Previously, another study found that miR-21 was significantly increased after infection by DENV-2 in HepG2 cells. However, the exact mechanism by which miR-21 facilitates the replication process is unclear, but it can be rationalized that it might target the NS1 protein of the DENV [185,186]. Another midgut-specific miRNA, miR-281, was expressed after DENV-2 infection in C6/36 cells. It was observed that miR-281 promotes viral infection by targeting the 5′-UTR of the viral genomic RNA, thereby enhancing the replication process of DENV [184].
In both human peripheral blood and primary monocytes, an increase in miRNA, specifically miR-146a, was detected upon infection with DENV. This miRNA was found to suppress the production of IFN-β by binding to tumor necrosis factor receptor-associated factor 6 (TRAF6) [187]. The TLR signaling pathway is enabled by TRAF6 and interleukin-1 receptor-associated kinase (IRAK1), which miR-146a targets. As a result, the production of IFN-I is diminished, ultimately allowing the pathogen to avoid being attacked by the host’s immune system [188]. This study confirms that neutralizing miR-146a restores the optimal IFN-I production level. Another miRNA, namely miR-378, hinders the production of GrzB in the NK cells and thus promotes DENV replication [189].
3.3.6. Dengue Fever and Its Association with Other Disease Outcomes
Cancer: A nationwide population-based cohort study has been published correlating the risk of developing leukemia with preceding dengue virus infection [190]. The observation of abnormal hematologic profiles and bone marrow suppression in leukemia patients led researchers to question whether there is a connection between dengue viral infection and the onset of leukemia. Infected patients’ bone marrow has also yielded dengue virus, and research suggests that hematopoietic progenitor cells are particularly vulnerable to dengue viral infection [190]. Dengue fever was determined to be present in 28 different patients. A total of 9 (32.14%) patients had hematological malignancies, leaving 19 (67.85%) patients with solid tumors. In 23 patients (82.14%), chemotherapy was still being administered, and 5 patients (17.85%) were being monitored [191].
Cardiovascular diseases: DENV-infected patients are found to have higher levels of proprotein convertase subtilisin/Kexin type 9 (PCSK9) in their plasma [192]. PCSK9 has also been reported to be associated with coronary artery disease, particularly with myocardial infarction resulting due to the rupture of the plaques [193]. In a study by Miranda et al., 12 out of 81 subjects had elevated cardiac biomarkers during dengue infections [194]. DENV could infect myocardial tissue and myocytes, thus altering calcium metabolism and causing myocarditis in pediatric patients [195]. ECG abnormalities, such as ST segment changes, and bradyarrhythmia, including atrioventricular block ventricular tachycardia, are reported [196,197]. Chaturvedi et al. experimentally proved that cardiac injury follows DENV infection [198].
Neurological disorders: In 2009, the World Health Organization (WHO) categorized several neurological disorders, including encephalopathy, encephalitis, dengue muscle dysfunction, and neuro-ophthalmic disorders, as potential complications resulting from severe dengue infection. A case study conducted in Brazil involving 150 patients with severe dengue infections reported diverse neurological symptoms. Additionally, DENV particles were detected in 48.8% of dengue-positive cases, thus providing further evidence linking neurological changes with DENV infection. Clinical manifestations of infection, such as meningitis (19.5%), encephalitis (46.3%), and meningoencephalitis (34.1%), were common [199]. The neuro-pathogenesis of DENV infection can be correlated with the presence of viral particles in cerebrospinal fluid (CSF) and damage to the blood–brain barrier (BBB) [200]. Dengue viruses are also reported to infect various immune cells and murine neuronal cells directly [201]. Recent studies have suggested the role of DENV NS1 antigen in triggering macrophages via TLR4, leading to the release of proinflammatory cytokines such as TNF-α, IL-12, and IL-4 and damaging the blood–brain barrier [202]. Strokes associated with DENV infection, either ischemic or hemorrhagic, are uncommon but not absent. Hemorrhages result from plasma leakage and vasculitis, but the bleeding is mainly intracranial [203]. Several studies also reported parkinsonism associated with DENV infection, mainly after recovery from acute illness. Patients developed symptoms such as bradykinesia, bradyphonia, and cogwheel rigidity, which could result from immune-mediated reactions [204]. In a prospective case-control study on 5400 children admitted with dengue hemorrhagic fever in Vietnam, 27 showed features of neurological syndromes. Seizures were reported in nearly 77% of individuals with clinical neurological manifestations [205]. Autoimmune conditions of neuromyelitis optica spectrum disorder (NOSD) were reported in two dengue-infected patients with aquaporin 4 antibodies in their serum [206]. Similarly, autoimmune conditions such as acute disseminated encephalomyelitis (ADEM) are also found among dengue-related neurological disorders. The most common manifestation was altered consciousness, followed by seizures and vision problems. The pathophysiology of this autoimmune reaction is unknown, but an auto-reactive attack on myelin could be due to imbalanced T cell tolerance [207] (Figure 4).
Renal disorders: Acute kidney injury in DENV-infected patients is considered to be an expanded dengue syndrome. A study by Nair et al. using the KIDGO criteria reported 69.4% dengue-associated AKI patients out of 85 subjects [208]. The correlation between dengue hemorrhagic fever (DHF) and kidney damage was studied with kidney specimens from fatal DHF patients. More sickle erythrocytes were located in glomeruli and inflammatory infiltrates. High numbers of IL17 and IL18+ showed the possibility of local lesions and enhanced vascular permeability [209]. Urine abnormalities were also diagnosed in DENV-infected patients. Increased blood and protein levels were reported among hospitalized dengue patients in Australia [210]. Jayarajah et al. conducted a prospective study among 170 DHF/DF-positive patients and measured the urine albumin/creatine ratio. They found the mean ratio to be 177 ug/mL and concluded that there was a high occurrence of microalbuminuria in DENV-infected patients [211]. A rare case where DENV infection was followed by the autoimmune conditions of lupus and nephritis has been documented in Maharashtra, India. Upon the microscopic evaluation of a kidney tissue biopsy, glomerulonephritis and segmental sclerosis (Stage IIIC) were observed. The possible reason for lupus nephritis can be described as a classic immune complex-mediated autoimmune reaction [212]. Hence, dengue-triggered immune reactions could form pathogenic autoantibodies against host neutrophils and the glomerular basement membrane [213].
3.4. Zika Virus and Associated Immune Evasion Strategies
Zika was first identified in Uganda and has since expanded extensively into the Americas and Western Pacific, with 86 countries that have reported active ZIKV by 2019 [214]. During pregnancy, Zika virus infection may cause congenital disorders such as CNS malformations, microcephaly, cerebral palsy, and encephalitis in fetuses and neonates, leading to brain anomalies [215]. The Zika virus employs various strategies to invade the vertebrate immune response and cause disease. Here, we summarize some of its immune evasion tactics.
The ZIKV structural E protein is responsible for the ability of the virus to bind to a different receptor and simultaneously inhibit IFN production [216]. Viral genome and sub-genome RNAs inhibit IFN signaling. An experimental approach involving the temporary production of ZIKV-3’ UTR and the transfection of RIG-I or MDA-5 agonists revealed that ZIKV sfRNAs obstruct RIG-I- and MDA-5-mediated IFN induction [217]. ZIKA-infected cells show a significant reduction in the expression of IFN and ISG. The viral proteins NS1, NS4A, and NS5 have been identified as major inhibitors of type 1 IFN. Additionally, it has been observed that the amount of antiviral transcriptional activator is decreased in ZIKAV-infected cells due to STAT2 degradation by NS5.
3.4.1. Hijacking the RNA Interference (RNAi) Pathway
Zika virus alters the RNAi pathway, thereby suppressing RNA silencing. In mouse brain tissue and hNSCs (human neuronal stem cells), ZIKV infection upregulated miR-124-3p and let-7c, which downregulated transferrin receptor (TFRC) and HMGA2 (high-mobility group AT-hook 2) mRNAs, respectively [218]. Additionally, ZIKV infection elevated miR-125a-5p and miR-125a-3p, exhibiting inhibitory effects on MAVS, a vital component of the innate immune system’s RIG-I and type I IFN response pathways [219].
3.4.2. Zika Virus Escapes NK Cell and DC Detection
Zika virus infection is detected by RIGI and IRF3, which cause the release of type I IFN and the overexpression of MHC class I and CEACAM1. MHC I expression can be dramatically decreased by blocking IFN-beta [220]. Through this IFN-beta-blocking mechanism, the Zika virus can escape NK cell detection. DCs are essential for detecting viral infections and coordinating both immediate and long-term antiviral responses. ZIKV promotes the expression of lipid metabolism genes by increasing SREBP TF binding and transcriptional initiation. By lowering the amount of cellular cholesterol, SREBP2 suppression by medication or genetics reduces the amount of ZIKV infection in monocyte-derived DCs (moDCs), possibly due to the reduction of ZIKV infection during the replication, assembly, and/or budding phases. To infect human moDCs, ZIKV thus encourages the activation of SREBP2-dependent cholesterol production, providing a unique treatment approach for ZIKV [221].
3.4.3. Immune Evasion Strategies for Various Zika Virus Strains
Despite being a single serotype, genetic diversity among different strains of ZIKV can lead to variations in surface proteins, helping the virus evade neutralizing antibodies and the host’s immune system [222]. Additionally, ZIKV can infect cells in immune-privileged sites such as the placenta, testes, and central nervous system, avoiding immune surveillance and establishing persistent infections [223]. ZIKV can also suppress host cell apoptosis, maintaining a cellular environment conducive to viral replication and effectively evading immune defenses [224]. The virus can hijack the autophagy pathway, a cellular process responsible for maintaining homeostasis, thus promoting its replication and evading the host’s immune response [225].
3.4.4. Zika Virus Infection and Its Association with Other Disease Outcomes
Cardiovascular diseases: Although mosquito-borne Zika infection is mainly associated with neurological syndromes, myocarditis was found to be associated with Zika infection owing to increased troponin I and creatine phosphokinase [198]. Brasil et al. reported a potential association of ZIKAV infection with congenital heart disorders in infants [226]. In a recent study, Rashid et al. reported that ZIKAV infection of human Sertoli cells significantly altered several proteins involved in cardiac hypertrophy [133].
Neurological disorders: ZIKV is known to preferentially infect a wide range of neural cells, including neural stem cells (NSCs), oligodendrocyte precursors, astrocytes, and microglia [227]. Symptomatic Zika infection is mainly restricted to mild and self-limiting febrile disease, but recent shreds of evidence link Zika to microcephaly in fetuses. Microcephaly can be classified into primary and secondary microcephaly. The former is characterized by small brain size owing to defects during embryonic development, while the latter reflects normal embryonic development, but brain damage restricts brain development [228,229]. A case–control study conducted in eight hospitals in Brazil from January to May 2016 linked microcephaly and Zika virus infection. This study analyzed cerebrospinal fluid sampling and performed an RT-PCR analysis of 32 cases and 64 controls. They found that 41% of neonates with microcephaly were positive for Zika infection, whereas the rest were uninfected. Zika virus was reported to infect neural progenitor cells (NPCs), and the increased number of viral particles damaged the cells, causing cellular apoptosis and leading to transcriptional attenuation [230]. The dysregulation of normal cellular functions in NPCs could lead to developmental defects. In organoid models, Zika virus infection was associated with a reduced number of neural progenitor cells. TLR-3 was reported to be upregulated in this model, and organoid shrinkage was observed. TLR-3 was thought to be a potential trigger for cellular apoptosis and thought to inhibit neurogenesis after Zika infection [230]. Activation of TLR-3 by an RNA virus mimetic poly (I: C) was found to modulate NDMA receptors and was associated with impaired neurodevelopment and abnormal arrangements of synaptic proteins [231]. A potential clinical link between ZIKV infection with autism spectrum disorder (ASD) was also reported in Brazil. The child in this case study was positive for Zika IgG, and ASD was diagnosed by neurologists and confirmed by the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) and Autism Spectrum Quotient (ASQ). Although exome sequencing did not reveal any pathogenic variant in genes related to ASD, the possibility of in utero infection by ZIKV must be clinically considered [232]. Epilepsy surveillance using a questionnaire and video electroencephalography in a cohort of normocephalic and ZIKV-exposed children and unexposed controls suggested a modest occurrence (IR: 2.8%, 95% CI: 0.34–9.81%) of epilepsy in exposed subjects. Although this study has a small sample size and low incidence of epilepsy, the neurodevelopmental assessment of children with suspected or confirmed in utero ZIKV infection should consider epilepsy surveillance during their first two years of life [233]. Kung et al. profiled the transcriptome of primary neuron cultures infected by ZIKV and used unbiased gene ontology enrichment analysis and in vivo confirmation in an immunocompetent mouse model. An altered expression of genes associated with bipolar disorder and schizophrenia was reported. In vivo validation using mice suggested that TNF-α signaling is a major switch for ZIKV-induced neurologic gene expression [234]. A probable link between neurodegenerative Alzheimer’s disease (AD) and ZIKV infection was studied using brain organoids. An upregulation of key Alzheimer’s pathologies, including amyloid beta (Aβ) and p-tau, was noticed in both AD and WT organoids exposed to ZIKV infection. The mechanism of neurodegenerative pathology was identified as a stress response in the endoplasmic reticulum and subsequent activation of the PERK-eIF2α axis [235]. Inhibition of PERK-mediated signaling reduced neurodegenerative phenotypes, including Aβ and p-tau [236]. In addition to its direct effect in causing neuropathologies, ZIKV infection is also associated with epigenetic alteration in the brain. Janssens et al. reported the altered DNA methylome of NPCs, astrocytes, and differentiated neurons in an organoid model. However, the mechanism is not well defined, and the question remains whether viral infection dysregulates the epigenetic regulators before any epigenetic shift [237]. The autoimmune condition Guillain–Barre syndrome (GBS) is also correlated with ZIKV-infected patients. In a case–control study of patients experiencing Guillain–Barre syndrome, 41 (98%) out of 42 patients were found to have anti-Zika IgG or IgM antibodies [238]. This may be due to a possibility of molecular mimicry where ZIKV cross-reacts with some neurological proteins and leads to an autoimmune catastrophe [239].
Renal disorders: The incidence of ZIKV infecting renal proximal tubular epithelial cells was demonstrated by Chen et al. using a C57BL/6 mouse. The authors found the glomerulus to be more infected and reported swelling of kidneys and apoptosis of renal cells [240]. Liu et al. reported that ZIKV infection causes AKI in mouse models by analyzing AKI-related biomarkers such as serum creatine kinase, kidney injury molecular-1 (Kim-1), and neutrophil gelatinase-associated lipocalin (NGAL). They proposed a mechanism where infection triggered the inflammasome’s Nod-like receptor 3 (NLR3) and apoptosis by suppressing Bcl-2 [241]. Congenital Zika syndrome patients were also found to be associated with a neurogenic bladder. The overactive bladder was evident in 21 of the 22 individuals evaluated, along with decreased bladder capacity and increased detrusor filling pressures [242] (Figure 5).
3.5. Chikungunya Virus and Associated Immune Evasion Strategies
CHIKV infection is responsible for long-term febrile illness, arthritis, and other complications such as chronic synovitis due to its persistence in joint-associated tissues [243]. CHIKV has acquired several mechanisms to hijack the host’s immune system. CHIKV non-structural protein-2 (NSP2) regulates the IFN-induced JAK-STAT signaling pathway [244]. CHIKV infection antagonizes the antiviral IFN response by creating mutations in the KR649AA site in the NLS of NSP2 or redirecting the NSP2 C-terminal methyltransferase-like domain into the nucleus [245]. Recent findings demonstrate that the NSP2 of VEEV specifically interacts with the nuclear importin molecule karyopherin α-1 (KPNA1) and downregulates STAT1-dependent type I and II IFN signaling [246]. The co-expression of MDA5/RIG-I along with NSP2, E1, and E2 inhibits more than 80% of the MDA5/RIG-I mediated IFN-beta promoter activity in the presence of viral protein [247]. Bone marrow stromal antigen 2 (BST-2), a well-characterized antiviral IFN stimulatory gene (ISG), prevents viral release. Upregulation of NSP1 counteracts the effect of BST-2, thereby enabling the release of viral particles from infected cells [248]. CHIKV can evade immune surveillance by suppressing the expression of zinc-finger antiviral protein (ZAP), an IFN-induced antiviral factor [249].
Virus replication can be inhibited by the transcription and expression of the cGAS-STING innate immune pathway. The CHIKV NSP1 either inhibits IFN-β promotor activation by interacting with STING or degrades cGAS to stabilize the viral protein [250]. CD8+ T cells are critical for the clearance of viral infection from lymphoid tissue but not joint-associated tissue. In contrast to humoral immunity, its role is poorly understood. Therefore, different studies suggest that CHIKV establishes and maintains a persistent infection in joint-associated tissue by evading CD8+ T cell immunity, but the underlying mechanism involved is still unclear [243]. The major immune checkpoints reported in CHKV infection are IFN-a, IL-10, and CCL2 [251]. RNAi is one of the major antiviral defense mechanisms present in eukaryotes. NSP2 and NSP3 are the two non-structural proteins that exhibit RNAi suppressor activity by separating double-stranded RNAs [252]. Other studies believe the capsid protein interacts with the viral RNA to prevent their interaction with RISC, which contributes to the inhibition of RNA-mediated viral RNA degradation [253].
Immune Evasion Strategies of Various Chikungunya Virus Strains
Immune response mechanisms to chikungunya virus infection are generally conserved across different strains. The genetic differences between the strains can influence virus transmission, spread, and virulence [254]. However, the immune response to CHIKV infection is primarily directed against the conserved viral antigens shared among different strains [255].
Nevertheless, the unique characteristics of each genotype can affect the disease’s epidemiology and severity [256]. The West African genotype originates in West Africa and has no major adaptive mutations identified. It is characterized by limited outbreaks, mainly occurring within the sylvatic cycle (mosquitoes-primates-mosquitoes). The ECSA (East/Central/South African) genotype is found in Eastern, Central, and Southern Africa, as well as the Indian Ocean islands. It has an A226V mutation in the E1 glycoprotein, which increases transmission by Aedes albopictus. This genotype is responsible for major outbreaks in the Indian Ocean islands, India, and Southeast Asia and is associated with both urban and sylvatic cycles. The Asian genotype is prevalent in Asia and the Pacific, with no major adaptive mutations identified, but it has caused major outbreaks in the Pacific region between 2013 and 2014. This genotype is associated with an urban cycle (mosquitoes-humans-mosquitoes) [257].
3.6. Yellow Fever Virus and Associated Immune Evasion Strategies
YFV, the causative agent of yellow fever, is a prototypic member of the genus Flaviviridae and is known to cause one of the most severe infectious diseases [9]. A wide range of clinical symptoms is associated with YFV infection, ranging from entirely asymptomatic hemorrhagic fever to a high mortality rate [258]. After disease remission, 20–60% of patients went through a more lethal stage of the illness, marked by hemorrhagic fever, jaundice, thrombocytopenia, liver failure, and renal failure [11]. In the liver, viral replication plays a major role in disease establishment (Figure 6).
YFV has evolved several immune evasion mechanisms, including hepatocyte apoptosis, to manifest infection, which is discussed briefly. MDA5 and LGP2 constitute part of the RIG-1-like receptor family. Upon the recognition of intracellular RNA by pattern recognition receptors, namely RIG-1, an interferon response to infection is mediated [259]. RIG-1 generally recognizes dsRNA with triphosphate and U or A-rich motifs [260]. Long dsRNA is mainly recognized by MDA5 [260]. YFV infections are restricted by both RIG-1 and MDA5, which play important roles in mediating antiviral effects [261]. The cap 1-2′O-methylation of RNA prevents RIG-1 recognition [262]. This genomic alteration helps in viral immune evasion and survival inside the host.
When STAT1 is phosphorylated via type I IFN signaling, NS5 (non-structural) protein binds with STAT2; this binding and subsequent suppression of IFN signaling is mediated by the E3 ubiquitin ligase TRIM23, which polyubiquitinates NS5 [262]. In this way, YFV represses IFN signaling and blocks the antiviral effects of IFN-1 [263]. YFV actively replicates inside the liver and is responsible for hepatocytotoxicity. The pathogenesis of yellow fever is mediated by the apoptosis of hepatocytes [11]. The disease progression is maintained by a systemic and unbalanced cytokine storm [264]. CD4+ Th1 and Th3 cells have been noticed in livers infected with YFV [265,266]. The CD4+ T cell-expressed TNF-α cytokines mediate hepatic damage and CD8+ T cell cytosis [10]. Accordingly, Th3 CD4+ T cells express the pro-apoptotic cytokine TGF-β, an anti-inflammatory protein and a pro-apoptotic inducer [265,266]. The generation of TNF-α and TGF-β indicates that the immunopathogenesis of YFV may involve an imbalanced pro- and anti-inflammatory cytokine response [267]. This cytokine imbalance in the liver displays immune evasion mechanisms, leading to the progression of the viral infection inside the human host.
Immune Evasion by YFV Strains
Currently, there are limited data on how the different genotypes of YFV affect immune responses in humans. However, the immune responses to YFV generally seem to be consistent across genotypes [268]. This is evidenced by the fact that the yellow fever vaccine, based on the live-attenuated 17D strain, has been effective against all YFV genotypes [269].
The immune system combats YFV through both innate and adaptive immune responses, as mentioned earlier. While the specific immune response may vary slightly among genotypes due to genetic differences, these variations are not well understood and are likely to be minor [270]. Despite genetic differences among the YFV genotypes, the immune response is generally consistent across genotypes. Individual factors can influence the severity of the disease and the effectiveness of the immune response, but these are likely unrelated to the specific genotype of the virus. More research is needed to understand the potential variations in immune responses to different YFV genotypes.
3.7. Rift Valley Fever Virus and Associated Immune Evasion Strategies
RVFV primarily affects domestic animals and occasionally humans, with an intermediate mortality rate. Mosquitoes transmit the virus, and outbreaks are most associated with periods of heavy rainfall and flooding, which create ideal breeding conditions for the mosquitoes that carry the virus. While most human infections are mild or asymptomatic, in rare cases, RVFV can cause severe disease, including hemorrhagic fever and encephalitis [271]. The entry of the RNA virus into the host is followed by a complex set of innate immune responses [272]. Retinoic acid-inducible gene I (RIG-I), melanoma differentiation factor 5 (MDA5), and Toll-like receptors (TLRs) are among the pattern recognition receptors (PRR) that recognize this virus [273]. RIG-I also triggers an interferon response inside the host cell and maintains an antiviral state [272]. The RVFV has developed sophisticated strategies to evade the immune system and dysregulate the immunological pathways to successfully initiate infection and manifest into a clinical condition, which will be discussed briefly.
