
Molecular Structural Analysis of Porcine CMAH–Native Ligand Complex and High Throughput Virtual Screening to Identify Novel Inhibitors

Abstract
:1. Introduction
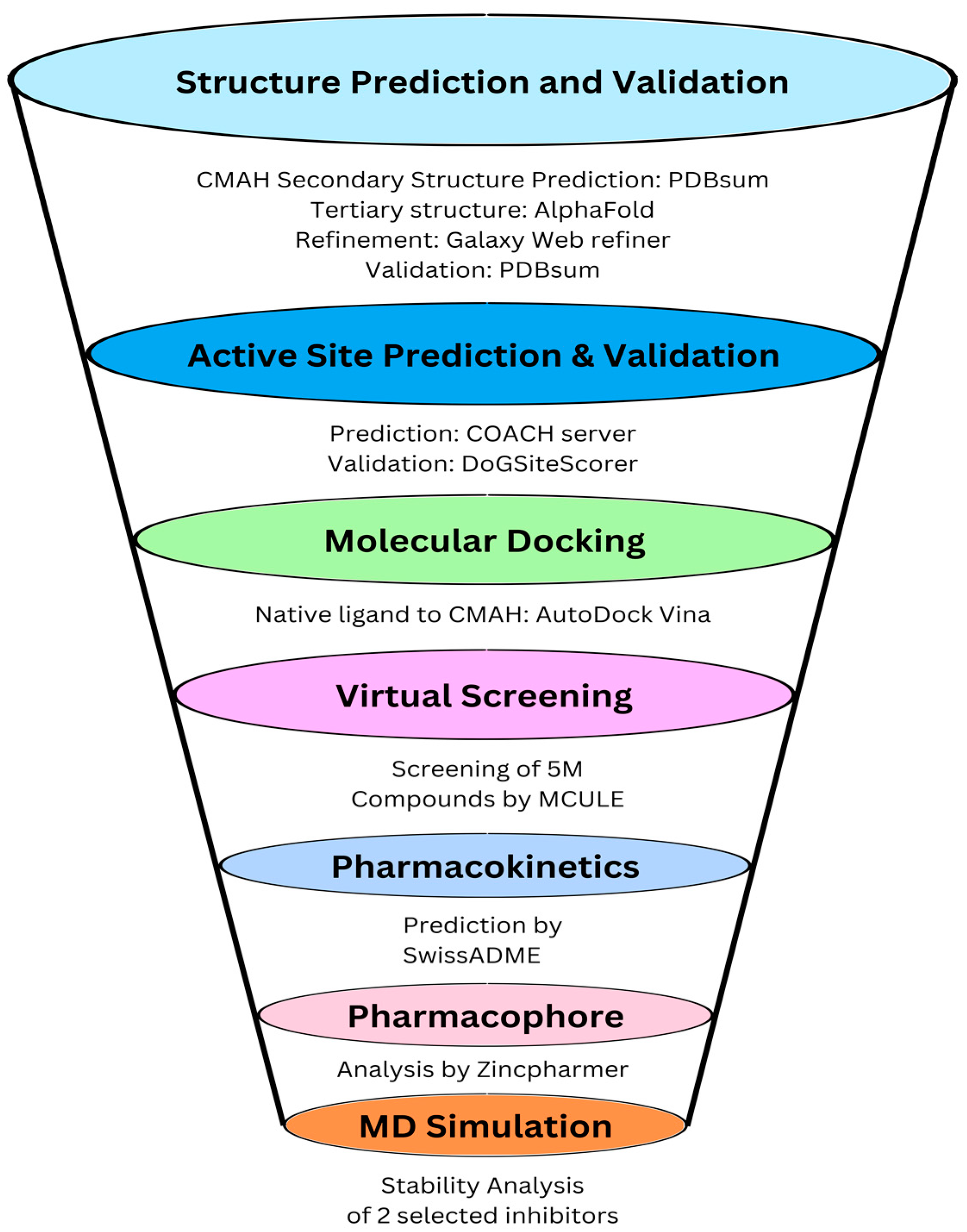
2. Materials and Methods
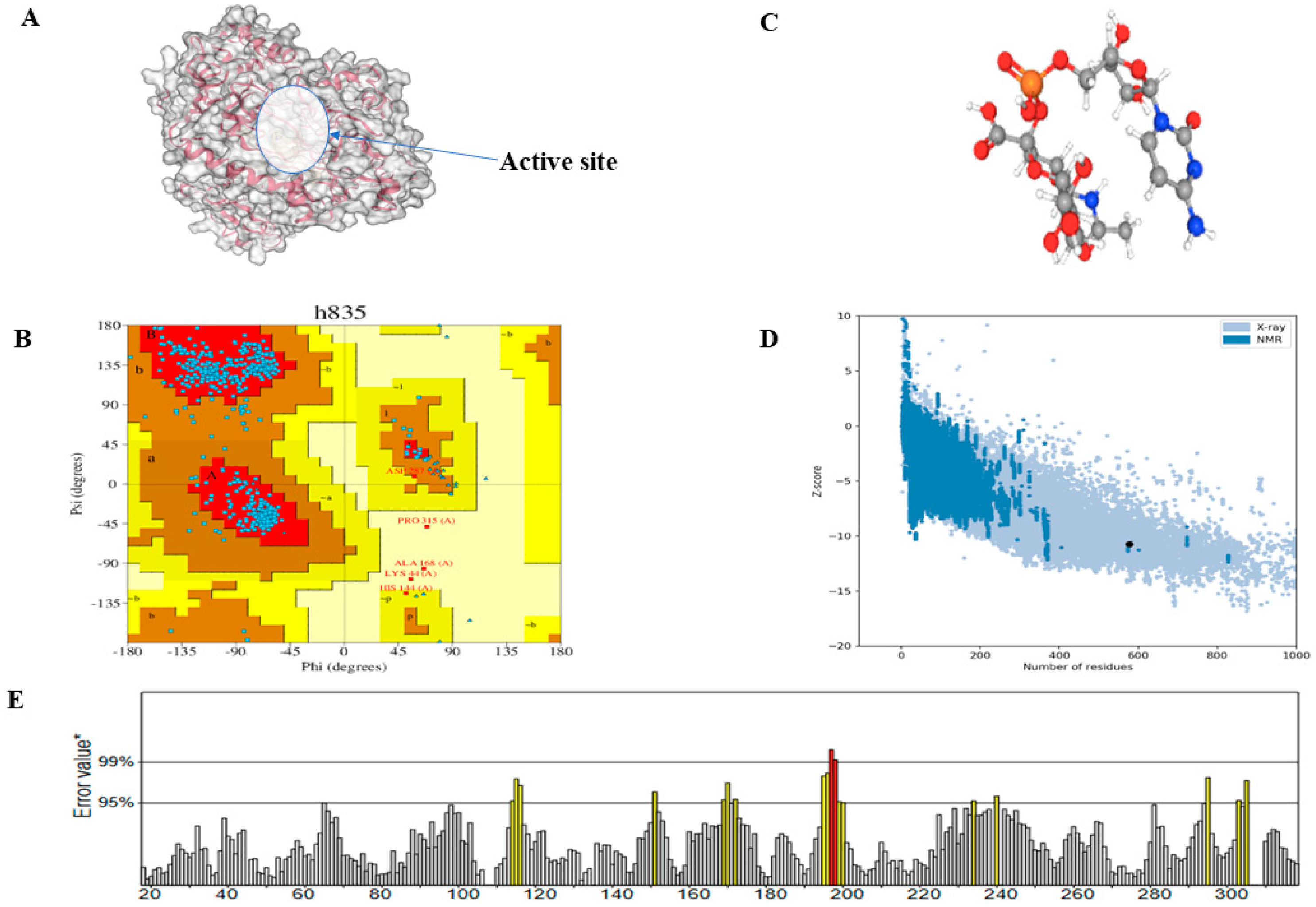
2.1. Prediction, Refinement, and Validation of Tertiary Structure of CMAH Protein