The post-transcriptional alternative splicing mechanism is altered by this viral invasion [274,275]. The progression of RVFV inside the host is inhibited by RIO kinase 3 (RIOK3), which plays a significant role in the production of IFN-1 through PRR signaling mediated by RIG-I-like receptors [276]. The antiviral effects of RIOK3 are stably maintained by the interaction of TRA2-β with fixed regions of RIOK3 pre-mRNA, which promotes the constitutive splicing of RIOK-3 mRNA [277]. Upon viral infection, TRA2-β is alternatively spliced, decreasing its cellular levels, forcing the RIOK-3 mRNA to undergo alternative splicing and produce the variant isoform RIOK-3 X2 [278]. This spliced isoform RIOK-3 X2 lowers the expression of interferon and helps in the successful evasion of the host’s immune pathways [277]. RIOK-3 X2 also plays a role in increasing the inflammatory NF-κβ response [272].
3.7.1. The Role of RVFV Non-Structural Proteins in Immune Evasion
The S segment of the RVFV genome encodes the non-structural proteins, which can suppress the immune system [279]. NSs form a complex with Sin3A-associated protein 30 (SAP30) and transcription factor Yin Yang 1 protein (YY1) and suppress the activation of the IFN-β promoter [280]. NSs also contribute to viral evasion by preventing the IFN-β promoter from being activated. NSs mediate the inhibition of host transcription and the activation of viral translation. Eukaryotic transcription factor IIH helps in the initiation of transcription by eukaryotic RNA polymerase II. The core complex of TF IIH comprises XPD, XPB, p44, p62, p8, p34, and p52 [272]. RVFV NSs can competitively bind to p44 and prevent it from binding with XPD [281]. P62 is also degraded by the ubiquitin–proteasomal pathway, where NSs work as an adaptor protein in the cullin 1-Skp1-FBXO3 E3 ligase complex [282,283]. The host transcription is halted by these two mechanisms, which affects the recruitment of the TF IIH complex in the nucleus.
RVFV-infected cells have shown nuclear deformities such as micronuclei and lobulated nuclei mainly due to defects in chromosomal cohesion and segregation [284]. These damages occur due to the accumulation of NSs filaments inside the nucleus [272]. The filaments also play a key role in arresting the cell cycle at the S or G0/G1 phases [285]. The chromatin DNA cohesion and segregation are affected by the SAP30-YY1 complex formed by NSs proteins [272]. Therefore, RVFV impairs the host cell replication machinery via NSs-mediated DNA damage. From the recent findings, it can be said that the virus has established effective immune evasion mechanisms to counteract the host’s immune response and has also developed ways to attack various cellular processes and pathways, which aids in the successful manifestation of infection and contributes to viral pathogenicity.
3.7.2. Immune Evasion by Different RVFV Strains
RVFV strains evade the immune system through various mechanisms, including antigenic variation, inhibition of the interferon response, modulation of host cell machinery, immune evasion proteins, and suppression of dendritic cell function. Different strains use different combinations of these strategies, making developing a single, effective vaccine or treatment challenging. Ongoing research aims to uncover immune evasion mechanisms to create targeted therapies and control outbreaks [272].
3.8. Japanese Encephalitis Virus and Associated Immune Evasion Strategies
JEV is known for its single-stranded RNA genome, which mutates faster and spreads through mosquitoes. Although the immune sentinels of the host are always ready to shoot any invading JEV, the latter has smartly evolved to rule out the host’s defense. Invasion of JEV triggers the RIG-I/IRF-3 and PI3K/NF-kB axis, facilitating the release of inflammatory cytokines and chemical cues that trigger more immune cells [286]. JEV was found to activate cellular autophagy, which eventually promoted its survival but downregulated the innate antiviral response. JEV-infected cells with inactivated autophagy produced more IFN [287]. A plausible reason could be the low clearance of mitochondrial antiviral signaling protein (MAVS) and its high aggregation, which switches INF3 and NF-kB [287]. Another strategy for blocking INF signaling was reported by Lin et al., where NS5 acts as a phosphatase and blocks INF-induced JAK-STAT signaling [288]. JEV NS1 also interacts with cyclin-dependent kinase 1 (CDK1) and prolongs the phosphorylated state, activating CREB. This CREB acts as a transcription factor, activates miR-22, and reduces INF-I production by targeting MAVS [289].
Immune evasion leading to reduced IgM and neutralizing antibodies against JEV was reported in a mouse model where virus infection elevated myeloid-derived suppressor cells (MDSCs). In this study, MDSCs were found to suppress the T follicular helper cell differentiation that brought down the splenic B cell population and plasma cells, leading to a shunted humoral immune response [290]. JEV also infects the DCs and impairs these antigen-presenting cells along with the production of IL-10, an anti-inflammatory cytokine. IL-10 production promotes JEV survival by reducing costimulatory activity and CD8+ T cell priming [291]. Similarly, JEV infection has been reported to modulate immune responses in a mouse monocyte cell line, RAW264.7. JEV infects monocytes and induces an anti-inflammatory environment by inhibiting histone deacetyl transferase. In macrophages, JEVI was found to modulate the suppressors of cytokine signaling (SOCS) to mute the pro-inflammatory nature of the host’s immune response [292]. Another reason for immune evasion might be the overexpression of indoleamine 2,3-dioxygenase (IDO) in antigen-presenting cells, followed by reduced antigen presentation [293].
JEV, similar to any other virus, modulates the cellular repertoire of microRNAs (miRNA) to invade and successfully establish its pathogenesis in the host. The role of miR-22 in JEV infection and IFN downregulation has been discussed previously. Another microRNA-432 was found to be downregulated in JEVI human brain microglial cells (CHME3). This miR-432 is associated with the phosphorylation of STAT-1 and the immune-mediated inflammatory response during viral infection. However, the downregulation is involved with SOCS upregulation, which eventually silences the host defense. Likewise, the upregulation of miR-155 during JEVI suppresses proinflammatory responses via the miRNA-based targeting of Pellino protein 1 (PELI1) [294]. These silenced PELI1 were originally responsible for inducing a pro-inflammatory response by acting as a positive regulator of MYD88 [295]. Similarly, miR-301a was found to target SOCS5 and IRF1 and repress it at the translational level [296]. Short-fragment ncRNA (sfRNA) are hallmarks of flavivirus infection and are known for blocking IFN responses, thus favoring viral pathogenesis [297] (Table 2).
Immune Evasion Strategies of JEV Strains
JEV differs in their geographical distribution and virulence among the five genotypes. The immune response to these genotypes is generally consistent. The immune system combats JEV through both innate and adaptive immune responses, as previously reported elsewhere [298]. Regardless of the genotype, the innate immune system is the first line of defense against JEV. It deploys interferons, cytokines, and other signaling molecules to limit the spread of the virus. Pattern recognition receptors such as TLRs, RIG-I, and MDA5 recognize viral RNA from different genotypes and activate antiviral responses accordingly [299,300].
The adaptive immune response is also consistent across JEV genotypes. B cells produce antibodies, including neutralizing antibodies that target the viral E protein, which is conserved among the genotypes. T cells, particularly CD8+ cytotoxic T cells, recognize and destroy virus-infected cells. CD4+ helper T cells support the overall immune response by producing cytokines [175]. Based on the live-attenuated SA14-14-2 strain (genotype III), the Japanese encephalitis vaccine is effective against all JEV genotypes, suggesting that the immune response is consistent across genotypes [301].
It is important to note that individual factors, such as age, immune system function, and underlying health conditions, can influence the severity of the disease and the effectiveness of the immune response. These factors might have a more significant impact on the outcome of the infection than the specific genotype of the virus [302]. The immune system combats the virus through both innate and adaptive immune responses, which effectively limit the virus’s spread and clear the infection, regardless of the specific genotype [303].
3.9. West Nile Viruses and Associated Immune Evasion Strategies
WNV is a neurotropic virus that, upon successful infection to the human host, activates various antiviral mechanisms in the host, including the production of IFN and pro-inflammatory cytokines followed by the expression of the anti-viral genes [304]. WNV also developed various mechanisms that facilitate the process of immune evasion. RLR signaling induces type 1 IFN production by recognizing the virus particle through PAMPs. After WNV infection, there is an inconsistency between the viral protein accumulation and IRF3 activation, indicating a possible hijacking through which WNV evades the RLRs detection [305]. Another possible mechanism is masking viral RNA, thereby allowing WNV to establish and synthesize new virus particles that eventually prevent various signaling pathways, including IFN-1 and TLR3 [306]. The viral infection also hinders IFN type I by preventing the phosphorylation of tyrosine kinase 2 (TYK2) and inhibiting the activation of STAT1 and STAT2 [307]. Furthermore, the non-structural proteins of WNV also degrade the interferon-alpha/beta receptor complex (IFNAR1) through proteasome- and lysosome-dependent pathways. However, whether this IFNAR1 degradation and phosphorylation inhibition are directly linked or act as separate events is unclear. Thus, this needs more attention to resolve the mechanism through which WNV inhibits IFN-I signaling [308]. The viral proteins, including both the structural and non-structural, that alter the IFN-I signaling process have not been identified clearly. WNV infection evades the anti-viral properties of interferon-induced protein with tetratricopeptide repeats 1 (IFIT1), an interferon-stimulated gene (ISG) protein that is highly activated after the viral infection. The virus uses the 2′-O-methyltransferase activity to escape the effects of IFIT1 as in normal scenarios; it interacts with the viral genetic material lacking the 2′-O-methylation and containing the 5′ triphosphate, thereby eventually blocking the functions of the viral proteins [309,310].
In addition to these above evading mechanisms, WNV uses other strategies, including the complement activation’s escape mechanism. The non-structural protein 1 (NS1) prevents the activation of the alternate pathway through the complement regulator factor H and facilitates the inactivation of C3b by recruiting factor I [311]. The C3b degradation leads to a less-than-optimal production of membrane attack complexes on the cell surface. The NS1 protein also hinders the activation of lectin and classical pathways by targeting the C4b-binding protein (C4BP) [312]. The higher concentration of NS1 in the extracellular space and lower levels of complement concentration eventually facilitate the inactivation of the complement system. Thus, in this manner, the viral non-structural proteins ease the evasion process from the host IFN system and the complement-dependent activation processes, thereby successfully causing the viral infection.
Immune Evasion Strategies by WNV Strains
The immune response to the West Nile virus can vary depending on the lineage and the individual’s immune system. However, there is limited information on the differences in immune invasion between Lineage 1 and Lineage 2 strains of WNV [313]. Some studies suggest that Lineage 2 strains may be less virulent than Lineage 1 strains, resulting in less severe disease in humans. However, more research is needed to confirm these findings and better understand the differences in pathogenicity between the two lineages [314]. In general, the immune system plays a critical role in controlling and clearing West Nile virus infection. Both innate and adaptive immune responses are involved in combating the virus. The innate immune response, which includes interferons, cytokines, and other signaling molecules, is the first line of defense against the virus [315]. The adaptive immune response, which includes T cells and antibodies, is essential for clearing the virus and providing long-term protection [316]. The differences in immune invasion between Lineage 1 and Lineage 2 strains of WNV are poorly understood. However, individual immune system strength plays a crucial role in determining the severity of the disease, regardless of the lineage.

{kind=link}
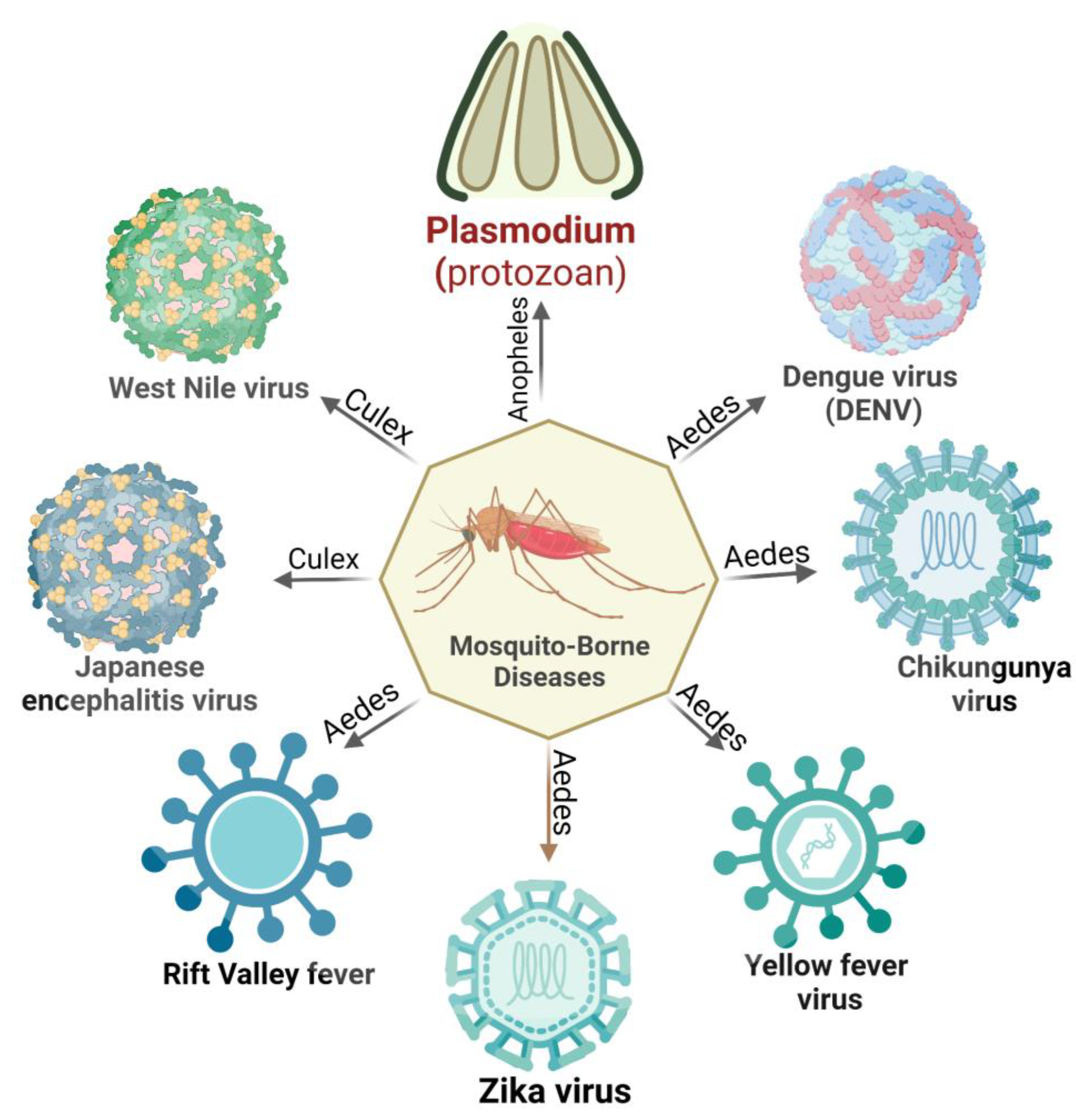{kind=link}
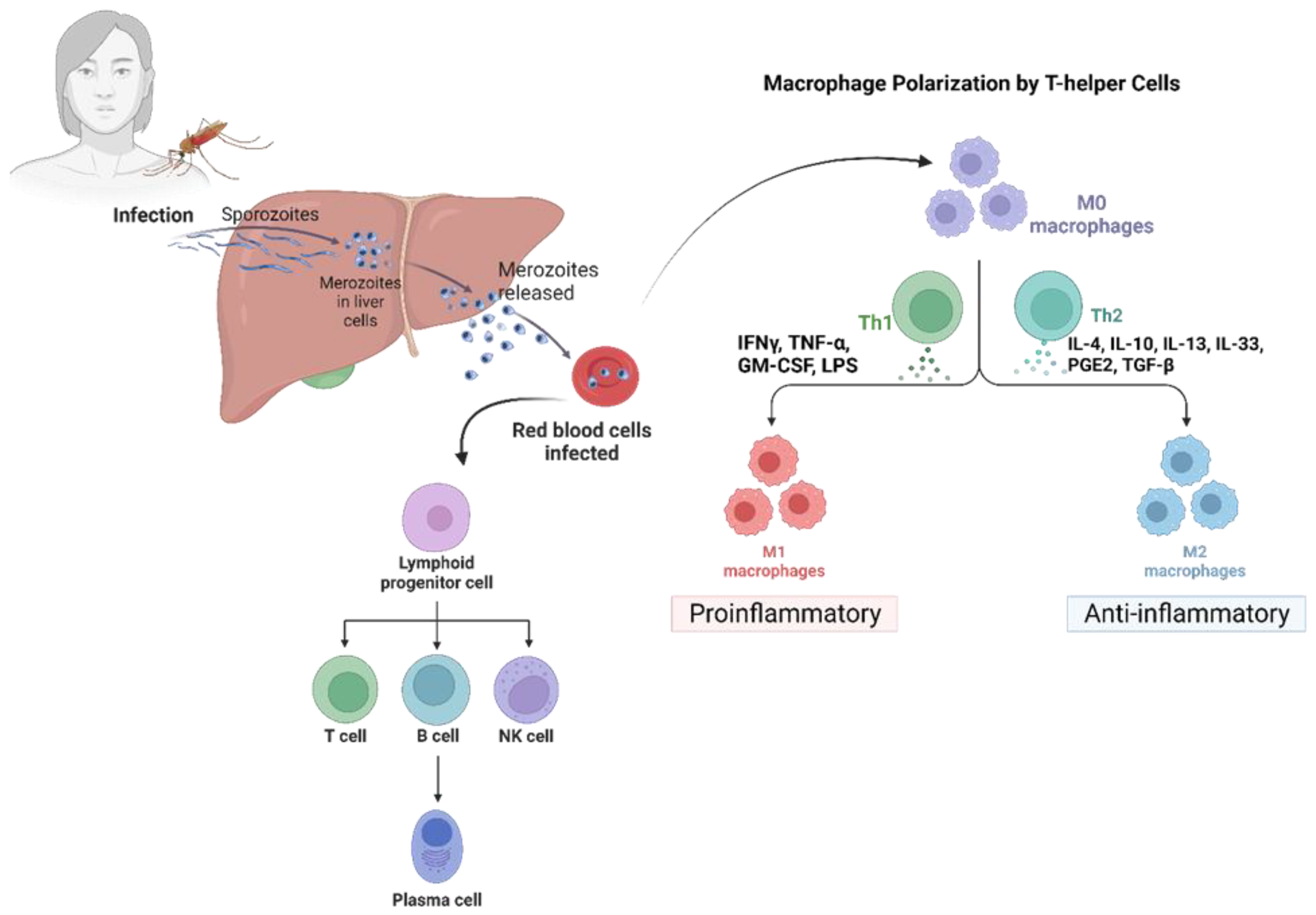{kind=link}
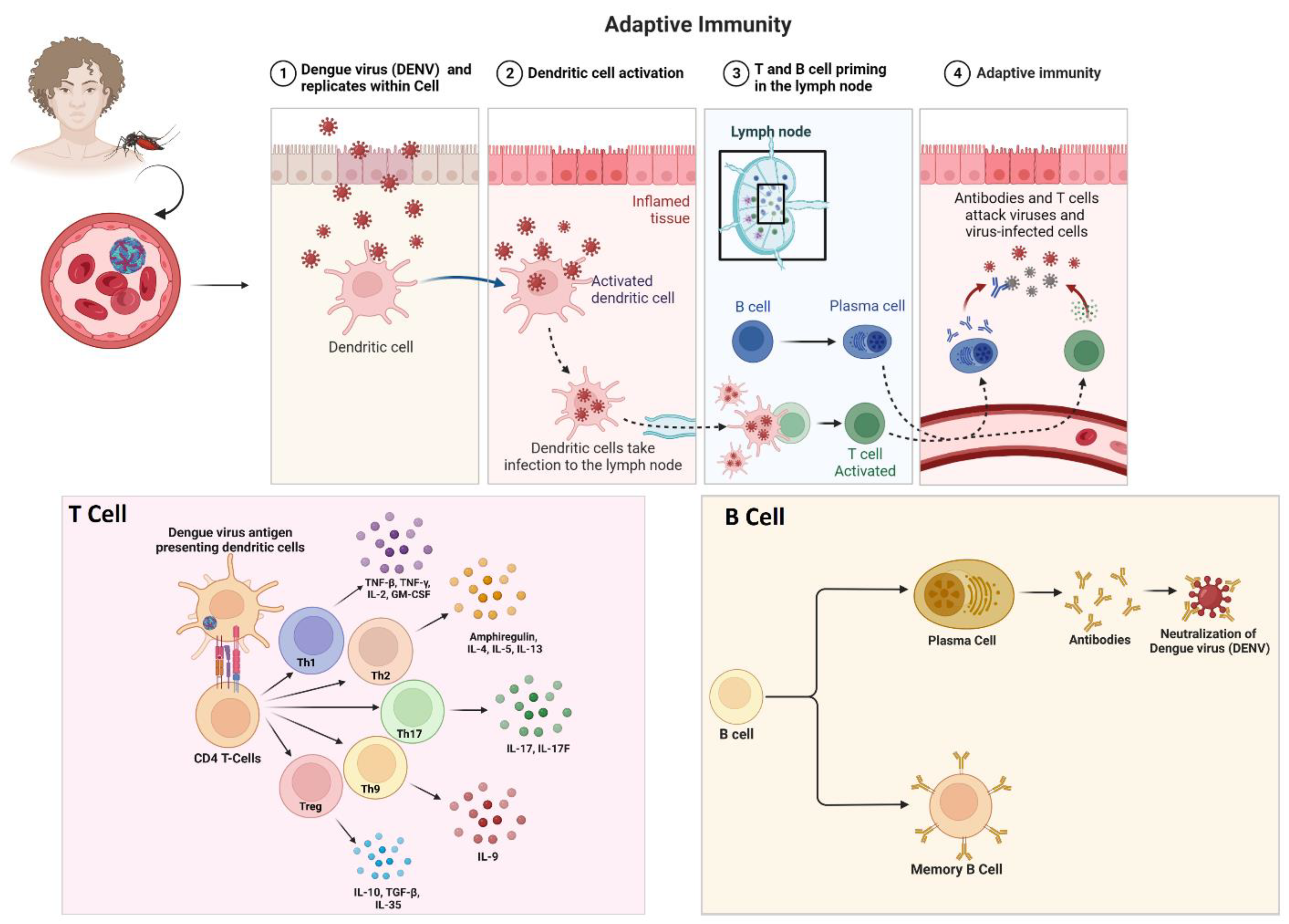{kind=link}
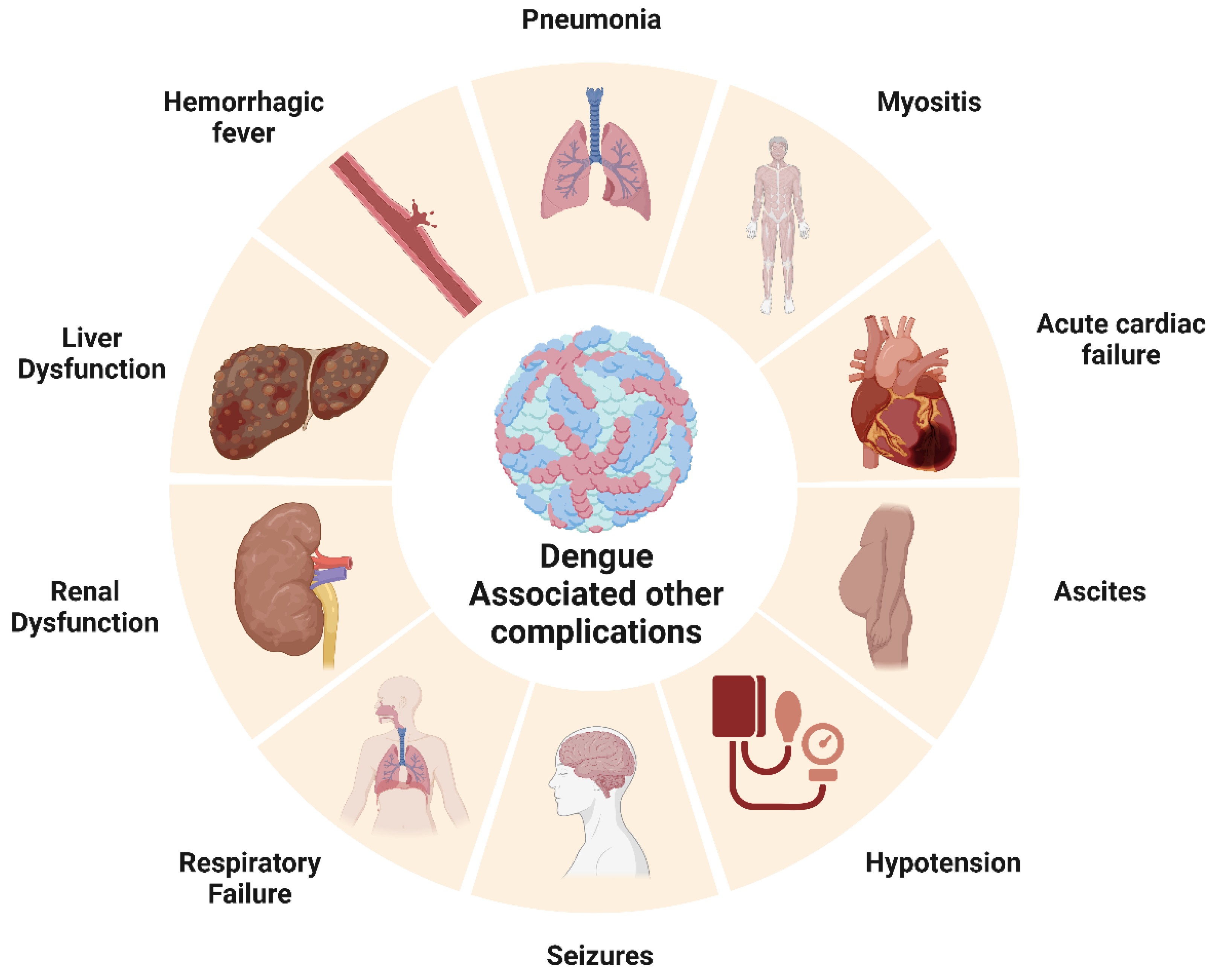{kind=link}
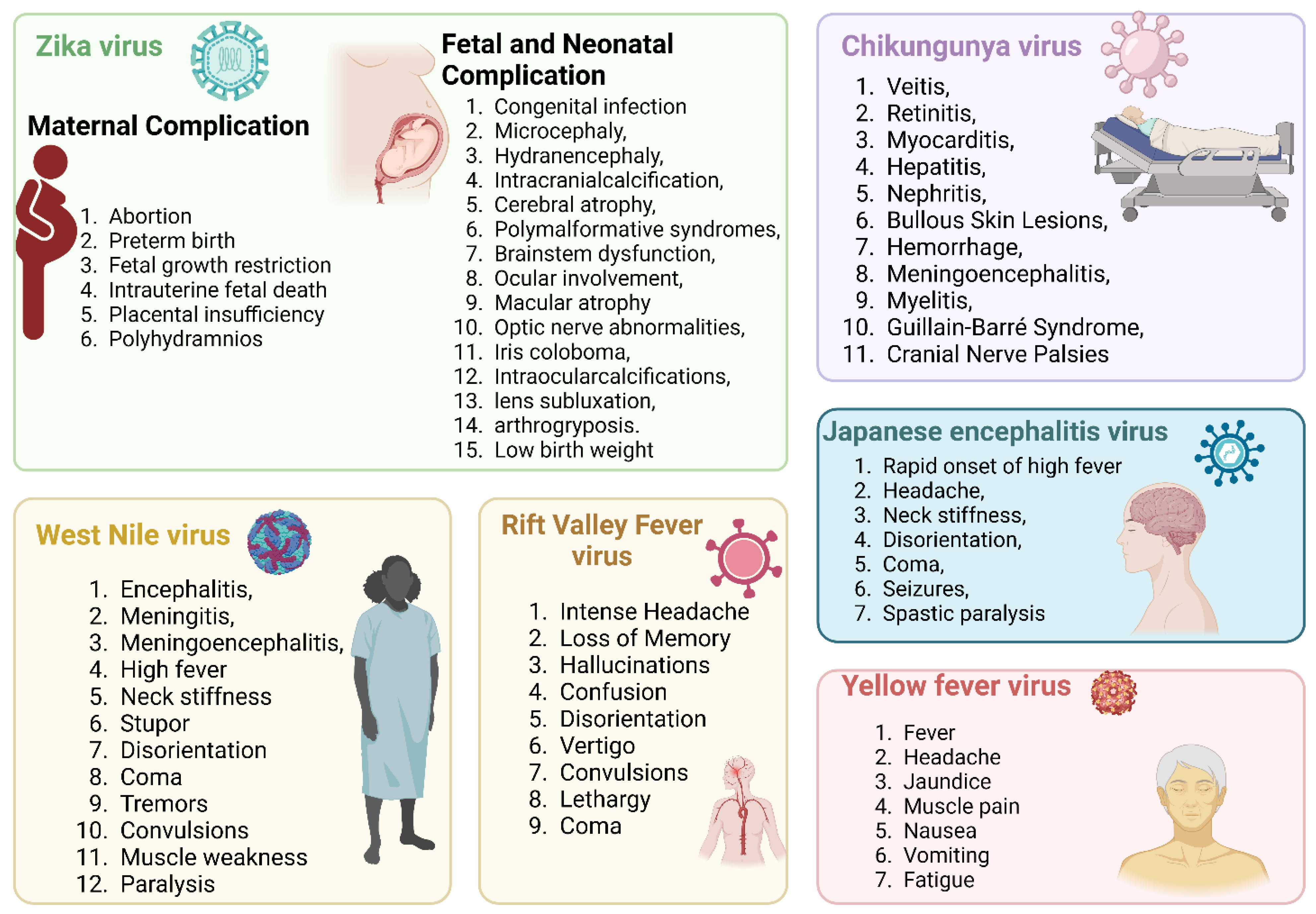{kind=link}
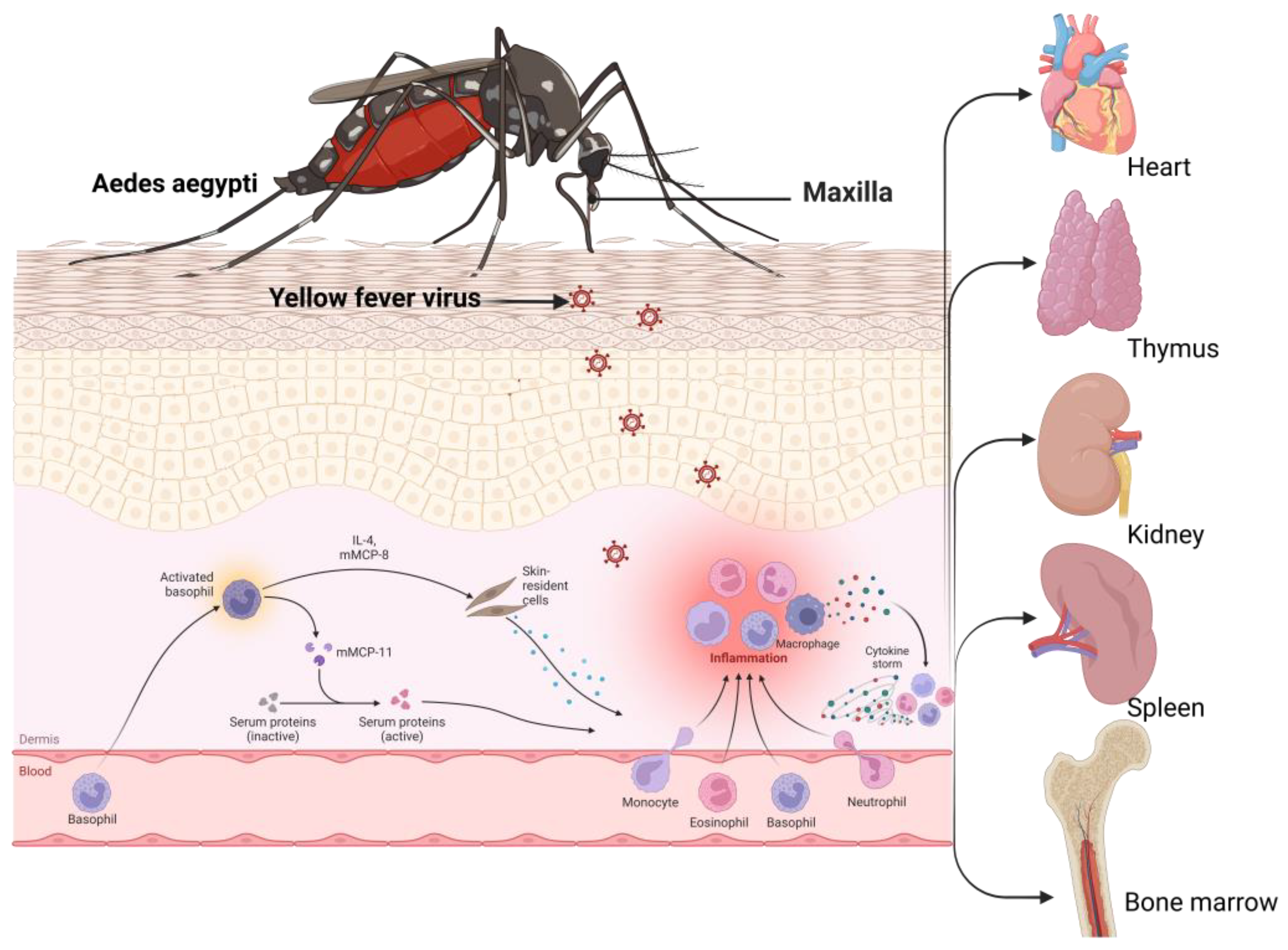{kind=link}
Table 2.
Mosquito-borne diseases and their link with post-infection other metabolic co-morbidities.
| Mosquito-Borne Diseases | Year | Location | Type of Study | Method of Assessment | Disease Manifestation | Reference |
|---|---|---|---|---|---|---|
| Chikungunya | 2005–2006 | Réunion | Prospective study | Fulfilled International Encephalitis Consortium criteria for encephalitis | Perinatal encephalopathy | [317] |
| Chikungunya | 2005–2006 | Mayotte | Prospective study | Unknown | Perinatal encephalopathy | [318] |
| Chikungunya | 2005–2006 | Réunion | Prospective observational study | Seizures; EEG consistent with encephalitis | Perinatal encephalopathy | [319] |
| Chikungunya | 2010 | India | Prospective study | Altered sensorium, apnoeic seizures | Perinatal encephalopathy | [320] |
| Chikungunya | 2014–2015 | Colombia | Case series study | EEG | Perinatal encephalopathy | [321] |
| Chikungunya | 2014–2015 | Colombia | Prospective study | EEG | Perinatal encephalopathy | [322] |
| Chikungunya | 2015 | Brazil | Case study | Seizures; abnormal brain MRI | Perinatal encephalopathy | [323] |
| Chikungunya | 2015 | Honduras | Retrospective study | Unknown | Perinatal encephalopathy | [324] |
| Chikungunya | 2016 | India | Case study | Dizygotic twins; both had seizures, required ventilation, thrombocytopenia; abnormal brain MRI | Perinatal encephalopathy | [325] |
| Chikungunya | 2016 | Brazil | Case study | Prostration, lethargy, seizures, required ventilation, thrombocytopenia; abnormal brain MRI and EEG | Perinatal encephalopathy | [326] |
| Chikungunya | 2005–2006 | Réunion | Retrospective study | DIC, transient scattered parenchymal petechiae, cerebellar hematoma | Perinatal brain hemorrhage | [327] |
| Chikungunya | 2005–2006 | Réunion | Retrospective descriptive study | Unknown | Perinatal brain hemorrhage | [328] |
| Chikungunya | 2015 | Brazil | Case study | Intraventricular bleeding (cranial US), lethargy | Perinatal brain hemorrhage | [329] |
| Chikungunya | 2005–2006 | Réunion | Case study | Seizures; hypotonia | Perinatal other | [328] |
| Chikungunya virus | 2008 | France | Case study | ECG and cardiac MRI | Myopericarditis, pericardial effusion | [330] |
| CHIKV infection | 2015 | Honduras | Retrospective study | CSF analysis, EEG, CAT (brain) scan, MRI (brain) | Meningoencephalitis, seizures. CSF analysis shows enhanced leukocyte presence. | [324] |
| CHIKV infection | Not specified | India | Case study | Neurologic examination, sensory examination, electrophysiological studies | Global areflexia and quadriparesis. Guillain–Barre syndrome | [331] |
| CHIKV infection | 2020 | India | Case study | MRI, nerve conduction studies | Sensorimotor axonopathy, myelopathy. | [332] |
| Dengue | 1987 to 1998 | Thailand | Observational study | MRI and autopsy | Seizure, mental confusion, nuchal rigidity, spasticity of limbs, positive clonus, hyponatremia, abnormal liver enzymes and CSF pleocytosis, hemiplegia and positive Kernig | [333] |
| Dengue | 2017 | Brazil | Case study | Brain MRI | Loss of vision, generalized myalgia, severe headache, retro-ocular pain, and cutaneous rash | [206] |
| Dengue | 1997–1999 | Vietnam | Case–control study | MRI and ultrasound | Cerebral edema, seizures, hemiplegia, hemorrhage, hyponatremia, hepatic failure, and microcapillary | [205] |
| Dengue | 2019 | India | Case study | MRI (brain), cerebrospinal fluid (CSF) examination | Bradyphonia, unclear speech, gait instability, cogwheel rigidity, mask-like facies and stooped posture while walking | [204] |
| Dengue | 2017 | Sri Lanka | Case study | Electroencephalogram and cerebrospinal fluid (CSF) examination | Encephalitis | [334] |
| Dengue | 2019 | India | Prospective study | Echocardiogram, ECG | Asymptomatic sinus bradycardia, symptomatic bradyarrhythmias, left ventricular systolic dysfunction (ejection fraction 35–45%), pericardial effusion, atrial fibrillation | [335] |
| Dengue | 1998 | India | Cohort study | Echocardiogram, ECG | Ejection fraction, global hypokinesia, ST and T changes in the ECG | [336] |
| Dengue | 1998 | India | Observational study | Echocardiogram | Sinus bradycardia | [337] |
| Dengue | 2007 | Sri Lanka | Cohort study | Echocardiogram, ECG | T inversion, ST depression and bundle branch blocks, hypotension and tachycardia and bradycardia, suggestive of significant cardiac dysfunction | [338] |
| Dengue | 2012 | Brazil | Case series study | Echocardiogram, ECG | Ileo-femoral deep vein thrombosis, pulmonary thromboembolism, mesenteric vein thrombosis | [339] |
| Dengue | 2014 | México | Case report | Echocardiogram, ECG | Myocarditis characterised by: S3 gallop rhythm, generalized lung rales and shock; ECG showed sinus tachycardia, ST depression in V1-V3, and ST elevation in a VR and aVL | [340] |
| Dengue | 2018 | Sri Lanka | Case series | CT angiography, ECG | Tachycardia in three cases. Myocarditis confirmed by troponin estimation and echocardiogram in one case, and in the other two, this was also confirmed by histopathology | [341] |
| Dengue | 2013 | Singapore | Case reports | Echocardiogram, ECG | Myocarditis | [342] |
| Dengue | 2015 | Vietnam | Case report | Echocardiogram, ECG | Acute cardiac failure | [343] |
| Dengue | 2008 | India | Case report | Echocardiogram, ECG | Myocarditis, Takotsubo cardiomyopathy, ECG showed sinus bradycardia | [344] |
| Dengue | 2016 | Taiwan | Case report | Echocardiogram, cardiac biomarkers | Elevated cardiac enzymes | [345] |
| Dengue | 2015 | Singapore | Case report | Echocardiogram, ECG | Acute myocardial infarction, elevated serum troponin I levels | [346] |
| Dengue | 2011 | France | Case report | Echocardiogram, ECG | Acute pericarditis, Consulting with acute chest pain; ECG revealed negative anterolateral T waves with long QT segment | [347] |
| Dengue | 2011 | Thailand | Prospective study | Holtrer | Sinus pause, first-degree and Mobitz type I second-degree atrioventricular block (Wenckebach) and atrial and ventricular ectopic beats | [196] |
| Dengue | 2016 | India | Case report | CT angiography, ECG | Repeated symptomatic episodes of a high-degree atrioventricular block with ventricular asystole | [348] |
| Dengue | 2015 | Sri Lanka | Case report | Echocardiogram, ECG | Bradycardia and heart block with atrioventricular dissociation | [349] |
| Dengue | 2013 | India | Case series | Echocardiography | Sinus bradycardia, echocardiography showed a decreased ejection fraction | [350] |
| Dengue | 2000 | Thailand | Case reports | CT angiography, ECG | Morbitz type I second-degree atrioventricular block | [351] |
| Dengue | 2004 | Thailand | Case report | Echocardiography and ECG | Myocarditis with bradycardia | [351] |
| Dengue | 2010 | India | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation, atrial fibrillation | [352] |
| Dengue | 2009 | Malaysia | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation | [353] |
| Dengue | 2003 | Brazil | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation | [354] |
| Dengue | 2008 | Saudi Arabia | Cohort study | Not specified | Acute kidney injury | [355] |
| Dengue | 2008 | Taiwan | Retrospective study | eGFR estimation | Acute kidney injury | [356] |
| Dengue | 2009 | Taiwan | Retrospective study | SCr > 2 mg/dL | Acute kidney injury | [357] |
| Dengue | 2010 | Thailand | Retrospective study | SCr > 2 mg/dL | Acute kidney injury | [358] |
| Dengue | 2011 | India | Prospective study | RIFLE | Acute kidney injury | [359] |
| Dengue | 2012 | Pakistan | Case series study | AKIN | Acute kidney injury | [360] |
| Dengue | 2012 | Taiwan | Retrospective case–control study | SCr increase ≥ 0.5 mg/dL | Acute kidney injury | [361] |
| Dengue | 2012 | India | Prospective study | AKIN | Acute kidney injury | [362] |
| Dengue | 2015 | Taiwan | Retrospective study | AKIN | Acute kidney injury | [363] |
| Dengue | 2015 | Malaysia | Retrospective study | AKIN | Acute kidney injury | [364] |
| Dengue | 2016 | India | Cohort study | KDIGO | Acute kidney injury | [208] |
| Dengue | 2017 | Taiwan | Retrospective case–control study | SCr > 2 mg/dL | Acute kidney injury | [365] |
| Dengue | 2017 | India | Observational study | SCr > 1.2 mg/dL | Acute kidney injury | [366] |
| Dengue | 2018 | India | Retrospective study | eGFR < 60 mL/min/1.73 m2 | Acute kidney injury | [367] |
| Dengue | 2018 | Taiwan | Retrospective study | KDIGO | Acute kidney injury | [368] |
| Dengue | 2018 | Malaysia | Follow-up prospective cohort | AKIN | Acute kidney injury | [369] |
| Dengue | 2018 | India | Observational prospective study | AKIN | Acute kidney injury | [370] |
| Dengue | 2019 | Thailand | Retrospective single-center study | KDIGO | Acute kidney injury | [371] |
| Dengue | 2019 | India | Retrospective single-center study | Not specified | Acute kidney injury | [372] |
| Dengue | 2019 | India | Retrospective study | AKIN | Acute kidney injury | [373] |
| Japanese encephalitis | 2014 | China | Case study | EEG, MRI, electromyography, and CSF analysis | Guillain–Barre syndrome | [374] |
| Japanese encephalitis | 2012 | India | Case study | MRI (cervico-thoracic spine) | Acute transverse myelitis | [375] |
| Malaria | 2014 | India | Observational study | Neurological and neuropsychiatric evaluation by MSME (Mini-Mental Status Examination) and BRPS (Brief Psychiatric Rating Scale) | Cerebellar ataxia, psychosis, convolution | [376] |
| Malaria | 2011 | Sierra Leone | Case study | MRI and autoimmune screen | Extensive abnormal high signal and swelling in brainstem and spinal cord | [377] |
| Malaria | 1965–1994, 2010–2015 | Ghana and Uganda | Case–control study | Multiplex Luminex bead assay, histological and cytological investigations | Burkitt Lymphoma | [378] |
| Malaria | 2005–2010 | Malawi | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [379] |
| Malaria | 2011–2015 | Uganda | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [380] |
| Malaria | 2010–2016 | Uganda, Tanzania, and Kenya | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [380] |
| Malaria | 1987–2015 | Sweden | Cohort study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [381] |
| Malaria | 2017 | South Korea | Case Study | Brain MRI | Parkinsonian features | [382] |
| Malaria | 2015 | India | Case study | CT scan of the brain, MRI, and electroencephalography | Cognitive dysfunction, calcification (lesion) in the tempo-parietal region, temporal and hippocampal hyperintensities, microhaemorrhages | [383] |
| Malaria | 1999 | India | Case study | Neurological (nerve conduction) examination | Guillain–Barre syndrome | [384] |
| West Nile Virus | 2022 | USA | Case study | ECG, transthoracic echocardiogram (TTE), X-ray study | Reduced LV systolic | [385] |
| West Nile Virus | 2022 | USA | Case study | TTE and X-ray | Left ventricular hypokinesis | [386] |
| West Nile virus (WNV) | 2016 | USA | Prospective cohort study | Biomicroscopy | Retinopathy and Neurological and Neurocognitive Sequelae | [387] |
| West Nile virus (WNV) | 2002–2004 | USA | Prospective cohort study | ELISA and anti-WNV IgM antibody detection | Encephalitis/meningoencephalitis/encephalomyelitis (WNE) | [388] |
| Yellow fever virus | 2016 | Portugal | Case study | CT scan; CSF investigation | Acute onset expression aphasia, agraphia, dyscalculia | [389] |
| Yellow fever virus | 2020 | Brazil | Experimental study | Histological and cytological laboratory investigations | Hepatic injury | [390] |
| ZIKV infection | 2016 | France | Case study | MRI (Spinal), electromyography, and CSF examination | Acute myelitis and lesions of the cervical and thoracic spinal cord | [391] |
| ZIKV infection | 2017 | Brazil | Case study | Brain MRI, electroencephalography, and autopsy | Meningoencephalitis. | [392] |
| ZIKV infection | 2015 | Colombia | Observational study | Electromyography and nerve conduction studies, CSF examination | Guillain–Barre syndrome (inflammatory demyelinating polyneuropathy) | [393] |
| ZIKV infection | 2017 | Colombia | Case–control study | MRI and electrophysiological examination | Encephalitis, peripheral facial palsy, thoracolumbosacral myelopathy, and transverse myelitis | [394] |
| ZIKV infection | 2017 | Brazil | Cross-sectional study | MRI (brain) and EEG examination | Epilepsy tended to be early and refractory | [395] |
4. Immunological Aspects Facilitating Therapeutic Development
Developing an effective vaccine against malaria has been challenging, as the parasite has evolved various mechanisms to evade the host’s immune response. However, recent studies have provided new insights into the immune mechanisms involved in malaria and potential targets for vaccine development [396]. However, the high genetic diversity of Plasmodium parasites poses a challenge to developing a vaccine that targets a single antigen. Therefore, researchers are exploring the use of multi-antigen vaccines or vaccines that target conserved regions of the parasite’s genome [397]. Another approach for vaccine development is to target the host’s immune response. Studies have shown that the immune response to malaria is complex and involves various components of both the innate and adaptive immune systems. For example, the production of type 1 interferons by dendritic cells has been shown to be important in controlling the parasite in animal models. Targeting these immune response components may lead to the development of more effective vaccines [398]. Additionally, recent studies have focused on understanding the role of the gut microbiome in modulating the host’s immune response to malaria. The gut microbiome has been shown to play a critical role in shaping the host’s immune system, and perturbations in the gut microbiome have been associated with increased susceptibility to malaria. Therefore, targeting the gut microbiome may also provide a novel avenue for vaccine development [399]. New insights into the immune mechanisms involved in malaria and the role of the gut microbiome in modulating the host’s immune response have provided potential targets for vaccine development. Recently, a randomized clinical trial was conducted for “R21 in adjuvant Matrix-M” in Burkina Faso. Its efficacy surpasses the previously most effective vaccine candidate, RTS,S/AS01 [400]. One major immune mechanism involved in malaria is the production of antibodies that target specific parasitic antigens [401].
The potential defenses against flavivirus infection, including DENVs, were thought to depend on whether the NS1 inductive antibodies had a complement-fixing function. The NS1 protein of DENV has many unique properties that make it special for vaccine development. Early studies suggested that the purified NS1 protein could serve as a subunit vaccine candidate for a variety of flaviviruses. Another target against dengue could be creating a recombinant subunit vaccine utilizing the E protein, which may result in a long-lasting protective immune response [402].
The glycoproteins E2 and E1, which are found on the envelope of CHIKV, facilitate the development of subunit vaccines [403]. Animal studies on YFV showed that YFV NS1 inductive antibodies with complement-fixing (CF) activity showed a protective function, while those without CF activity did not, pointing to the crucial role that vaccine-induced CF antibodies play in protection [404].
Vaccine development strategies for the Zika virus include mRNA vaccines, DNA vaccines, and virus-like particle vaccines. DNA vaccines are generally encoded from the viral coding sequence of prM-E regions [405]. As vaccine candidates for various infectious diseases, in vitro transcribed mRNAs that substitute 1-methyl pseudouridine for uridine or 5-methylcytidine for cytidine have now been used [406]. By using this strategy, various RNA vaccines have been developed against ZENV [407].
Furthermore, the DNA vaccine strategy for developing vaccines against RVFV includes encoding of the M segment at the second or fourth methionine that codes for Gn and Gc with (RVFV + NSm DNA) or without (RVFV-NSm DNA) the Nsm protein, respectively [408].
5. Conclusions
Mosquito-borne diseases are major issues in tropical countries. These diseases are caused by various pathogens, such as viruses (DENV, CHIKV, ZIKV, WNV, JEV, YFV, and RVFV) and protozoa (Plasmodium). The initial immune response against mosquito-borne infections is nearly the same across various species of parasites and viruses. In contrast, certain strains at different stages of infection are found to alter the immune microenvironment according to their pathological needs. The infection stimulates the host’s immune system, leading to other metabolic co-morbidities, such as cardiovascular, neurological, neurodegenerative, and renal disorders and carcinoma. This highly complex process involves both the adaptive and innate immune systems, which exert their effects through various molecular and regulatory pathways through changes in the basic nature of cells. The intricate lifecycle and ever-changing antigens present in malaria pose significant challenges to creating an effective vaccine. To improve the potency of treatment, adopting a dual approach that bolsters T cell-mediated immunity during the liver stages and enhances antibody-driven immunity in the blood stages may be beneficial. In the context of mosquito-borne viral diseases, strategies often focus on addressing viral structural and non-structural proteins. Through this review, we have discussed the immune metabolic pathways of each disease in depth so that in the future, we can deepen our understanding of the suitability of each disease for vaccine discovery. We can also increase our understanding of finding new avenues for suitable drug targets for mosquito-borne diseases.
Author Contributions
Concept and design: R.K.P. and M.K.; data acquisition, analysis, and interpretation: S.B., D.G., R.S.(Rounak Saha), R.S.(Rima Sarkar) and S.K.; manuscript drafting: S.B., D.G., R.S.(Rounak Saha), R.S.(Rima Sarkar) and S.K.; diagrammatic presentation: M.K.; manuscript revision: R.K.P.; project supervision: R.K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the manuscript.
Acknowledgments
We would like to acknowledge Biorender for their excellent platform, which we utilized to create professional-quality diagrams for this review article.
Conflicts of Interest
The authors have no relevant financial or non-financial interest to declare.
References
- World Health Organization. Ethics and Vector-Borne Diseases: WHO Guidance; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Tolle, M.A. Mosquito-borne Diseases. Curr. Probl. Pediatr. Adolesc. Health Care 2009, 39, 97–140. [Google Scholar] [CrossRef]
- Oakley, M.S.; Gerald, N.; McCutchan, T.F.; Aravind, L.; Kumar, S. Clinical and molecular aspects of malaria fever. Trends Parasitol. 2011, 27, 442–449. [Google Scholar] [CrossRef]
- Collins, W.E.; Jeffery, G.M. Plasmodium malariae: Parasite and disease. Clin. Microbiol. Rev. 2007, 20, 579–592. [Google Scholar] [CrossRef]
- Hwang, J.; Cullen, K.A.; Kachur, S.P.; Arguin, P.M.; Baird, J.K. Severe morbidity and mortality risk from malaria in the United States, 1985–2011. Open Forum Infect. Dis. 2014, 1, ofu034. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Lindsay, S.W.; Scott, T.W.; Ooi, E.E.; Gubler, D.J.; Das, P. The Lancet Commission on dengue and other Aedes-transmitted viral diseases. Lancet 2020, 395, 1890–1891. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Sherman, M.B.; Weaver, S.C. Chikungunya virus: Evolution and genetic determinants of emergence. Curr. Opin. Virol. 2011, 1, 310–317. [Google Scholar] [CrossRef]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Monath, T.P.; Vasconcelos, P.F.C. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Pagliari, C.; Medeiros, D.B.A.; Duarte, M.I.S.; Vasconcelos, P.F.C. Immunity and immune response, pathology and pathologic changes: Progress and challenges in the immunopathology of yellow fever. Rev. Med. Virol. 2013, 23, 305–318. [Google Scholar] [CrossRef]
- Lequime, S.; Lambrechts, L. Vertical transmission of arboviruses in mosquitoes: A historical perspective. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 28, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Davies, F. Rift Valley fever. Dis. Cattle Trop. Econ. Zoonotic Relev. 1981, 23, 153–165. [Google Scholar]
- Van den Eynde, C.; Sohier, C.; Matthijs, S.; De Regge, N. Japanese Encephalitis Virus Interaction with Mosquitoes: A Review of Vector Competence, Vector Capacity and Mosquito Immunity. Pathogens 2022, 11, 317. [Google Scholar] [CrossRef]
- Girard, Y.A.; Klingler, K.A.; Higgs, S. West Nile virus dissemination and tissue tropisms in orally infected Culex pipiens quinquefasciatus. Vector Borne Zoonotic Dis. 2004, 4, 109–122. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus infection: A balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.F.; Zhang, R.; Wang, T.; Qin, C.F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482–486. [Google Scholar] [CrossRef]
- Boyer, S.; Calvez, E.; Chouin-Carneiro, T.; Diallo, D.; Failloux, A.B. An overview of mosquito vectors of Zika virus. Microbes Infect. 2018, 20, 646–660. [Google Scholar] [CrossRef]
- Hanley, K.A.; Monath, T.P.; Weaver, S.C.; Rossi, S.L.; Richman, R.L.; Vasilakis, N. Fever versus fever: The role of host and vector susceptibility and interspecific competition in shaping the current and future distributions of the sylvatic cycles of dengue virus and yellow fever virus. Infect. Genet. Evol. 2013, 19, 292–311. [Google Scholar] [CrossRef]
- Caron, M.; Paupy, C.; Grard, G.; Becquart, P.; Mombo, I.; Nso, B.B.; Kassa Kassa, F.; Nkoghe, D.; Leroy, E.M. Recent introduction and rapid dissemination of Chikungunya virus and Dengue virus serotype 2 associated with human and mosquito coinfections in Gabon, central Africa. Clin. Infect. Dis. 2012, 55, e45–e53. [Google Scholar] [CrossRef]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1167–1173. [Google Scholar] [CrossRef]
- Shaw, W.R.; Catteruccia, F. Vector biology meets disease control: Using basic research to fight vector-borne diseases. Nat. Microbiol. 2019, 4, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.I.; Richardson, J.H.; Sánchez-Vargas, I.; Olson, K.E.; Beaty, B.J. Dengue virus type 2: Replication and tropisms in orally infected Aedes aegypti mosquitoes. BMC Microbiol. 2007, 7, 9. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Cox, J.; Vanlandingham, D.L.; Feitosa, F.M.; Cheng, G.; Kurscheid, S.; Wang, P.; Krishnan, M.N.; Higgs, S.; Fikrig, E. Alterations in the Aedes aegypti Transcriptome during Infection with West Nile, Dengue and Yellow Fever Viruses. PLoS Pathog. 2011, 7, e1002189. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Ramirez, J.L.; Dimopoulos, G. The Aedes aegypti Toll Pathway Controls Dengue Virus Infection. PLoS Pathog. 2008, 4, e1000098. [Google Scholar] [CrossRef]
- Dong, Y.; Das, S.; Cirimotich, C.; Souza-Neto, J.A.; McLean, K.J.; Dimopoulos, G. Engineered anopheles immunity to Plasmodium infection. PLoS Pathog. 2011, 7, e1002458. [Google Scholar] [CrossRef]
- Clayton, A.M.; Dong, Y.; Dimopoulos, G. The Anopheles innate immune system in the defense against malaria infection. J. Innate Immun. 2014, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Ramphul, U.N.; Garver, L.S.; Molina-Cruz, A.; Canepa, G.E.; Barillas-Mury, C. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1273–1280. [Google Scholar] [CrossRef]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.; Sauerwein, R.W.; Taylor-Salmon, E.; et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef]
- Sim, S.; Jupatanakul, N.; Ramirez, J.L.; Kang, S.; Romero-Vivas, C.M.; Mohammed, H.; Dimopoulos, G. Transcriptomic Profiling of Diverse Aedes aegypti Strains Reveals Increased Basal-level Immune Activation in Dengue Virus-refractory Populations and Identifies Novel Virus-vector Molecular Interactions. PLoS Negl. Trop. Dis. 2013, 7, e2295. [Google Scholar] [CrossRef]
- Behura, S.K.; Gomez-Machorro, C.; Harker, B.W.; deBruyn, B.; Lovin, D.D.; Hemme, R.R.; Mori, A.; Romero-Severson, J.; Severson, D.W. Global Cross-Talk of Genes of the Mosquito Aedes aegypti in Response to Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e1385. [Google Scholar] [CrossRef]
- Sánchez-Vargas, I.; Scott, J.C.; Poole-Smith, B.K.; Franz, A.W.E.; Barbosa-Solomieu, V.; Wilusz, J.; Olson, K.E.; Blair, C.D. Dengue Virus Type 2 Infections of Aedes aegypti Are Modulated by the Mosquito’s RNA Interference Pathway. PLoS Pathog. 2009, 5, e1000299. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Trinidad, L.; Voysey, R.; Duchemin, J.-B.; Walker, P.J. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 18915–18920. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.; Desprès, P.; Choumet, V.; Missé, D. Role of skin immune cells on the host susceptibility to mosquito-borne viruses. Virology 2014, 464–465, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Sinnis, P.; Zavala, F. The skin: Where malaria infection and the host immune response begin. Semin. Immunopathol. 2012, 34, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar] [CrossRef]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Cleary, E.; Hetzel, M.W.; Clements, A.C.A. A review of malaria epidemiology and control in Papua New Guinea 1900 to 2021: Progress made and future directions. Front. Epidemiol. 2022, 2, 795. [Google Scholar] [CrossRef]
- Gomes, P.S.; Bhardwaj, J.; Rivera-Correa, J.; Freire-De-Lima, C.G.; Morrot, A. Immune Escape Strategies of Malaria Parasites. Front. Microbiol. 2016, 7, 1617. [Google Scholar] [CrossRef]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Yam, X.Y.; Preiser, P.R. Host immune evasion strategies of malaria blood stage parasite. Mol. BioSyst. 2017, 13, 2498–2508. [Google Scholar] [CrossRef]
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, 17050. [Google Scholar] [CrossRef]
- Buffet, P.A.; Safeukui, I.; Deplaine, G.; Brousse, V.; Prendki, V.; Thellier, M.; Turner, G.D.; Mercereau-Puijalon, O. The pathogenesis of Plasmodium falciparum malaria in humans: Insights from splenic physiology. Blood 2011, 117, 381–392. [Google Scholar] [CrossRef]
- White, N.J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar. J. 2011, 10, 297. [Google Scholar] [CrossRef]
- Price, R.N.; Tjitra, E.; Guerra, C.A.; Yeung, S.; White, N.J.; Anstey, N.M. Vivax malaria: Neglected and not benign. Am. J. Trop. Med. Hyg. 2007, 77, 79–87. [Google Scholar] [CrossRef]
- Lopez, C.; Yepes-Perez, Y.; Hincapie-Escobar, N.; Diaz-Arevalo, D.; Patarroyo, M.A. What Is Known about the Immune Response Induced by Plasmodium vivax Malaria Vaccine Candidates? Front. Immunol. 2017, 8, 126. [Google Scholar] [CrossRef]
- Popa, G.L.; Popa, M.I. Recent Advances in Understanding the Inflammatory Response in Malaria: A Review of the Dual Role of Cytokines. J. Immunol. Res. 2021, 2021, 7785180. [Google Scholar] [CrossRef] [PubMed]
- Funaro, A.; Spagnoli, G.C.; Ausiello, C.M.; Alessio, M.; Roggero, S.; Delia, D.; Zaccolo, M.; Malavasi, F. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J. Immunol. 1990, 145, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Burel, J.G.; Apte, S.H.; McCarthy, J.S.; Doolan, D.L. Plasmodium vivax but Not Plasmodium falciparum Blood-Stage Infection in Humans Is Associated with the Expansion of a CD8+ T Cell Population with Cytotoxic Potential. PLoS Negl. Trop. Dis. 2016, 10, e0005031. [Google Scholar] [CrossRef]
- Flannery, E.L.; Kangwanrangsan, N.; Chuenchob, V.; Roobsoong, W.; Fishbaugher, M.; Zhou, K.; Billman, Z.P.; Martinson, T.; Olsen, T.M.; Schafer, C.; et al. Plasmodium vivax latent liver infection is characterized by persistent hypnozoites, hypnozoite-derived schizonts, and time-dependent efficacy of primaquine. Mol. Ther. Methods Clin. Dev. 2022, 26, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Renia, L.; Goh, Y.S. Malaria Parasites: The Great Escape. Front. Immunol. 2016, 7, 463. [Google Scholar] [CrossRef]
- Mandala, W.L.; Harawa, V.; Dzinjalamala, F.; Tembo, D. The role of different components of the immune system against Plasmodium falciparum malaria: Possible contribution towards malaria vaccine development. Mol. Biochem. Parasitol. 2021, 246, 111425. [Google Scholar] [CrossRef]
- Belachew, E.B. Immune Response and Evasion Mechanisms of Plasmodium falciparum Parasites. J. Immunol. Res. 2018, 2018, 6529681. [Google Scholar] [CrossRef]
- Vallejo, A.F.; Read, R.C.; Arevalo-Herrera, M.; Herrera, S.; Elliott, T.; Polak, M.E. Malaria systems immunology: Plasmodium vivax induces tolerance during primary infection through dysregulation of neutrophils and dendritic cells. J. Infect. 2018, 77, 440–447. [Google Scholar] [CrossRef]
- Zheng, H.; Tan, Z.; Xu, W. Immune Evasion Strategies of Pre-Erythrocytic Malaria Parasites. Mediat. Inflamm. 2014, 2014, 362605. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, B.M.; Fidock, D.A.; Kyle, D.E.; Kappe, S.H.I.; Alonso, P.L.; Collins, F.H.; Duffy, P.E. Malaria: Progress, perils, and prospects for eradication. J. Clin. Investig. 2008, 118, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Meis, J.F.G.M.; Verhave, J.P.; Jap, P.H.K.; Meuwissen, J.H.E.T. An ultrastructural study on the role of Kupffer cells in the process of infection by Plasmodium berghei sporozoites in rats. Parasitology 2009, 86, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, M.; Nakashima, H.; Kinoshita, M.; Sato, A.; Nakashima, M.; Miyazaki, H.; Nishiyama, K.; Yamamoto, J.; Seki, S. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J. Leukoc. Biol. 2013, 94, 1325–1336. [Google Scholar] [CrossRef]
- Steers, N.; Schwenk, R.; Bacon, D.J.; Berenzon, D.; Williams, J.; Krzych, U. The immune status of Kupffer cells profoundly influences their responses to infectious Plasmodium berghei sporozoites. Eur. J. Immunol. 2005, 35, 2335–2346. [Google Scholar] [CrossRef]
- Mota, M.M.; Pradel, G.; Vanderberg, J.P.; Hafalla, J.C.R.; Frevert, U.; Nussenzweig, R.S.; Nussenzweig, V.; Rodríguez, A. Migration of Plasmodium Sporozoites through Cells before Infection. Science 2001, 291, 141–144. [Google Scholar] [CrossRef]
- Carrolo, M.; Giordano, S.; Cabrita-Santos, L.; Corso, S.; Vigário, A.M.; Silva, S.; Leirião, P.; Carapau, D.; Armas-Portela, R.; Comoglio, P.M.; et al. Hepatocyte growth factor and its receptor are required for malaria infection. Nat. Med. 2003, 9, 1363–1369. [Google Scholar] [CrossRef]
- Leirião, P.; Albuquerque, S.S.; Corso, S.; Van Gemert, G.-J.; Sauerwein, R.W.; Rodriguez, A.; Giordano, S.; Mota, M.M. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell. Microbiol. 2005, 7, 603–609. [Google Scholar] [CrossRef]
- Coppi, A.; Pinzon-Ortiz, C.; Hutter, C.; Sinnis, P. The Plasmodium circumsporozoite protein is proteolytically processed during cell invasion. J. Exp. Med. 2005, 201, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Coppi, A.; Tewari, R.; Bishop, J.R.; Bennett, B.L.; Lawrence, R.; Esko, J.D.; Billker, O.; Sinnis, P. Heparan Sulfate Proteoglycans Provide a Signal to Plasmodium Sporozoites to Stop Migrating and Productively Invade Host Cells. Cell Host Microbe 2007, 2, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Buscaglia, C.A.; Wang, Q.; Levay, A.; Nussenzweig, D.R.; Walker, J.R.; Winzeler, E.A.; Fujii, H.; Fontoura, B.M.A.; Nussenzweig, V. Plasmodium Circumsporozoite Protein Promotes the Development of the Liver Stages of the Parasite. Cell 2007, 131, 492–504. [Google Scholar] [CrossRef]
- Frevert, U.; Galinski, M.R.; Hügel, F.-U.; Allon, N.; Schreier, H.; Smulevitch, S.; Shakibaei, M.; Clavijo, P. Malaria circumsporozoite protein inhibits protein synthesis in mammalian cells. EMBO J. 1998, 17, 3816–3826. [Google Scholar] [CrossRef]
- Kaushansky, A.; Ye, A.S.; Austin, L.S.; Mikolajczak, S.A.; Vaughan, A.M.; Camargo, N.; Metzger, P.G.; Douglass, A.N.; MacBeath, G.; Kappe, S.H. Suppression of Host p53 Is Critical for Plasmodium Liver-Stage Infection. Cell Rep. 2013, 3, 630–637. [Google Scholar] [CrossRef]
- Prudêncio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.; Amino, R.; van de Sand, C.; Regen, T.; Retzlaff, S.; Rennenberg, A.; Krueger, A.; Pollok, J.-M.; Menard, R.; Heussler, V.T. Manipulation of Host Hepatocytes by the Malaria Parasite for Delivery into Liver Sinusoids. Science 2006, 313, 1287–1290. [Google Scholar] [CrossRef]
- Garg, S.; Agarwal, S.; Kumar, S.; Shams Yazdani, S.; Chitnis, C.E.; Singh, S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat. Commun. 2013, 4, 1736. [Google Scholar] [CrossRef]
- Howard, R.J.; Barnwell, J.W.; Kao, V. Antigenic variation of Plasmodium knowlesi malaria: Identification of the variant antigen on infected erythrocytes. Proc. Natl. Acad. Sci. USA 1983, 80, 4129–4133. [Google Scholar] [CrossRef]
- Su, X.Z.; Heatwole, V.M.; Wertheimer, S.P.; Guinet, F.; Herrfeldt, J.A.; Peterson, D.S.; Ravetch, J.A.; Wellems, T.E. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 1995, 82, 89–100. [Google Scholar] [CrossRef]
- Smith, J.D.; Chitnis, C.E.; Craig, A.G.; Roberts, D.J.; Hudson-Taylor, D.E.; Peterson, D.S.; Pinches, R.; Newbold, C.I.; Miller, L.H. Switches in expression of plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 1995, 82, 101–110. [Google Scholar] [CrossRef]
- Handunnetti, S.M.; Mendis, K.N.; David, P.H. Antigenic variation of cloned Plasmodium fragile in its natural host Macaca sinica. Sequential appearance of successive variant antigenic types. J. Exp. Med. 1987, 165, 1269–1283. [Google Scholar] [CrossRef]
- Roberts, D.J.; Craig, A.G.; Berendt, A.R.; Pinches, R.; Nash, G.; Marsh, K.; Newbold, C.I. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature 1992, 357, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Baruch, D.I.; Pasloske, B.L.; Singh, H.B.; Bi, X.; Ma, X.C.; Feldman, M.; Taraschi, T.F.; Howard, R.J. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 1995, 82, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Chan, J.-A.; Howell, K.B.; Reiling, L.; Ataide, R.; Mackintosh, C.L.; Fowkes, F.J.I.; Petter, M.; Chesson, J.M.; Langer, C.; Warimwe, G.M.; et al. Targets of antibodies against Plasmodium falciparum–infected erythrocytes in malaria immunity. J. Clin. Investig. 2012, 122, 3227–3238. [Google Scholar] [CrossRef]
- Fernandez, V.; Hommel, M.; Chen, Q.; Hagblom, P.; Wahlgren, M. Small, Clonally Variant Antigens Expressed on the Surface of the Plasmodium falciparum–Infected Erythrocyte Are Encoded by the rif Gene Family and Are the Target of Human Immune Responses. J. Exp. Med. 1999, 190, 1393–1404. [Google Scholar] [CrossRef]
- Kyes, S.A.; Rowe, J.A.; Kriek, N.; Newbold, C.I. Rifins: A second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1999, 96, 9333–9338. [Google Scholar] [CrossRef]
- Petter, M.; Haeggström, M.; Khattab, A.; Fernandez, V.; Klinkert, M.-Q.; Wahlgren, M. Variant proteins of the Plasmodium falciparum RIFIN family show distinct subcellular localization and developmental expression patterns. Mol. Biochem. Parasitol. 2007, 156, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.; Petter, M.; Tilly, A.-K.; Biller, L.; Uliczka, K.A.; Duffy, M.F.; Tannich, E.; Bruchhaus, I. Temporal Expression and Localization Patterns of Variant Surface Antigens in Clinical Plasmodium falciparum Isolates during Erythrocyte Schizogony. PLoS ONE 2012, 7, e49540. [Google Scholar] [CrossRef]
- Hiller, N.L.; Bhattacharjee, S.; van Ooij, C.; Liolios, K.; Harrison, T.; Lopez-Estraño, C.; Haldar, K. A Host-Targeting Signal in Virulence Proteins Reveals a Secretome in Malarial Infection. Science 2004, 306, 1934–1937. [Google Scholar] [CrossRef]
- Howard, R.J.; Barnwell, J.W.; Rock, E.P.; Neequaye, J.; Ofori-Adjei, D.; Lee Maloy, W.; Lyon, J.A.; Saul, A. Two approximately 300 kilodalton Plasmodium falciparum proteins at the surface membrane of infected erythrocytes. Mol. Biochem. Parasitol. 1988, 27, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Biggs, B.A.; Goozé, L.; Wycherley, K.; Wollish, W.; Southwell, B.; Leech, J.H.; Brown, G.V. Antigenic variation in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1991, 88, 9171–9174. [Google Scholar] [CrossRef]
- Bull, P.C.; Abdi, A.I. The role of PfEMP1 as targets of naturally acquired immunity to childhood malaria: Prospects for a vaccine. Parasitology 2016, 143, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Deitsch, K.W.; Hviid, L. Variant surface antigens, virulence genes and the pathogenesis of malaria. Trends Parasitol. 2004, 20, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.L.; Cooke, B.M.; Coppel, R.L.; Nunomura, W.; Mohandas, N. Mapping the Binding Domains Involved in the Interaction between the Plasmodium falciparum Knob-associated Histidine-rich Protein (KAHRP) and the Cytoadherence Ligand P. falciparum Erythrocyte Membrane Protein 1 (PfEMP1). J. Biol. Chem. 1999, 274, 23808–23813. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.; Lavstsen, T.; Berger, S.S.; Wang, C.W.; Petersen, J.E.V.; Avril, M.; Brazier, A.J.; Freeth, J.; Jespersen, J.S.; Nielsen, M.A.; et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 2013, 498, 502–505. [Google Scholar] [CrossRef]
- Howell, D.P.-G.; Levin, E.A.; Springer, A.L.; Kraemer, S.M.; Phippard, D.J.; Schief, W.R.; Smith, J.D. Mapping a common interaction site used by Plasmodium falciparum Duffy binding-like domains to bind diverse host receptors. Mol. Microbiol. 2008, 67, 78–87. [Google Scholar] [CrossRef]
- Fried, M.; Duffy, P.E. Adherence of Plasmodium falciparum to Chondroitin Sulfate A in the Human Placenta. Science 1996, 272, 1502–1504. [Google Scholar] [CrossRef]
- Baruch, D.I.; Gormely, J.A.; Ma, C.; Howard, R.J.; Pasloske, B.L. Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1996, 93, 3497–3502. [Google Scholar] [CrossRef]
- Vogt, A.M.; Barragan, A.; Chen, Q.; Kironde, F.; Spillmann, D.; Wahlgren, M. Heparan sulfate on endothelial cells mediates the binding ofPlasmodium falciparum–infected erythrocytes via the DBL1α domain of PfEMP1. Blood 2003, 101, 2405–2411. [Google Scholar] [CrossRef]
- Stevenson, L.; Laursen, E.; Cowan, G.J.; Bandoh, B.; Barfod, L.; Cavanagh, D.R.; Andersen, G.R.; Hviid, L. α2-Macroglobulin Can Crosslink Multiple Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) Molecules and May Facilitate Adhesion of Parasitized Erythrocytes. PLoS Pathog. 2015, 11, e1005022. [Google Scholar] [CrossRef]
- Ghumra, A.; Semblat, J.-P.; McIntosh, R.S.; Raza, A.; Rasmussen, I.B.; Braathen, R.; Johansen, F.-E.; Sandlie, I.; Mongini, P.K.; Rowe, J.A.; et al. Identification of Residues in the Cμ4 Domain of Polymeric IgM Essential for Interaction with Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1)1. J. Immunol. 2008, 181, 1988–2000. [Google Scholar] [CrossRef] [PubMed]
- Adams, Y.; Kuhnrae, P.; Higgins, M.K.; Ghumra, A.; Rowe, J.A. Rosetting Plasmodium falciparum-Infected Erythrocytes Bind to Human Brain Microvascular Endothelial Cells In Vitro, Demonstrating a Dual Adhesion Phenotype Mediated by Distinct P. falciparum Erythrocyte Membrane Protein 1 Domains. Infect. Immun. 2014, 82, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.-M.; Rowe, J.A. Plasmodium falciparum: Rosettes do not protect merozoites from invasion-inhibitory antibodies. Exp. Parasitol. 2006, 112, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, M.; Carlson, J.; Udomsangpetch, R.; Perlmann, P. Why do Plasmodium falciparumm-infected erythrocytes form spontaneous erythrocyte rosettes? Parasitol. Today 1989, 5, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Yam, X.Y.; Niang, M.; Madnani, K.G.; Preiser, P.R. Three Is a Crowd–New Insights into Rosetting in Plasmodium falciparum. Trends Parasitol. 2017, 33, 309–320. [Google Scholar] [CrossRef]
- Rowe, J.A.; Moulds, J.M.; Newbold, C.I.; Miller, L.H.P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature 1997, 388, 292–295. [Google Scholar] [CrossRef]
- Chen, Q.; Barragan, A.; Fernandez, V.; Sundström, A.; Schlichtherle, M.; Sahlén, A.; Carlson, J.; Datta, S.; Wahlgren, M. Identification of Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) as the Rosetting Ligand of the Malaria Parasite, P. falciparum. J. Exp. Med. 1998, 187, 15–23. [Google Scholar] [CrossRef]
- Albrecht, L.; Moll, K.; Blomqvist, K.; Normark, J.; Chen, Q.; Wahlgren, M. var gene transcription and PfEMP1 expression in the rosetting and cytoadhesive Plasmodium falciparum clone FCR3S1.2. Malar. J. 2011, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Vigan-Womas, I.; Guillotte, M.; Juillerat, A.; Vallieres, C.; Lewit-Bentley, A.; Tall, A.; Baril, L.; Bentley, G.A.; Mercereau-Puijalon, O. Allelic Diversity of the Plasmodium falciparum Erythrocyte Membrane Protein 1 Entails Variant-Specific Red Cell Surface Epitopes. PLoS ONE 2011, 6, e16544. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, M.; Goel, S.; Akhouri, R.R. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat. Rev. Microbiol. 2017, 15, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Akhouri, R.R.; Goel, S.; Furusho, H.; Skoglund, U.; Wahlgren, M. Architecture of Human IgM in Complex with P. falciparum Erythrocyte Membrane Protein 1. Cell Rep. 2016, 14, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Palmkvist, M.; Moll, K.; Joannin, N.; Lara, P.; R Akhouri, R.; Moradi, N.; Öjemalm, K.; Westman, M.; Angeletti, D.; et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat. Med. 2015, 21, 314–317. [Google Scholar] [CrossRef]
- Moll, K.; Palmkvist, M.; Ch’ng, J.; Kiwuwa, M.S.; Wahlgren, M. Evasion of Immunity to Plasmodium falciparum: Rosettes of Blood Group A Impair Recognition of PfEMP1. PLoS ONE 2016, 10, e0145120. [Google Scholar] [CrossRef]
- Niang, M.; Bei, A.K.; Madnani, K.G.; Pelly, S.; Dankwa, S.; Kanjee, U.; Gunalan, K.; Amaladoss, A.; Yeo, K.P.; Bob, N.S.; et al. STEVOR Is a Plasmodium falciparum Erythrocyte Binding Protein that Mediates Merozoite Invasion and Rosetting. Cell Host Microbe 2014, 16, 81–93. [Google Scholar] [CrossRef]
- Singh, H.; Madnani, K.; Lim, Y.B.; Cao, J.; Preiser, P.R.; Lim, C.T. Expression dynamics and physiologically relevant functional study of STEVOR in asexual stages of Plasmodium falciparum infection. Cell. Microbiol. 2017, 19, e12715. [Google Scholar] [CrossRef]
- Sylla, B.S.; Wild, C.P. A million africans a year dying from cancer by 2030: What can cancer research and control offer to the continent? Int. J. Cancer 2012, 130, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, K.; Tongren, J.E.; Riley, E.M. The war between the malaria parasite and the immune system: Immunity, immunoregulation and immunopathology. Clin. Exp. Immunol. 2003, 133, 145–152. [Google Scholar] [CrossRef]
- Yao, S.; Hong, C.-C.; Ruiz-Narváez, E.A.; Evans, S.S.; Zhu, Q.; Schaefer, B.A.; Yan, L.; Coignet, M.V.; Lunetta, K.L.; Sucheston-Campbell, L.E.; et al. Genetic ancestry and population differences in levels of inflammatory cytokines in women: Role for evolutionary selection and environmental factors. PLoS Genet. 2018, 14, e1007368. [Google Scholar] [CrossRef]
- Wang, J.; Ou, Z.L.; Hou, Y.F.; Luo, J.M.; Shen, Z.Z.; Ding, J.; Shao, Z.M. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene 2006, 25, 7201–7211. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Chaudhuri, A.; Watabe, M.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006, 12, 933–938. [Google Scholar] [CrossRef]
- Bentley, G.A.; Gamain, B. How does Plasmodium falciparum stick to CSA? Let’s see in the crystal. Nat. Struct. Mol. Biol. 2008, 15, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Cooney, C.A.; Jousheghany, F.; Yao-Borengasser, A.; Phanavanh, B.; Gomes, T.; Kieber-Emmons, A.M.; Siegel, E.R.; Suva, L.J.; Ferrone, S.; Kieber-Emmons, T.; et al. Chondroitin sulfates play a major role in breast cancer metastasis: A role for CSPG4 and CHST11gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011, 13, R58. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.; Deitsch, K.W.; Duca, K.A.; Torgbor, C. The Link between Plasmodium falciparum Malaria and Endemic Burkitt’s Lymphoma-New Insight into a 50-Year-Old Enigma. PLoS Pathog. 2016, 12, e1005331. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef]
- Malagon, F.; Gonzalez-Angulo, J.; Carrasco, E.; Robert, L. Etiopathogenesis of Burkitt’s lymphoma: A lesson from a BL-like in CD1 mouse immune to Plasmodium yoelii yoelii. Infect. Agents Cancer 2011, 6, 10. [Google Scholar] [CrossRef]
- Alencar Filho, A.C.; Lacerda, M.V.; Okoshi, K.; Okoshi, M.P. Malaria and vascular endothelium. Arq. Bras. Cardiol. 2014, 103, 165–169. [Google Scholar] [CrossRef]
- Holm, A.E.; Gomes, L.C.; Marinho, C.R.F.; Silvestre, O.M.; Vestergaard, L.S.; Biering-Sorensen, T.; Brainin, P. Prevalence of Cardiovascular Complications in Malaria: A Systematic Review and Meta-Analysis. Am. J. Trop. Med. Hyg. 2021, 104, 1643–1650. [Google Scholar] [CrossRef]
- Brainin, P.; Mohr, G.H.; Modin, D.; Claggett, B.; Silvestre, O.M.; Shah, A.; Vestergaard, L.S.; Jensen, J.U.S.; Hviid, L.; Torp-Pedersen, C.; et al. Heart failure associated with imported malaria: A nationwide Danish cohort study. ESC Heart Fail. 2021, 8, 3521–3529. [Google Scholar] [CrossRef]
- Pain, A.; Ferguson, D.J.P.; Kai, O.; Urban, B.C.; Lowe, B.; Marsh, K.; Roberts, D.J. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc. Natl. Acad. Sci. USA 2001, 98, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Wennicke, K.; Debierre-Grockiego, F.; Wichmann, D.; Brattig, N.W.; Pankuweit, S.; Maisch, B.; Schwarz, R.T.; Ruppert, V. Glycosylphosphatidylinositol-induced cardiac myocyte death might contribute to the fatal outcome of Plasmodium falciparum malaria. Apoptosis 2008, 13, 857–866. [Google Scholar] [CrossRef]
- Huber, S.A.; Sartini, D. Roles of Tumor Necrosis Factor Alpha (TNF-α) and the p55 TNF Receptor in CD1d Induction and Coxsackievirus B3-Induced Myocarditis. J. Virol. 2005, 79, 2659–2665. [Google Scholar] [CrossRef]
- Tatsumi, T.; Akashi, K.; Keira, N.; Matoba, S.; Mano, A.; Shiraishi, J.; Yamanaka, S.; Kobara, M.; Hibino, N.; Hosokawa, S.; et al. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J. Mol. Cell. Cardiol. 2004, 37, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Colomba, C.; Trizzino, M.; Gioè, C.; Coelho, F.; Lopo, I.; Pinheiro, P.; Sousa, J.; Cascio, A. Malaria and the heart: Two rare case reports of Plasmodium falciparumassociated pericarditis. J. Vector Borne Dis. 2017, 54, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, A.H.; Green, S.T.; West, J.N.; McKendrick, M.W. Myocarditis Associated with Plasmodium falciparum Malaria: A Case Report and a Review of the Literature. J. Travel Med. 2006, 8, 219–220. [Google Scholar] [CrossRef]
- Cantalupo, A.; Gargiulo, A.; Dautaj, E.; Liu, C.; Zhang, Y.; Hla, T.; Lorenzo, A.D. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 2017, 70, 426–434. [Google Scholar] [CrossRef]
- Dhangadamajhi, G.; Singh, S. Malaria link of hypertension: A hidden syndicate of angiotensin II, bradykinin and sphingosine 1-phosphate. Hum. Cell 2021, 34, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Kingston, H.W.F.; Ghose, A.; Rungpradubvong, V.; Satitthummanid, S.; Herdman, M.T.; Plewes, K.; Leopold, S.J.; Ishioka, H.; Mohanty, S.; Maude, R.J.; et al. Reduced Cardiac Index Reserve and Hypovolemia in Severe Falciparum Malaria. J. Infect. Dis. 2019, 221, 1518–1527. [Google Scholar] [CrossRef]
- Akide Ndunge, O.B.; Kilian, N.; Salman, M.M. Cerebral Malaria and Neuronal Implications of Plasmodium Falciparum Infection: From Mechanisms to Advanced Models. Adv. Sci. 2022, 9, e2202944. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.-U.; Lao, Y.; Spicer, V.; Coombs, K.M. Zika Virus Infection of Sertoli Cells Alters Protein Expression Involved in Activated Immune and Antiviral Response Pathways, Carbohydrate Metabolism and Cardiovascular Disease. Viruses 2022, 14, 377. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nguyen, F.N.; Manfredsson, F.P.; Kondrikova, G.; Sullivan, L.F.; Meyers, C.; Chen, W.; Mandel, R.J.; Muzyczka, N. α-Synuclein Expression in Rat Substantia Nigra Suppresses Phospholipase D2 Toxicity and Nigral Neurodegeneration. Mol. Ther. 2010, 18, 1758–1768. [Google Scholar] [CrossRef]
- Gandarilla-Martínez, N.; Carbayo-Viejo, Á.; Sánchez-Gómez, A.; Valldeoriola-Serra, F. Parkinsonism in a patient with Plasmodium falciparum malaria. Park. Relat. Disord. 2020, 80, 181–183. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; van der Pol, S.M.A.; Jansen, Q.; Witte, M.E.; van der Valk, P.; Rozemuller, A.J.M.; Drukarch, B.; de Vries, H.E.; Van Horssen, J. Association of Parkinson disease-related protein PINK1 with Alzheimer disease and multiple sclerosis brain lesions. Free Radic. Biol. Med. 2011, 50, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Kuhle, J.; Rüegg, S.; Lindberg, R.L.P.; Petzold, A.; Sulejczak, D.; Papuc, E.; Rejdak, R.; Stelmasiak, Z.; Grieb, P. Neurofilament heavy chain and heat shock protein 70 as markers of seizure-related brain injury. Epilepsia 2012, 53, 922–927. [Google Scholar] [CrossRef]
- Anstey, N.M.; Douglas, N.M.; Poespoprodjo, J.R.; Price, R.N. Chapter Three—Plasmodium vivax: Clinical Spectrum, Risk Factors and Pathogenesis. In Advances in Parasitology; Hay, S.I., Price, R., Baird, J.K., Eds.; Academic Press: New York, NY, USA, 2012; Volume 80, pp. 151–201. [Google Scholar]
- de Silva, H.J.; Hoang, P.; Dalton, H.; de Silva, N.R.; Jewell, D.P.; Peiris, J.B. Immune activation during cerebellar dysfunction following Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 129–131. [Google Scholar] [CrossRef]
- Bergmark, B.; Bergmark, R.; Beaudrap, P.D.; Boum, Y.; Mwanga-Amumpaire, J.; Carroll, R.; Zapol, W. Inhaled Nitric Oxide and Cerebral Malaria: Basis of a Strategy for Buying Time for Pharmacotherapy. Pediatr. Infect. Dis. J. 2012, 31, e250–e254. [Google Scholar] [CrossRef]
- Turner, G.D.; Morrison, H.; Jones, M.; Davis, T.M.; Looareesuwan, S.; Buley, I.D.; Gatter, K.C.; Newbold, C.I.; Pukritayakamee, S.; Nagachinta, B.; et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am. J. Pathol. 1994, 145, 1057–1069. [Google Scholar]
- Castaldo, N.; Tascini, C.; Della Siega, P.; Peghin, M.; Pecori, D. Clinical presentation and immunological features of Post-Malaria Neurologic Syndrome: A case report and review of literature. Malar. J. 2020, 19, 419. [Google Scholar] [CrossRef]
- Carter, J.A.; Neville, B.G.R.; White, S.; Ross, A.J.; Otieno, G.; Mturi, N.; Musumba, C.; Newton, C.R.J.C. Increased Prevalence of Epilepsy Associated with Severe Falciparum Malaria in Children. Epilepsia 2004, 45, 978–981. [Google Scholar] [CrossRef]
- Birbeck, G.L.; Molyneux, M.E.; Kaplan, P.W.; Seydel, K.B.; Chimalizeni, Y.F.; Kawaza, K.; Taylor, T.E. Blantyre Malaria Project Epilepsy Study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: A prospective cohort study. Lancet Neurol. 2010, 9, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Katsoulis, O.; Georgiadou, A.; Cunnington, A.J. Immunopathology of Acute Kidney Injury in Severe Malaria. Front. Immunol. 2021, 12, 651739. [Google Scholar] [CrossRef]
- Eiam-Ong, S.; Sitprija, V. Falciparum malaria and the kidney: A model of inflammation. Am. J. Kidney Dis. 1998, 32, 361–375. [Google Scholar] [CrossRef]
- Plewes, K.; Turner, G.D.H.; Dondorp, A.M. Pathophysiology, clinical presentation, and treatment of coma and acute kidney injury complicating falciparum malaria. Curr. Opin. Infect. Dis. 2018, 31, 69–77. [Google Scholar] [CrossRef]
- Kanodia, K.V.; Shah, P.R.; Vanikar, A.V.; Kasat, P.; Gumber, M.; Trivedi, H.L. Malaria induced acute renal failure: A single center experience. Saudi J. Kidney Dis. Transplant. 2010, 21, 1088–1091. [Google Scholar]
- Lyke, K.E.; Burges, R.; Cissoko, Y.; Sangare, L.; Dao, M.; Diarra, I.; Kone, A.; Harley, R.; Plowe, C.V.; Doumbo, O.K.; et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 2004, 72, 5630–5637. [Google Scholar] [CrossRef]
- Sinniah, R.; Rui-Mei, L.; Kara, A. Up-regulation of cytokines in glomerulonephritis associated with murine malaria infection. Int. J. Exp. Pathol. 1999, 80, 87–95. [Google Scholar] [CrossRef]
- Barber, B.E.; Grigg, M.J.; Piera, K.A.; William, T.; Cooper, D.J.; Plewes, K.; Dondorp, A.M.; Yeo, T.W.; Anstey, N.M. Intravascular haemolysis in severe Plasmodium knowlesi malaria: Association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, P.H.; Morris-Jones, S.D.; Hviid, L.; Theander, T.G.; Høier-Madsen, M.; Bayoumi, R.A.; Greenwood, B.M. Anti-phospholipid antibodies in patients with Plasmodium falciparum malaria. Immunology 1993, 79, 653–657. [Google Scholar] [PubMed]
- de Alwis, R.; Williams, K.L.; Schmid, M.A.; Lai, C.Y.; Patel, B.; Smith, S.A.; Crowe, J.E.; Wang, W.K.; Harris, E.; de Silva, A.M. Dengue viruses are enhanced by distinct populations of serotype cross-reactive antibodies in human immune sera. PLoS Pathog. 2014, 10, e1004386. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Gale, M., Jr. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Nasirudeen, A.M.A.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.-P.; Liu, D.X. RIG-I, MDA5 and TLR3 Synergistically Play an Important Role in Restriction of Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, S.; Li, Y.; He, L.; Wu, M.; Jiang, L.; Feng, L.; Zhang, P.; Huang, X. Activation of Toll-Like Receptor 3 Impairs the Dengue Virus Serotype 2 Replication through Induction of IFN-β in Cultured Hepatoma Cells. PLoS ONE 2011, 6, e23346. [Google Scholar] [CrossRef]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-H.; Wang, M.-Y.; Huang, C.-Y.; Wu, C.-H.; Hung, L.-F.; Yang, C.-Y.; Ke, P.-Y.; Luo, S.-F.; Liu, S.-J.; Ho, L.-J. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, A.; Manoharan, M. Chapter 16—Dengue Virus. In Emerging and Reemerging Viral Pathogens; Ennaji, M.M., Ed.; Academic Press: New York, NY, USA, 2020; pp. 281–359. [Google Scholar]
- Reich, N.G.; Shrestha, S.; King, A.A.; Rohani, P.; Lessler, J.; Kalayanarooj, S.; Yoon, I.K.; Gibbons, R.V.; Burke, D.S.; Cummings, D.A. Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J. R. Soc. Interface 2013, 10, 20130414. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Shresta, S.; Sette, A. Immunodominance changes as a function of the infecting dengue virus serotype and primary versus secondary infection. J. Virol. 2014, 88, 11383–11394. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A. Plasma leakage in dengue haemorrhagic fever. Thromb. Haemost. 2009, 102, 1042–1049. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Chang, D.C.; Hoang, L.T.; Mohamed Naim, A.N.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.-Y. Evasion of early innate immune response by 2′-O-methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef]
- Dong, H.; Chang, D.C.; Hua, M.H.C.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.-M.; Dedon, P.C.; et al. 2′-O Methylation of Internal Adenosine by Flavivirus NS5 Methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Turtle, L.; Bali, T.; Buxton, G.; Chib, S.; Chan, S.; Soni, M.; Hussain, M.; Isenman, H.; Fadnis, P.; Venkataswamy, M.M.; et al. Human T cell responses to Japanese encephalitis virus in health and disease. J. Exp. Med. 2016, 213, 1331–1352. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.-M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The Mitochondrial Targeting Chaperone 14-3-3ε; Regulates a RIG-I Translocon that Mediates Membrane Association and Innate Antiviral Immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio 2015, 6, e00553-15. [Google Scholar] [CrossRef]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. P122 Dengue virus targets the adaptor protein MITA to subvert host innate immunity. Cytokine 2012, 59, 558. [Google Scholar] [CrossRef]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue Virus Co-opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Tambyah, P.A.; Ching, C.S.; Sepramaniam, S.; Ali, J.M.; Armugam, A.; Jeyaseelan, K. microRNA expression in blood of dengue patients. Ann. Clin. Biochem. 2016, 53, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Jiang, X.; Gu, D.; Zhang, Y.; Kong, S.K.; Jiang, C.; Xie, W. Dysregulated Serum MiRNA Profile and Promising Biomarkers in Dengue-infected Patients. Int. J. Med. Sci. 2016, 13, 195–205. [Google Scholar] [CrossRef]
- Kanokudom, S.; Vilaivan, T.; Wikan, N.; Thepparit, C.; Smith, D.R.; Assavalapsakul, W. miR-21 promotes dengue virus serotype 2 replication in HepG2 cells. Antivir. Res. 2017, 142, 169–177. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; He, L.; Li, Y.; Wang, T.; Feng, L.; Jiang, L.; Zhang, P.; Huang, X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J. Infect. 2013, 67, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.-C.; Yu, I.S.; Lu, L.-F.; Rudensky, A.; Chen, H.-Y.; Tsai, C.-W.; Chang, Y.-L.; Wu, C.-T.; Chang, L.-Y.; Shih, S.-R.; et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat. Commun. 2014, 5, 3344. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, L.; Zeng, Y.; Si, L.; Guo, X.; Zhou, J.; Fang, D.; Zeng, G.; Jiang, L. Suppressed expression of miR-378 targeting gzmb in NK cells is required to control dengue virus infection. Cell. Mol. Immunol. 2016, 13, 700–708. [Google Scholar] [CrossRef]
- Chien, Y.-W.; Wang, C.-C.; Wang, Y.-P.; Lee, C.-Y.; Perng, G.C. Risk of Leukemia after Dengue Virus Infection: A Population-Based Cohort Study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 558–564. [Google Scholar] [CrossRef]
- Talwar, V.; Goel, V.; Raina, S.; Talwar, J.; Patnaik, N.; Doval, D.C. Dengue fever in cancer patients: Retrospective analysis. Curr. Med. Res. Pract. 2016, 6, 157–159. [Google Scholar] [CrossRef]
- Gan, E.S.; Tan, H.C.; Le, D.H.T.; Huynh, T.T.; Wills, B.; Seidah, N.G.; Ooi, E.E.; Yacoub, S. Dengue virus induces PCSK9 expression to alter antiviral responses and disease outcomes. J. Clin. Investig. 2020, 130, 5223–5234. [Google Scholar] [CrossRef]
- Almontashiri, N.A.M.; Vilmundarson, R.O.; Ghasemzadeh, N.; Dandona, S.; Roberts, R.; Quyyumi, A.A.; Chen, H.-H.; Stewart, A.F.R. Plasma PCSK9 Levels Are Elevated with Acute Myocardial Infarction in Two Independent Retrospective Angiographic Studies. PLoS ONE 2014, 9, e106294. [Google Scholar] [CrossRef]
- Miranda, C.H.; Borges, M.d.C.; Matsuno, A.K.; Vilar, F.C.; Gali, L.G.; Volpe, G.J.; Schmidt, A.; Pazin-Filho, A.; da Silva, F.M.F.; de Castro-Jorge, L.A.; et al. Evaluation of Cardiac Involvement During Dengue Viral Infection. Clin. Infect. Dis. 2013, 57, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Salgado, D.M.; Eltit, J.M.; Mansfield, K.; Panqueba, C.; Castro, D.; Vega, M.R.; Xhaja, K.; Schmidt, D.; Martin, K.J.; Allen, P.D.; et al. Heart and skeletal muscle are targets of dengue virus infection. Pediatr. Infect. Dis. J. 2010, 29, 238–242. [Google Scholar] [CrossRef]
- La-Orkhun, V.; Supachokchaiwattana, P.; Lertsapcharoen, P.; Khongphatthanayothin, A. Spectrum of cardiac rhythm abnormalities and heart rate variability during the convalescent stage of dengue virus infection: A Holter study. Ann. Trop. Paediatr. 2011, 31, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Thacker, M.M.; Makwana, P.V. Ventricular Tachycardia in Primary Dengue Fever. J. Assoc. Physicians India 2018, 66, 100. [Google Scholar] [PubMed]
- Chaturvedi, U.C.; Mathur, A.; Mehrotra, R.M. Experimentally produced cardiac injury following dengue virus infection. Indian J. Pathol. Bacteriol. 1974, 17, 218–220. [Google Scholar]
- Araújo, F.M.C.; Araújo, M.S.; Nogueira, R.M.R.; Brilhante, R.S.N.; Oliveira, D.N.; Rocha, M.F.G.; Cordeiro, R.A.; Araújo, R.M.C.; Sidrim, J.J.C. Central nervous system involvement in dengue. Neurology 2012, 78, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, U.C.; Dhawan, R.; Khanna, M.; Mathur, A. Breakdown of the blood-brain barrier during dengue virus infection of mice. J. Gen. Virol. 1991, 72, 859–866. [Google Scholar] [CrossRef]
- Bordignon, J.; Strottmann, D.M.; Mosimann, A.L.; Probst, C.M.; Stella, V.; Noronha, L.; Zanata, S.M.; Dos Santos, C.N. Dengue neurovirulence in mice: Identification of molecular signatures in the E and NS3 helicase domains. J. Med. Virol. 2007, 79, 1506–1517. [Google Scholar] [CrossRef]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef]
- Mathew, S.; Pandian, J.D. Stroke in Patients with Dengue. J. Stroke Cerebrovasc. Dis. 2010, 19, 253–256. [Google Scholar] [CrossRef]
- Panda, P.K.; Sharawat, I.K.; Bolia, R.; Shrivastava, Y. Case Report: Dengue Virus-Triggered Parkinsonism in an Adolescent. Am. J. Trop. Med. Hyg. 2020, 103, 851–854. [Google Scholar] [CrossRef]
- Cam, B.V.; Fonsmark, L.; Hue, N.B.; Phuong, N.T.; Poulsen, A.; Heegaard, E.D. Prospective case-control study of encephalopathy in children with dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 2001, 65, 848–851. [Google Scholar] [CrossRef]
- Lana-Peixoto, M.A.; Pedrosa, D.; Talim, N.; Amaral, J.; Horta, A.; Kleinpaul, R. Neuromyelitis optica spectrum disorder associated with dengue virus infection. J. Neuroimmunol. 2018, 318, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.G.; Nam, N.T.; Han, N.H.B.; El-Shabouny, A.-E.; Makram, A.-E.M.; Abd-Elhay, F.A.-E.; Dang, T.N.; Hieu, N.L.T.; Huong, V.T.Q.; Tung, T.H.; et al. Post-dengue acute disseminated encephalomyelitis: A case report and meta-analysis. PLoS Negl. Trop. Dis. 2017, 11, e0005715. [Google Scholar] [CrossRef]
- Nair, J.J.; Bhat, A.; Prabhu, M.V. A Clinical Study of Acute Kidney Injury in Tropical Acute Febrile Illness. J. Clin. Diagn. Res. 2016, 10, OC01–OC05. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, C.; Simões Quaresma, J.A.; Kanashiro-Galo, L.; de Carvalho, L.V.; Vitoria, W.O.; da Silva, W.L.F.; Penny, R.; Vasconcelos, B.C.B.; da Costa Vasconcelos, P.F.; Duarte, M.I.S. Human kidney damage in fatal dengue hemorrhagic fever results of glomeruli injury mainly induced by IL17. J. Clin. Virol. 2016, 75, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.L.; McBride, W.J.H.; Hanna, J.N. Clinical Features of Hospitalized Patients During Dengue-3 Epidemic in Far North Queensland, 1997–1999. Dengue Bull. 2002, 23, 24–29. [Google Scholar]
- Jayarajah, U.; Madarasinghe, M.; Hapugoda, D.; Dissanayake, U.; Perera, L.; Kannangara, V.; Udayangani, C.; Peiris, R.; Yasawardene, P.; De Zoysa, I.; et al. Clinical and Biochemical Characteristics of Dengue Infections in Children From Sri Lanka. Glob. Pediatr. Health 2020, 7, 2333794X20974207. [Google Scholar] [CrossRef] [PubMed]
- Talib, S.; Bhattu, S.; Bhattu, R.; Deshpande, S.; Dahiphale, D. Dengue fever triggering systemic lupus erythematosus and lupus nephritis: A case report. Int. Med. Case Rep. J. 2013, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, K.J.; Florindez, J.A.; Daftarian, P.; Andrews, D.M.; Ortega, L.M.; Mendoza, J.M.; Contreras, G.N.; Nayer, A. Anti-GBM disease and ANCA during dengue infection. Clin. Nephrol. 2015, 83, 104–110. [Google Scholar] [CrossRef]
- Gutiérrez-Bugallo, G.; Piedra, L.A.; Rodriguez, M.; Bisset, J.A.; Lourenço-de-Oliveira, R.; Weaver, S.C.; Vasilakis, N.; Vega-Rúa, A. Vector-borne transmission and evolution of Zika virus. Nat. Ecol. Evol. 2019, 3, 561–569. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonça, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Kuhn, R.J. Zika Virus Structure, Maturation, and Receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira França, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef]
- Dang, J.W.; Tiwari, S.K.; Qin, Y.; Rana, T.M. Genome-wide Integrative Analysis of Zika-Virus-Infected Neuronal Stem Cells Reveals Roles for MicroRNAs in Cell Cycle and Stemness. Cell Rep. 2019, 27, 3618–3628.e5. [Google Scholar] [CrossRef] [PubMed]
- Baril, M.; Racine, M.-E.; Penin, F.; Lamarre, D. MAVS Dimer Is a Crucial Signaling Component of Innate Immunity and the Target of Hepatitis C Virus NS3/4A Protease. J. Virol. 2009, 83, 1299–1311. [Google Scholar] [CrossRef]
- Glasner, A.; Oiknine-Djian, E.; Weisblum, Y.; Diab, M.; Panet, A.; Wolf, D.G.; Mandelboim, O. Zika Virus Escapes NK Cell Detection by Upregulating Major Histocompatibility Complex Class I Molecules. J. Virol. 2017, 91, e00785-17. [Google Scholar] [CrossRef]
- Branche, E.; Wang, Y.-T.; Viramontes, K.M.; Valls Cuevas, J.M.; Xie, J.; Ana-Sosa-Batiz, F.; Shafee, N.; Duttke, S.H.; McMillan, R.E.; Clark, A.E.; et al. SREBP2-dependent lipid gene transcription enhances the infection of human dendritic cells by Zika virus. Nat. Commun. 2022, 13, 5341. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis. Microbiol. Mol. Biol. Rev. 2017, 81, e00055-16. [Google Scholar] [CrossRef] [PubMed]
- Estevez-Herrera, J.; Perez-Yanes, S.; Cabrera-Rodriguez, R.; Marquez-Arce, D.; Trujillo-Gonzalez, R.; Machado, J.D.; Madrid, R.; Valenzuela-Fernandez, A. Zika Virus Pathogenesis: A Battle for Immune Evasion. Vaccines 2021, 9, 294. [Google Scholar] [CrossRef]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef]
- Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandao, L.; Crovella, S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. Int. J. Mol. Sci. 2019, 20, 1048. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Pereira, J.P.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.-A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Barron, T.F.; Vannucci, S.J. Craniometric measures of microcephaly using MRI. Early Hum. Dev. 2012, 88, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; Smith, R.A.; Darlington, L.G.; Stone, T.W. Prenatal activation of Toll-like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Mol. Brain 2012, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; Riesgo, R.S.; Quincozes-Santos, A.; Schuler-Faccini, L.; Tureta, E.F.; Rosa, R.L.; Berger, M.; Oliveira, A.C.C.; Beltrão-Braga, P.C.B.; Souza, D.O.; et al. Zika Virus Infection Associated with Autism Spectrum Disorder: A Case Report. Neuroimmunomodulation 2021, 28, 229–232. [Google Scholar] [CrossRef]
- Blackmon, K.; Waechter, R.; Landon, B.; Noël, T.; Macpherson, C.; Donald, T.; Cudjoe, N.; Evans, R.; Burgen, K.S.; Jayatilake, P.; et al. Epilepsy surveillance in normocephalic children with and without prenatal Zika virus exposure. PLoS Negl. Trop. Dis. 2020, 14, e0008874. [Google Scholar] [CrossRef]
- Kung, P.-L.; Chou, T.-W.; Lindman, M.; Chang, N.P.; Estevez, I.; Buckley, B.D.; Atkins, C.; Daniels, B.P. Zika virus-induced TNF-α signaling dysregulates expression of neurologic genes associated with psychiatric disorders. J. Neuroinflamm. 2022, 19, 100. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Choi, H.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.-Y.; Kim, M.-J.; Choi, S.W.; Kim, Y.B.; Kang, K.-S. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discov. 2022, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Schotsaert, M.; Karnik, R.; Balasubramaniam, V.; Dejosez, M.; Meissner, A.; García-Sastre, A.; Zwaka, T.P. Zika Virus Alters DNA Methylation of Neural Genes in an Organoid Model of the Developing Human Brain. mSystems 2018, 3, e00219-17. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Lucchese, G.; Kanduc, D. Zika virus and autoimmunity: From microcephaly to Guillain-Barré syndrome, and beyond. Autoimmun. Rev. 2016, 15, 801–808. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.-F.; Chen, J.; Zhou, X.; Dong, Z.; Chen, T.; Yang, Y.; Zou, P.; Jiang, B.; Hu, Y.; et al. Zika virus infects renal proximal tubular epithelial cells with prolonged persistency and cytopathic effects. Emerg. Microbes Infect. 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tang, L.; Tang, H.; Pu, J.; Gong, S.; Fang, D.; Zhang, H.; Li, Y.-P.; Zhu, X.; Wang, W.; et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front. Immunol. 2019, 10, 1925. [Google Scholar] [CrossRef]
- Costa Monteiro, L.M.; Cruz, G.N.d.O.; Fontes, J.M.; Saad Salles, T.R.D.; Boechat, M.C.B.; Monteiro, A.C.; Moreira, M.E.L. Neurogenic bladder findings in patients with Congenital Zika Syndrome: A novel condition. PLoS ONE 2018, 13, e0193514. [Google Scholar] [CrossRef]
- Davenport, B.J.; Bullock, C.; McCarthy, M.K.; Hawman, D.W.; Murphy, K.M.; Kedl, R.M.; Diamond, M.S.; Morrison, T.E. Chikungunya Virus Evades Antiviral CD8+ T Cell Responses To Establish Persistent Infection in Joint-Associated Tissues. J. Virol. 2020, 94, e02036-19. [Google Scholar] [CrossRef]
- Simmons, J.D.; White, L.J.; Morrison, T.E.; Montgomery, S.A.; Whitmore, A.C.; Johnston, R.E.; Heise, M.T. Venezuelan Equine Encephalitis Virus Disrupts STAT1 Signaling by Distinct Mechanisms Independent of Host Shutoff. J. Virol. 2009, 83, 10571–10581. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.P.; McNally, K.L.; Robertson, S.J.; Best, S.M.; Pijlman, G.P.; Fros, J.J. The Methyltransferase-Like Domain of Chikungunya Virus nsP2 Inhibits the Interferon Response by Promoting the Nuclear Export of STAT1. J. Virol. 2018, 92, e01008-18. [Google Scholar] [CrossRef]
- Burdeinick-Kerr, R.; Govindarajan, D.; Griffin, D.E. Noncytolytic Clearance of Sindbis Virus Infection from Neurons by Gamma Interferon Is Dependent on Jak/Stat Signaling. J. Virol. 2009, 83, 3429–3435. [Google Scholar] [CrossRef]
- Bae, S.; Lee, J.Y.; Myoung, J. Chikungunya Virus-Encoded nsP2, E2 and E1 Strongly Antagonize the Interferon-β Signaling Pathway. J. Microbiol. Biotechnol. 2019, 29, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.J.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; Jong, Y.P.d.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93, e00715-19. [Google Scholar] [CrossRef]
- Webb, L.G.; Veloz, J.; Pintado-Silva, J.; Zhu, T.; Rangel, M.V.; Mutetwa, T.; Zhang, L.; Bernal-Rubio, D.; Figueroa, D.; Carrau, L.; et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 2020, 16, e1008999. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Mathur, K.; Anand, A.; Dubey, S.K.; Sanan-Mishra, N.; Bhatnagar, R.K.; Sunil, S. Analysis of chikungunya virus proteins reveals that non-structural proteins nsP2 and nsP3 exhibit RNA interference (RNAi) suppressor activity. Sci. Rep. 2016, 6, 38065. [Google Scholar] [CrossRef]
- Qian, Q.; Zhou, H.; Shu, T.; Mu, J.; Fang, Y.; Xu, J.; Li, T.; Kong, J.; Qiu, Y.; Zhou, X. The Capsid Protein of Semliki Forest Virus Antagonizes RNA Interference in Mammalian Cells. J. Virol. 2020, 94, e01233-19. [Google Scholar] [CrossRef]
- Rangel, M.V.; McAllister, N.; Dancel-Manning, K.; Noval, M.G.; Silva, L.A.; Stapleford, K.A. Emerging Chikungunya Virus Variants at the E1-E1 Interglycoprotein Spike Interface Impact Virus Attachment and Inflammation. J. Virol. 2022, 96, e0158621. [Google Scholar] [CrossRef]
- Tanabe, I.S.B.; Tanabe, E.L.L.; Santos, E.C.; Martins, W.V.; Araujo, I.; Cavalcante, M.C.A.; Lima, A.R.V.; Camara, N.O.S.; Anderson, L.; Yunusov, D.; et al. Cellular and Molecular Immune Response to Chikungunya Virus Infection. Front. Cell. Infect. Microbiol. 2018, 8, 345. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.R.; Plante, K.S.; Heise, M.T. Chikungunya Virus: Current Perspectives on a Reemerging Virus. Microbiol. Spectr. 2016, 4, 143–161. [Google Scholar] [CrossRef]
- Kleinert, R.D.V.; Montoya-Diaz, E.; Khera, T.; Welsch, K.; Tegtmeyer, B.; Hoehl, S.; Ciesek, S.; Brown, R.J.P. Yellow Fever: Integrating Current Knowledge with Technological Innovations to Identify Strategies for Controlling a Re-Emerging Virus. Viruses 2019, 11, 960. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Suthar, M.S.; Aguirre, S.; Fernandez-Sesma, A. Innate Immune Sensing of Flaviviruses. PLoS Pathog. 2013, 9, e1003541. [Google Scholar] [CrossRef]
- Guo, H.-Y.; Zhang, X.-C.; Jia, R.-Y. Toll-Like Receptors and RIG-I-Like Receptors Play Important Roles in Resisting Flavivirus. J. Immunol. Res. 2018, 2018, 6106582. [Google Scholar] [CrossRef]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.-M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N12′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martínez-Romero, C.; Tenoever, B.R.; et al. The Interferon Signaling Antagonist Function of Yellow Fever Virus NS5 Protein Is Activated by Type I Interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef]
- ter Meulen, J.; Sakho, M.; Koulemou, K.; Magassouba, N.F.; Bah, A.; Preiser, W.; Daffis, S.; Klewitz, C.; Bae, H.-G.; Niedrig, M.; et al. Activation of the Cytokine Network and Unfavorable Outcome in Patients with Yellow Fever. J. Infect. Dis. 2004, 190, 1821–1827. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Pagliari, C.; Fernandes, E.R.; Guedes, F.; Takakura, C.F.H.; Andrade, H.F.; Vasconcelos, P.F.C.; Duarte, M.I.S. Revisiting the liver in human yellow fever: Virus-induced apoptosis in hepatocytes associated with TGF-β, TNF-α and NK cells activity. Virology 2006, 345, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Fernandes, E.R.; Pagliari, C.; Guedes, F.; Vasconcelos, P.F.d.C.; Andrade Junior, H.F.d.; Duarte, M.I.S. Immunohistochemical examination of the role of Fas ligand and lymphocytes in the pathogenesis of human liver yellow fever. Virus Res. 2006, 116, 91–97. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-Β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Stock, N.K.; Laraway, H.; Faye, O.; Diallo, M.; Niedrig, M.; Sall, A.A. Biological and phylogenetic characteristics of yellow fever virus lineages from West Africa. J. Virol. 2013, 87, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.D.; Barrett, A.D. Live Attenuated Yellow Fever 17D Vaccine: A Legacy Vaccine Still Controlling Outbreaks In Modern Day. Curr. Infect. Dis. Rep. 2017, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Azamor, T.; da Silva, A.M.V.; Melgaco, J.G.; Dos Santos, A.P.; Xavier-Carvalho, C.; Alvarado-Arnez, L.E.; Batista-Silva, L.R.; de Souza Matos, D.C.; Bayma, C.; Missailidis, S.; et al. Activation of an Effective Immune Response after Yellow Fever Vaccination Is Associated with the Genetic Background and Early Response of IFN-gamma and CLEC5A. Viruses 2021, 13, 96. [Google Scholar] [CrossRef]
- Siam, A.L.; Meegan, J.M.; Gharbawi, K.F. Rift Valley fever ocular manifestations: Observations during the 1977 epidemic in Egypt. Br. J. Ophthalmol. 1980, 64, 366–374. [Google Scholar] [CrossRef]
- Wang, X.; Yuan, Y.; Liu, Y.; Zhang, L. Arm race between Rift Valley fever virus and host. Front. Immunol. 2022, 13, 230. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Havranek, K.E.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. Transcriptome profiling in Rift Valley fever virus infected cells reveals modified transcriptional and alternative splicing programs. PLoS ONE 2019, 14, e0217497. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Jesus, P.D.D.; Su, V.; Han, S.; Gong, D.; Wu, N.C.; Tian, Y.; Li, X.; Wu, T.-T.; Chanda, S.K.; et al. RIOK3 Is an Adaptor Protein Required for IRF3-Mediated Antiviral Type I Interferon Production. J. Virol. 2014, 88, 7987–7997. [Google Scholar] [CrossRef]
- Bisom, T.C.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection. Viruses 2022, 14, 2064. [Google Scholar] [CrossRef]
- White, L.A.; Bisom, T.C.; Grimes, H.L.; Hayashi, M.; Lanchy, J.-M.; Lodmell, J.S. Tra2beta-Dependent Regulation of RIO Kinase 3 Splicing During Rift Valley Fever Virus Infection Underscores the Links between Alternative Splicing and Innate Antiviral Immunity. Front. Cell. Infect. Microbiol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.J.; Ikegami, T. Rift Valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. Virol. J. 2016, 13, 118. [Google Scholar] [CrossRef]
- Le May, N.; Mansuroglu, Z.; Léger, P.; Josse, T.; Blot, G.; Billecocq, A.; Flick, R.; Jacob, Y.; Bonnefoy, E.; Bouloy, M. A SAP30 Complex Inhibits IFN-β Expression in Rift Valley Fever Virus Infected Cells. PLoS Pathog. 2008, 4, e13. [Google Scholar] [CrossRef]
- Le May, N.; Dubaele, S.; De Santis, L.P.; Billecocq, A.; Bouloy, M.; Egly, J.-M. TFIIH Transcription Factor, a Target for the Rift Valley Hemorrhagic Fever Virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs Protein of Rift Valley Fever Virus Promotes Posttranslational Downregulation of the TFIIH Subunit p62. J. Virol. 2011, 85, 234–6243. [Google Scholar] [CrossRef]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence Factor NSs of Rift Valley Fever Virus Recruits the F-Box Protein FBXO3 To Degrade Subunit p62 of General Transcription Factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Josse, T.; Gilleron, J.; Billecocq, A.; Leger, P.; Bouloy, M.; Bonnefoy, E. Nonstructural NSs Protein of Rift Valley Fever Virus Interacts with Pericentromeric DNA Sequences of the Host Cell, Inducing Chromosome Cohesion and Segregation Defects. J. Virol. 2010, 84, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Austin, D.; Narayanan, A.; Popova, T.; Kainulainen, M.; Bailey, C.; Kashanchi, F.; Weber, F.; Kehn-Hall, K. Induction of DNA Damage Signaling upon Rift Valley Fever Virus Infection Results in Cell Cycle Arrest and Increased Viral Replication. J. Biol. Chem. 2012, 287, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-H.; Liao, C.-L.; Lin, Y.-L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-κB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar] [CrossRef]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, D.; Jia, F.; Zhang, L.; Ashraf, U.; Li, Y.; Duan, H.; Song, Y.; Chen, H.; Cao, S.; et al. Japanese Encephalitis Virus NS1′ Protein Interacts with Host CDK1 Protein to Regulate Antiviral Response. Microbiol. Spectr. 2021, 9, e01661-21. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, N. Myeloid-Derived Suppressor Cells Inhibit T Follicular Helper Cell Immune Response in Japanese Encephalitis Virus Infection. J. Immunol. 2017, 199, 3094–3105. [Google Scholar] [CrossRef]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef]
- Kundu, K.; Dutta, K.; Nazmi, A.; Basu, A. Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: Implications for the hosts’ innate immune response. Cell. Immunol. 2013, 285, 100–110. [Google Scholar] [CrossRef]
- Adhya, D.; Dutta, K.; Kundu, K.; Basu, A. Histone deacetylase inhibition by Japanese encephalitis virus in monocyte/macrophages: A novel viral immune evasion strategy. Immunobiology 2013, 218, 1235–1247. [Google Scholar] [CrossRef]
- Rastogi, M.; Singh, S.K. Japanese Encephalitis Virus exploits microRNA-155 to suppress the non-canonical NF-κB pathway in human microglial cells. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2020, 1863, 194639. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Hazra, B.; Chakraborty, S.; Basu, A. miR-301a mediated immune evasion by Japanese encephalitis virus. Oncotarget 2017, 8, 90620–90621. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.-Y.; Hsu, T.-W.; Chen, Y.-L.; Liu, S.-F.; Tsai, Y.-J.; Lin, Y.-T.; Chen, Y.-S.; Fan, Y.-H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef]
- Han, N.; Adams, J.; Chen, P.; Guo, Z.Y.; Zhong, X.F.; Fang, W.; Li, N.; Wen, L.; Tao, X.Y.; Yuan, Z.M.; et al. Comparison of genotypes I and III in Japanese encephalitis virus reveals distinct differences in their genetic and host diversity. J. Virol. 2014, 88, 11469–11479. [Google Scholar] [CrossRef] [PubMed]
- Khare, B.; Kuhn, R.J. The Japanese Encephalitis Antigenic Complex Viruses: From Structure to Immunity. Viruses 2022, 14, 2213. [Google Scholar] [CrossRef]
- Fadnis, P.R.; Ravi, V.; Desai, A.; Turtle, L.; Solomon, T. Innate immune mechanisms in Japanese encephalitis virus infection: Effect on transcription of pattern recognition receptors in mouse neuronal cells and brain tissue. Viral Immunol. 2013, 26, 366–377. [Google Scholar] [CrossRef]
- Fan, Y.C.; Chen, Y.Y.; Chen, J.M.; Huang, C.; Huang, M.; Chiou, S.S. Effectiveness of Live-Attenuated Genotype III Japanese Encephalitis Viral Vaccine against Circulating Genotype I Viruses in Swine. Viruses 2022, 14, 114. [Google Scholar] [CrossRef]
- Yun, S.I.; Lee, Y.M. Japanese encephalitis: The virus and vaccines. Hum. Vaccines Immunother. 2014, 10, 263–279. [Google Scholar] [CrossRef]
- Sharma, K.B.; Vrati, S.; Kalia, M. Pathobiology of Japanese encephalitis virus infection. Mol. Asp. Med. 2021, 81, 100994. [Google Scholar] [CrossRef]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Muüller, M.; Rana, S.; Shrestha, B.; Radford, J.; Van Rooijen, N.; Campbell, I.L.; King, N.J.C. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 2008, 205, 2319–2337. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Smith, M.; Katze, M.G.; Shi, P.-Y.; Gale, M. The Host Response to West Nile Virus Infection Limits Viral Spread through the Activation of the Interferon Regulatory Factor 3 Pathway. J. Virol. 2004, 78, 7737–7747. [Google Scholar] [CrossRef]
- Wilson, J.R.; Sessions, P.F.d.; Leon, M.A.; Scholle, F. West Nile Virus Nonstructural Protein 1 Inhibits TLR3 Signal Transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M. Resistance to Alpha/Beta Interferon Is a Determinant of West Nile Virus Replication Fitness and Virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Evans, J.D.; Crown, R.A.; Sohn, J.A.; Seeger, C. West Nile Virus Infection Induces Depletion of IFNAR1 Protein Levels. Viral Immunol. 2011, 24, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 Gene Family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.D.; Rangappa, M.; Satchidanandam, V. Virus-Specific Cytolytic Antibodies to Nonstructural Protein 1 of Japanese Encephalitis Virus Effect Reduction of Virus Output from Infected Cells. J. Virol. 2009, 83, 4766–4777. [Google Scholar] [CrossRef]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of Flavivirus Nonstructural Protein NS1 to C4b Binding Protein Modulates Complement Activation. J. Immunol. 2011, 187, 424–433. [Google Scholar] [CrossRef]
- Cho, H.; Diamond, M.S. Immune responses to West Nile virus infection in the central nervous system. Viruses 2012, 4, 3812–3830. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ivanics, E.; Erdelyi, K.; Ursu, K.; Ferenczi, E.; Weissenbock, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Arjona, A.; Wang, P.; Montgomery, R.R.; Fikrig, E. Innate immune control of West Nile virus infection. Cell. Microbiol. 2011, 13, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, T. Recent advances in understanding West Nile virus host immunity and viral pathogenesis. F1000Research 2018, 7, 338. [Google Scholar] [CrossRef]
- Gerardin, P.; Couderc, T.; Bintner, M.; Tournebize, P.; Renouil, M.; Lemant, J.; Boisson, V.; Borgherini, G.; Staikowsky, F.; Schramm, F.; et al. Chikungunya virus-associated encephalitis: A cohort study on La Reunion Island, 2005–2009. Neurology 2016, 86, 94–102. [Google Scholar] [CrossRef]
- Sissoko, D.; Malvy, D.; Giry, C.; Delmas, G.; Paquet, C.; Gabrie, P.; Pettinelli, F.; Sanquer, M.A.; Pierre, V. Outbreak of Chikungunya fever in Mayotte, Comoros archipelago, 2005–2006. Trans. R Soc. Trop. Med. Hyg. 2008, 102, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Robillard, P.Y.; Boumahni, B.; Gerardin, P.; Michault, A.; Fourmaintraux, A.; Schuffenecker, I.; Carbonnier, M.; Djemili, S.; Choker, G.; Roge-Wolter, M.; et al. Vertical maternal fetal transmission of the chikungunya virus. Ten cases among 84 pregnant women. Presse Med. 2006, 35, 785–788. [Google Scholar] [CrossRef]
- Shenoy, S.; Pradeep, G.C. Neurodevelopmental outcome of neonates with vertically transmitted Chikungunya fever with encephalopathy. Indian Pediatr. 2012, 49, 238–240. [Google Scholar] [PubMed]
- Villamil-Gomez, W.; Alba-Silvera, L.; Menco-Ramos, A.; Gonzalez-Vergara, A.; Molinares-Palacios, T.; Barrios-Corrales, M.; Rodriguez-Morales, A.J. Congenital Chikungunya Virus Infection in Sincelejo, Colombia: A Case Series. J. Trop. Pediatr. 2015, 61, 386–392. [Google Scholar] [CrossRef]
- Torres, J.R.; Falleiros-Arlant, L.H.; Duenas, L.; Pleitez-Navarrete, J.; Salgado, D.M.; Castillo, J.B. Congenital and perinatal complications of chikungunya fever: A Latin American experience. Int. J. Infect. Dis. 2016, 51, 85–88. [Google Scholar] [CrossRef]
- Bandeira, A.C.; Campos, G.S.; Sardi, S.I.; Rocha, V.F.; Rocha, G.C. Neonatal encephalitis due to Chikungunya vertical transmission: First report in Brazil. IDCases 2016, 5, 57–59. [Google Scholar] [CrossRef]
- Samra, J.A.; Hagood, N.L.; Summer, A.; Medina, M.T.; Holden, K.R. Clinical Features and Neurologic Complications of Children Hospitalized with Chikungunya Virus in Honduras. J. Child. Neurol. 2017, 32, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Karthiga, V.; Kommu, P.P.; Krishnan, L. Perinatal chikungunya in twins. J. Pediatr. Neurosci. 2016, 11, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.; Viana, R.; Brainer-Lima, A.; Flore Ancio, T.; Carvalho, M.D.; van der Linden, V.; Amorim, A.; Rocha, M.A.; Medeiros, F. Perinatal Chikungunya Virus-associated Encephalitis Leading to Postnatal-Onset Microcephaly and Optic Atrophy. Pediatr. Infect. Dis. J. 2018, 37, 94–95. [Google Scholar] [CrossRef]
- Gerardin, P.; Barau, G.; Michault, A.; Bintner, M.; Randrianaivo, H.; Choker, G.; Lenglet, Y.; Touret, Y.; Bouveret, A.; Grivard, P.; et al. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Reunion. PLoS Med. 2008, 5, e60. [Google Scholar] [CrossRef]
- Ramful, D.; Carbonnier, M.; Pasquet, M.; Bouhmani, B.; Ghazouani, J.; Noormahomed, T.; Beullier, G.; Attali, T.; Samperiz, S.; Fourmaintraux, A.; et al. Mother-to-child transmission of Chikungunya virus infection. Pediatr. Infect. Dis. J. 2007, 26, 811–815. [Google Scholar] [CrossRef]
- Lyra, P.P.; Campos, G.S.; Bandeira, I.D.; Sardi, S.I.; Costa, L.F.; Santos, F.R.; Ribeiro, C.A.; Jardim, A.M.; Santiago, A.C.; de Oliveira, P.M.; et al. Congenital Chikungunya Virus Infection after an Outbreak in Salvador, Bahia, Brazil. AJP Rep. 2016, 6, e299–e300. [Google Scholar] [CrossRef]
- Simon, F.; Paule, P.; Oliver, M. Chikungunya Virus–Induced Myopericarditis: Toward an Increase of Dilated Cardiomyopathy in Countries with Epidemics? Am. J. Trop. Med. Hyg. 2008, 78, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Vibha, D.; Srivastava, A.K.; Shukla, G.; Prasad, K. Guillain-Barre syndrome complicating chikungunya virus infection. J. Neurovirol. 2017, 23, 504–507. [Google Scholar] [CrossRef]
- Patwardhan, A.; Nalini, A.; Baishya, P.P.; Kulanthaivelu, K.; Krishnareddy, H.; Dutta, D.; Chawla, T.; Chowdary, R.M.; Yadav, R.; Vengalil, S. Case Report: Post-Chikungunya-Associated Myeloneuropathy. Am. J. Trop. Med. Hyg. 2021, 105, 942–945. [Google Scholar] [CrossRef]
- Pancharoen, C.; Thisyakorn, U. Neurological manifestations in dengue patients. Southeast Asian J. Trop. Med. Public Health 2001, 32, 341–345. [Google Scholar]
- Bopeththa, B.; Ralapanawa, U. Post encephalitic parkinsonism following dengue viral infection. BMC Res. Notes 2017, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Mitra, P. A Study on Cardiac Manifestations of Dengue Fever. J. Assoc. Physicians India 2019, 67, 14–16. [Google Scholar]
- Wali, J.P.; Biswas, A.; Chandra, S.; Malhotra, A.; Aggarwal, P.; Handa, R.; Wig, N.; Bahl, V.K. Cardiac involvement in Dengue Haemorrhagic Fever. Int. J. Cardiol. 1998, 64, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sheetal, S.; Jacob, E. A Study on the Cardiac Manifestations of Dengue. J. Assoc Physicians India 2016, 64, 30–34. [Google Scholar] [PubMed]
- Kularatne, S.A.; Pathirage, M.M.; Kumarasiri, P.V.; Gunasena, S.; Mahindawanse, S.I. Cardiac complications of a dengue fever outbreak in Sri Lanka, 2005. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 804–808. [Google Scholar] [CrossRef] [PubMed]
- da Costa, P.S.; Ribeiro, G.M.; Junior, C.S.; da Costa Campos, L. Severe thrombotic events associated with dengue fever, Brazil. Am. J. Trop. Med. Hyg. 2012, 87, 741–742. [Google Scholar] [CrossRef]
- Guadalajara-Boo, J.F.; Ruiz-Esparza, M.E.; Aranda Frausto, A.; Soto Abraham, M.V.; Gaspar-Hernandez, J. Histologic and angiographic imaging of acute shock dengue myocarditis. Rev. Esp. Cardiol. 2014, 67, 226–227. [Google Scholar] [CrossRef]
- Kularatne, S.A.M.; Rajapakse, M.M.; Ralapanawa, U.; Waduge, R.; Pathirage, L.; Rajapakse, R. Heart and liver are infected in fatal cases of dengue: Three PCR based case studies. BMC Infect. Dis. 2018, 18, 681. [Google Scholar] [CrossRef]
- Marques, N.; Gan, V.C.; Leo, Y.S. Dengue myocarditis in Singapore: Two case reports. Infection 2013, 41, 709–714. [Google Scholar] [CrossRef]
- Bich, T.D.; Pham, O.K.; Hai, D.H.; Nguyen, N.M.; Van, H.N.; The, T.D.; Wills, B.; Yacoub, S. A pregnant woman with acute cardiorespiratory failure: Dengue myocarditis. Lancet 2015, 385, 1260. [Google Scholar] [CrossRef]
- Naresh, G.; Kulkarni, A.V.; Sinha, N.; Jhamb, N.; Gulati, S. Dengue hemorrhagic fever complicated with encephalopathy and myocarditis: A case report. J. Commun. Dis. 2008, 40, 223–224. [Google Scholar] [PubMed]
- Chou, M.T.; Yu, W.L. Takotsubo cardiomyopathy in a patient with dengue fever. J. Formos. Med. Assoc. Taiwan Yi Zhi 2016, 115, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, K.; Teo, L.; Raymond, W.C.; MacLaren, G. Dengue Myopericarditis Mimicking Acute Myocardial Infarction. Circulation 2015, 131, e519–e522. [Google Scholar] [CrossRef]
- Tayeb, B.; Piot, C.; Roubille, F. Acute pericarditis after dengue fever. Ann. Cardiol. D’angeiologie 2011, 60, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.K.; Sanzgiri, P.S.; Nagvekar, V. High degree atrioventricular block with ventricular asystole in a case of dengue fever. Indian Heart J. 2016, 68 (Suppl. S2), S194–S197. [Google Scholar] [CrossRef] [PubMed]
- Navinan, M.R.; Yudhishdran, J.; Herath, S.; Liyanage, I.; Kugadas, T.; Kumara, D.; Kulatunga, A. Complete heart block in dengue complicating management of shock due to both bleeding and leakage: A case report. BMC Res. Notes 2015, 8, 68. [Google Scholar] [CrossRef]
- Sharda, M.; Gupt, A.; Nagar, D.; Soni, A.K. Dengue fever: An additional cause for bradycardia. J. Assoc. Physicians India 2014, 62, 362–363. [Google Scholar]
- Khongphatthallayothin, A.; Chotivitayatarakorn, P.; Somchit, S.; Mitprasart, A.; Sakolsattayadorn, S.; Thisyakorn, C. Morbitz type I second degree AV block during recovery from dengue hemorrhagic fever. Southeast Asian J. Trop. Med. Public Health 2000, 31, 642–645. [Google Scholar]
- Kaushik, J.S.; Gupta, P.; Rajpal, S.; Bhatt, S. Spontaneous resolution of sinoatrial exit block and atrioventricular dissociation in a child with dengue fever. Singap. Med. J. 2010, 51, e146–e148. [Google Scholar]
- Mahmod, M.; Darul, N.D.; Mokhtar, I.; Nor, N.M.; Anshar, F.M.; Maskon, O. Atrial fibrillation as a complication of dengue hemorrhagic fever: Non-self-limiting manifestation. Int. J. Infect. Dis. 2009, 13, e316–e318. [Google Scholar] [CrossRef]
- Horta Veloso, H.; Ferreira Junior, J.A.; Braga de Paiva, J.M.; Faria Honorio, J.; Junqueira Bellei, N.C.; Vicenzo de Paola, A.A. Acute atrial fibrillation during dengue hemorrhagic fever. Braz. J. Infect. Dis. 2003, 7, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Azhar, E.I.; El-Fiky, S.; Madani, H.H.; Abuljadial, M.A.; Ashshi, A.M.; Turkistani, A.M.; Hamouh, E.A. Clinical profile and outcome of hospitalized patients during first outbreak of dengue in Makkah, Saudi Arabia. Acta Trop. 2008, 105, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.C.; Lu, P.L.; Chang, J.M.; Lin, M.Y.; Tsai, J.J.; Chen, Y.H.; Chang, K.; Chen, H.C.; Hwang, S.J. Impact of renal failure on the outcome of dengue viral infection. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 1350–1356. [Google Scholar] [CrossRef]
- Lee, I.-K.; Liu, J.-W.; Yang, K.D. Clinical Characteristics, Risk Factors, and Outcomes in Adults Experiencing Dengue Hemorrhagic Fever Complicated with Acute Renal Failure. Am. J. Trop. Med. Hyg. 2009, 80, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Laoprasopwattana, K.; Pruekprasert, P.; Dissaneewate, P.; Geater, A.; Vachvanichsanong, P. Outcome of dengue hemorrhagic fever-caused acute kidney injury in Thai children. J. Pediatr. 2010, 157, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Basu, G.; Chrispal, A.; Boorugu, H.; Gopinath, K.G.; Chandy, S.; Prakash, J.A.; Thomas, K.; Abraham, A.M.; John, G.T. Acute kidney injury in tropical acute febrile illness in a tertiary care centre—RIFLE criteria validation. Nephrol. Dial. Transplant. 2011, 26, 524–531. [Google Scholar] [CrossRef]
- Khalil, M.A.; Sarwar, S.; Chaudry, M.A.; Maqbool, B.; Khalil, Z.; Tan, J.; Yaqub, S.; Hussain, S.A. Acute kidney injury in dengue virus infection. Clin. Kidney J. 2012, 5, 390–394. [Google Scholar] [CrossRef]
- Lee, I.K.; Liu, J.W.; Yang, K.D. Fatal dengue hemorrhagic fever in adults: Emphasizing the evolutionary pre-fatal clinical and laboratory manifestations. PLoS Negl. Trop. Dis. 2012, 6, e1532. [Google Scholar] [CrossRef]
- Mehra, N.; Patel, A.; Abraham, G.; Reddy, Y.N.; Reddy, Y.N. Acute kidney injury in dengue fever using Acute Kidney Injury Network criteria: Incidence and risk factors. Trop. Dr. 2012, 42, 160–162. [Google Scholar] [CrossRef]
- Huang, S.Y.; Lee, I.K.; Liu, J.W.; Kung, C.T.; Wang, L. Clinical features of and risk factors for rhabdomyolysis among adult patients with dengue virus infection. Am. J. Trop. Med. Hyg. 2015, 92, 75–81. [Google Scholar] [CrossRef]
- Mallhi, T.H.; Khan, A.H.; Adnan, A.S.; Sarriff, A.; Khan, Y.H.; Jummaat, F. Incidence, Characteristics and Risk Factors of Acute Kidney Injury among Dengue Patients: A Retrospective Analysis. PLoS ONE 2015, 10, e0138465. [Google Scholar] [CrossRef]
- Huang, H.S.; Hsu, C.C.; Ye, J.C.; Su, S.B.; Huang, C.C.; Lin, H.J. Predicting the mortality in geriatric patients with dengue fever. Medicine 2017, 96, e7878. [Google Scholar] [CrossRef] [PubMed]
- Vakrani, G.P.; Subramanyam, N.T. Acute Renal Failure in Dengue Infection. J. Clin. Diagn. Res. 2017, 11, OC10–OC13. [Google Scholar] [CrossRef]
- Basu, B.; Roy, B. Acute Renal Failure Adversely Affects Survival in Pediatric Dengue Infection. Indian J. Crit. Care Med. 2018, 22, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Huang, C.H.; Huang, W.C.; Chen, Y.C.; Tsai, C.Y.; Chang, K.; Chen, Y.H. Prognostic Factors in Adult Patients with Dengue: Developing Risk Scoring Models and Emphasizing Factors Associated with Death ≤7 Days after Illness Onset and ≤3 Days after Presentation. J. Clin. Med. 2018, 7, 396. [Google Scholar] [CrossRef]
- Mallhi, T.H.; Khan, A.H.; Adnan, A.S.; Sarriff, A.; Khan, Y.H.; Gan, S.H. Short-term renal outcomes following acute kidney injury among dengue patients: A follow-up analysis from large prospective cohort. PLoS ONE 2018, 13, e0192510. [Google Scholar] [CrossRef]
- Mehta, K.; Pajai, A.; Bhurke, S.; Shirkande, A.; Bhadade, R.; D’Souza, R. Acute Kidney Injury of Infectious Etiology in Monsoon Season: A Prospective Study Using Acute Kidney Injury Network Criteria. Indian J. Nephrol. 2018, 28, 143–152. [Google Scholar] [CrossRef]
- Diptyanusa, A.; Phumratanaprapin, W.; Phonrat, B.; Poovorawan, K.; Hanboonkunupakarn, B.; Sriboonvorakul, N.; Thisyakorn, U. Characteristics and associated factors of acute kidney injury among adult dengue patients: A retrospective single-center study. PLoS ONE 2019, 14, e0210360. [Google Scholar] [CrossRef]
- Padyana, M.; Karanth, S.; Vaidya, S.; Gopaldas, J.A. Clinical Profile and Outcome of Dengue Fever in Multidisciplinary Intensive Care Unit of a Tertiary Level Hospital in India. Indian J. Crit. Care Med. 2019, 23, 270–273. [Google Scholar] [CrossRef]
- Patel, M.L.; Himanshu, D.; Chaudhary, S.C.; Atam, V.; Sachan, R.; Misra, R.; Mohapatra, S.D. Clinical Characteristic and Risk Factors of Acute Kidney Injury among Dengue Viral Infections in Adults: A Retrospective Analysis. Indian J. Nephrol. 2019, 29, 15–21. [Google Scholar] [CrossRef]
- Xiang, J.Y.; Zhang, Y.H.; Tan, Z.R.; Huang, J.; Zhao, Y.W. Guillain-Barre syndrome associated with Japanese encephalitis virus infection in China. Viral Immunol. 2014, 27, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Praharaj, H.N.; Patil, T.B.; Giri, P. Acute transverse myelitis following Japanese encephalitis viral infection: An uncommon complication of a common disease. BMJ Case Rep. 2012, 2012, bcr2012007094. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Kumar, H.; Meena, B.L.; Chandra, S.; Agrawal, J.; Kanogiya, N. Neuropsychiatric Profile in Malaria: An Overview. J. Clin. Diagn. Res. 2016, 10, OC24–OC28. [Google Scholar] [CrossRef]
- Pace, A.A.; Edwards, S.; Weatherby, S. A new clinical variant of the post-malaria neurological syndrome. J. Neurol. Sci. 2013, 334, 183–185. [Google Scholar] [CrossRef]
- Derkach, A.; Otim, I.; Pfeiffer, R.M.; Onabajo, O.O.; Legason, I.D.; Nabalende, H.; Ogwang, M.D.; Kerchan, P.; Talisuna, A.O.; Ayers, L.W.; et al. Associations between IgG reactivity to Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) antigens and Burkitt lymphoma in Ghana and Uganda case-control studies. EBioMedicine 2019, 39, 358–368. [Google Scholar] [CrossRef]
- Johnston, W.T.; Mutalima, N.; Sun, D.; Emmanuel, B.; Bhatia, K.; Aka, P.; Wu, X.; Borgstein, E.; Liomba, G.N.; Kamiza, S.; et al. Relationship between Plasmodium falciparum malaria prevalence, genetic diversity and endemic Burkitt lymphoma in Malawi. Sci. Rep. 2014, 4, 3741. [Google Scholar] [CrossRef]
- Peprah, S.; Ogwang, M.D.; Kerchan, P.; Reynolds, S.J.; Tenge, C.N.; Were, P.A.; Kuremu, R.T.; Wekesa, W.N.; Sumba, P.O.; Masalu, N.; et al. Risk factors for Burkitt lymphoma in East African children and minors: A case-control study in malaria-endemic regions in Uganda, Tanzania and Kenya. Int. J. Cancer 2020, 146, 953–969. [Google Scholar] [CrossRef]
- Wyss, K.; Granath, F.; Wangdahl, A.; Djarv, T.; Fored, M.; Naucler, P.; Farnert, A. Malaria and risk of lymphoid neoplasms and other cancer: A nationwide population-based cohort study. BMC Med. 2020, 18, 296. [Google Scholar] [CrossRef]
- Park, J.E. Parkinsonism and Tremor in a Patient with Plasmodium vivax Malaria. Tremor Other Hyperkinetic Mov. 2017, 7, 471. [Google Scholar] [CrossRef]
- Mittal, S.; Misri, Z.K.; Rakshith, K.C.; Mittal, S. Post Malarial Neurological Syndrome: An Uncommon Cause. J. Neuroinfect. Dis. 2015, 1, 1–4. [Google Scholar] [CrossRef]
- Kanjalkar, M.; Karnad, D.R.; Narayana, R.V.; Shah, P.U. Guillain-Barre syndrome following malaria. J. Infect. 1999, 38, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Ji, W.; Bhaya, B.; Ahsan, C. A Rare Case of Cardiac Recovery after Acute Myocarditis from West Nile Virus Infection: A Review of the Current Literature. Case Rep. Cardiol. 2022, 2022, 8517728. [Google Scholar] [CrossRef]
- Gao, A.R.; Nichols, L.; Mannuru, D. A Rare Case of West Nile Virus-Associated Cardiomyopathy. Cureus 2022, 14, e28473. [Google Scholar] [CrossRef] [PubMed]
- Hasbun, R.; Garcia, M.N.; Kellaway, J.; Baker, L.; Salazar, L.; Woods, S.P.; Murray, K.O. West Nile Virus Retinopathy and Associations with Long Term Neurological and Neurocognitive Sequelae. PLoS ONE 2016, 11, e0148898. [Google Scholar] [CrossRef]
- Weatherhead, J.E.; Miller, V.E.; Garcia, M.N.; Hasbun, R.; Salazar, L.; Dimachkie, M.M.; Murray, K.O. Long-term neurological outcomes in West Nile virus-infected patients: An observational study. Am. J. Trop. Med. Hyg. 2015, 92, 1006–1012. [Google Scholar] [CrossRef]
- Beirao, P.; Pereira, P.; Nunes, A.; Antunes, P. Yellow fever vaccine-associated neurological disease, a suspicious case. BMJ Case Rep. 2017, 2017, bcr2016218706. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Kang, L.-I.; Zanella, L.G.F.D.A.B.D.; Silveira, C.G.T.; Ho, Y.-L.; Foquet, L.; Bial, G.; McCune, B.T.; Duarte-Neto, A.N.; Thomas, A.; et al. Consumptive coagulopathy of severe yellow fever occurs independently of hepatocellular tropism and massive hepatic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 32648–32656. [Google Scholar] [CrossRef]
- Mecharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef]
- Schwartzmann, P.V.; Ramalho, L.N.; Neder, L.; Vilar, F.C.; Ayub-Ferreira, S.M.; Romeiro, M.F.; Takayanagui, O.M.; Dos Santos, A.C.; Schmidt, A.; Figueiredo, L.T.; et al. Zika Virus Meningoencephalitis in an Immunocompromised Patient. Mayo Clin. Proc. 2017, 92, 460–466. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jimenez-Arango, J.A.; Zea-Vera, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Anaya, J.M.; Rodriguez, Y.; Monsalve, D.M.; Vega, D.; Ojeda, E.; Gonzalez-Bravo, D.; Rodriguez-Jimenez, M.; Pinto-Diaz, C.A.; Chaparro, P.; Gunturiz, M.L.; et al. A comprehensive analysis and immunobiology of autoimmune neurological syndromes during the Zika virus outbreak in Cucuta, Colombia. J. Autoimmun. 2017, 77, 123–138. [Google Scholar] [CrossRef]
- Nunes, M.L.; Esper, N.B.; Franco, A.R.; Radaelli, G.; Soder, R.B.; Bomfim, R.; Kalil Neto, F.; Gameleira, F.T.; Portuguez, M.W.; da Costa, J.C. Epilepsy after congenital zika virus infection: EEG and neuroimaging features. Seizure 2021, 84, 14–22. [Google Scholar] [CrossRef]
- Riley, E.M.; Stewart, V.A. Immune mechanisms in malaria: New insights in vaccine development. Nat. Med. 2013, 19, 168–178. [Google Scholar] [CrossRef]
- Takala, S.L.; Plowe, C.V. Genetic diversity and malaria vaccine design, testing and efficacy: Preventing and overcoming ‘vaccine resistant malaria’. Parasite Immunol. 2009, 31, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Sack, B.K.; King, C.R.; Nielsen, C.M.; Rayner, J.C.; Higgins, M.K.; Long, C.A.; Seder, R.A. Malaria Vaccines: Recent Advances and New Horizons. Cell Host Microbe 2018, 24, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Waide, M.L.; Schmidt, N.W. The gut microbiome, immunity, and Plasmodium severity. Curr. Opin. Microbiol. 2020, 58, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Datoo, M.S.; Natama, M.H.; Some, A.; Traore, O.; Rouamba, T.; Bellamy, D.; Yameogo, P.; Valia, D.; Tegneri, M.; Ouedraogo, F.; et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: A randomised controlled trial. Lancet 2021, 397, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Long, C.A.; Zavala, F. Immune Responses in Malaria. Cold Spring Harb. Perspect. Med. 2017, 7, a025577. [Google Scholar] [CrossRef]
- Pang, E.L.; Loh, H.S. Towards development of a universal dengue vaccine—How close are we? Asian Pac. J. Trop. Med. 2017, 10, 220–228. [Google Scholar] [CrossRef]
- Metz, S.W.; Geertsema, C.; Martina, B.E.; Andrade, P.; Heldens, J.G.; van Oers, M.M.; Goldbach, R.W.; Vlak, J.M.; Pijlman, G.P. Functional processing and secretion of Chikungunya virus E1 and E2 glycoproteins in insect cells. Virol. J. 2011, 8, 353. [Google Scholar] [CrossRef]
- Schlesinger, J.J.; Brandriss, M.W.; Walsh, E.E. Protection against 17D yellow fever encephalitis in mice by passive transfer of monoclonal antibodies to the nonstructural glycoprotein gp48 and by active immunization with gp48. J. Immunol. 1985, 135, 2805–2809. [Google Scholar] [CrossRef]
- Abbink, P.; Larocca, R.A.; De La Barrera, R.A.; Bricault, C.A.; Moseley, E.T.; Boyd, M.; Kirilova, M.; Li, Z.; Ng’ang’a, D.; Nanayakkara, O.; et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 2016, 353, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Dowd, K.A.; Chen, R.E.; Desai, P.; Foreman, B.; Burgomaster, K.E.; Himansu, S.; Kong, W.P.; Graham, B.S.; Pierson, T.C.; et al. Protective Efficacy of Nucleic Acid Vaccines Against Transmission of Zika Virus During Pregnancy in Mice. J. Infect. Dis. 2019, 220, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Spik, K.; Shurtleff, A.; McElroy, A.K.; Guttieri, M.C.; Hooper, J.W.; SchmalJohn, C. Immunogenicity of combination DNA vaccines for Rift Valley fever virus, tick-borne encephalitis virus, Hantaan virus, and Crimean Congo hemorrhagic fever virus. Vaccine 2006, 24, 4657–4666. [Google Scholar] [CrossRef]
Figure 1.
Different pathogens involved in mosquito-borne diseases include Plasmodium, dengue virus, chikungunya virus, yellow fever virus, Zika virus, Rift Valley fever virus, Japanese encephalitis virus, and West Nile virus.
Figure 1.
Different pathogens involved in mosquito-borne diseases include Plasmodium, dengue virus, chikungunya virus, yellow fever virus, Zika virus, Rift Valley fever virus, Japanese encephalitis virus, and West Nile virus.

Figure 2.
Plasmodium and infected red blood cells activate dendritic cells through parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) and are phagocytosed, and their antigens are presented to T cells. PRR signaling leads to the secretion of cytokines that initiate inflammation via Th1 and Th2 cell differentiation and macrophage polarization. Macrophages are responsible for the regulation of inflammation during the infection phase. T cells help with B cell differentiation and antibody secretion and secrete IFN-γ, which activates macrophages. IFN-γ-activated macrophages engulf opsonized cells.
Figure 2.
Plasmodium and infected red blood cells activate dendritic cells through parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) and are phagocytosed, and their antigens are presented to T cells. PRR signaling leads to the secretion of cytokines that initiate inflammation via Th1 and Th2 cell differentiation and macrophage polarization. Macrophages are responsible for the regulation of inflammation during the infection phase. T cells help with B cell differentiation and antibody secretion and secrete IFN-γ, which activates macrophages. IFN-γ-activated macrophages engulf opsonized cells.

Figure 3.
The adaptive immune responses combine forces to fight the dengue virus. B cells produce antibodies that specifically recognize and neutralize the foreign viral particles, and cytotoxic T cells recognize and kill cells that are infected with the dengue virus.
Figure 3.
The adaptive immune responses combine forces to fight the dengue virus. B cells produce antibodies that specifically recognize and neutralize the foreign viral particles, and cytotoxic T cells recognize and kill cells that are infected with the dengue virus.

Figure 4.
Dengue and its link with other disease complications after dengue virus infection and disease severity.
Figure 4.
Dengue and its link with other disease complications after dengue virus infection and disease severity.

Figure 5.
Mosquito-borne viral diseases (West Nile, chikungunya, Zika, yellow fever, Japanese encephalitis, Rift Valley fever) are recognized as multi-organ diseases with a broad spectrum of manifestations. Post-acute viral syndromes persist, presenting with prolonged effects with post-disease multiorgan complications.
Figure 5.
Mosquito-borne viral diseases (West Nile, chikungunya, Zika, yellow fever, Japanese encephalitis, Rift Valley fever) are recognized as multi-organ diseases with a broad spectrum of manifestations. Post-acute viral syndromes persist, presenting with prolonged effects with post-disease multiorgan complications.

Figure 6.
Representation of yellow fever (YF) pathway development. This figure illustrates the sequence of events in YF-related disease development, starting with the bite of an infected mosquito and ending with apoptosis and a heightened inflammatory reaction. The presence of the yellow fever virus (YFV) is indicated by a distinct color. It is hypothesized that intense viral replication in the liver sets off a series of molecular reactions, leading to considerable disruption in cytokine balance and an increased release of pro-inflammatory substances. Consequently, this results in substantial vascular impairment and multi-organ malfunction.
Figure 6.
Representation of yellow fever (YF) pathway development. This figure illustrates the sequence of events in YF-related disease development, starting with the bite of an infected mosquito and ending with apoptosis and a heightened inflammatory reaction. The presence of the yellow fever virus (YFV) is indicated by a distinct color. It is hypothesized that intense viral replication in the liver sets off a series of molecular reactions, leading to considerable disruption in cytokine balance and an increased release of pro-inflammatory substances. Consequently, this results in substantial vascular impairment and multi-organ malfunction.

Table 1.
Different Plasmodium sp. and its immune responses.
| Plasmodium Species | Severity | Distribution | Innate Immunity | Humoral Immunity | Major Involved Pathway | References |
|---|---|---|---|---|---|---|
| P. falciparum | High | Africa, Asia, and Latin America | Neutrophils, monocytes, and NK cells | Long-lived plasma B cells and memory B cells; antibodies | Toll, immune deficiency (Imd), Janus kinase (JNK), and signal transducers and activators of transcription (STAT) | [52,53] |
| P. vivax | Moderate | Asia, Latin America, Africa | Cytokines, dendritic cells | B cells and antibodies | IFN-α/IFN-γ and TCR pathway, MAPK6/MAPK4 signalling | [46,54] |
| P. ovale | Low–moderate | Africa, Asia | NA | NA | NA | |
| P. malariae | Low | Africa, Asia, and Latin America | NA | NA | NA | |
| P. knowlesi | Moderate–high | Southeast Asia | NA | NA | NA |
(NA = Not available).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bhattacharjee, S.; Ghosh, D.; Saha, R.; Sarkar, R.; Kumar, S.; Khokhar, M.; Pandey, R.K. Mechanism of Immune Evasion in Mosquito-Borne Diseases. Pathogens 2023, 12, 635. https://doi.org/10.3390/pathogens12050635
AMA Style
Bhattacharjee S, Ghosh D, Saha R, Sarkar R, Kumar S, Khokhar M, Pandey RK. Mechanism of Immune Evasion in Mosquito-Borne Diseases. Pathogens. 2023; 12(5):635. https://doi.org/10.3390/pathogens12050635
Chicago/Turabian StyleBhattacharjee, Swagato, Debanjan Ghosh, Rounak Saha, Rima Sarkar, Saurav Kumar, Manoj Khokhar, and Rajan Kumar Pandey. 2023. "Mechanism of Immune Evasion in Mosquito-Borne Diseases" Pathogens 12, no. 5: 635. https://doi.org/10.3390/pathogens12050635
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.