2.2. Prediction of Active Site Residues
2.3. Computational Docking of Porcine CMAH and the Native Ligand
2.4. High Throughput Structure-Based-Virtual Screening
2.5. Molecular Dynamic Simulations of the Complexes
3. Results
3.1. Predicted Active Site Residues and Tertiary Structure Validation
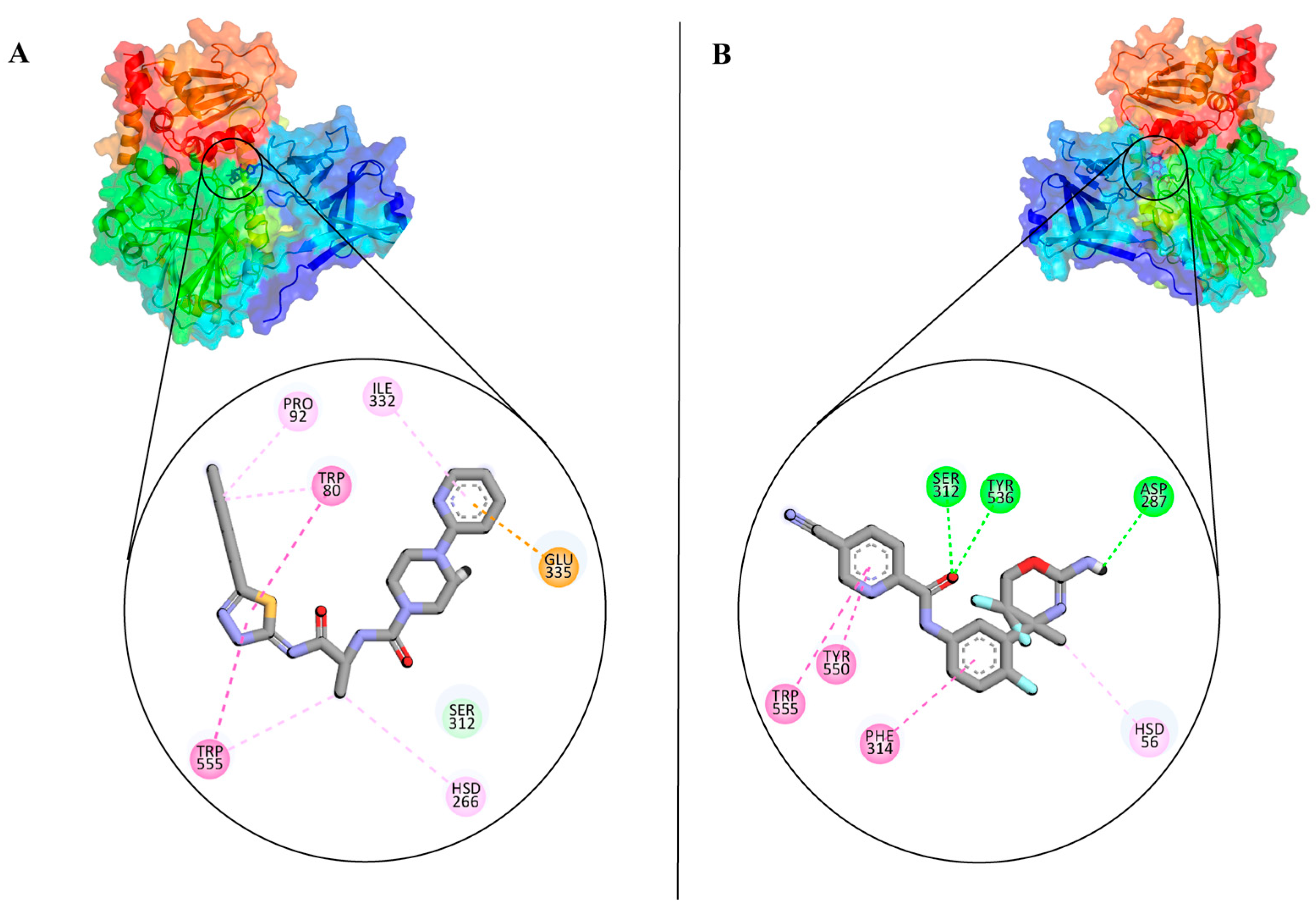
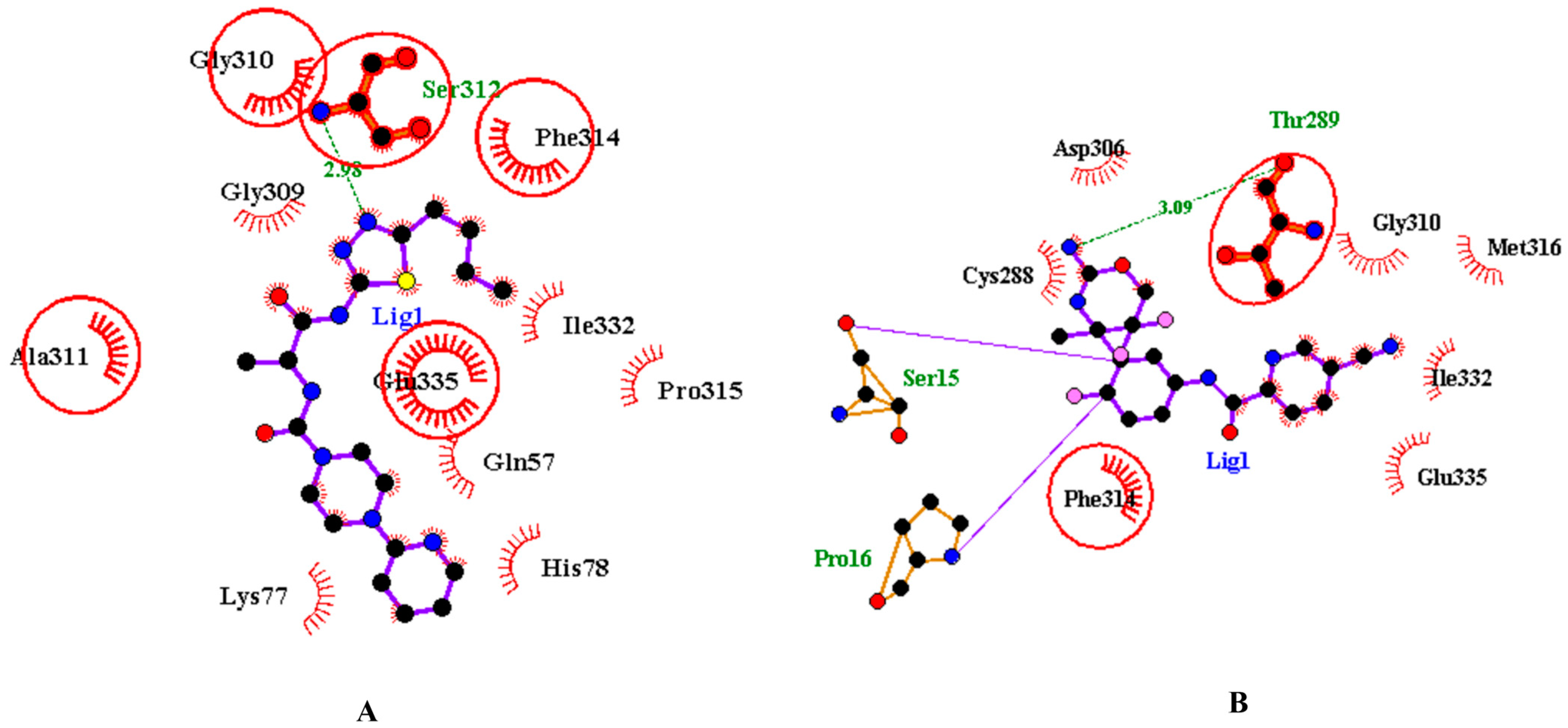
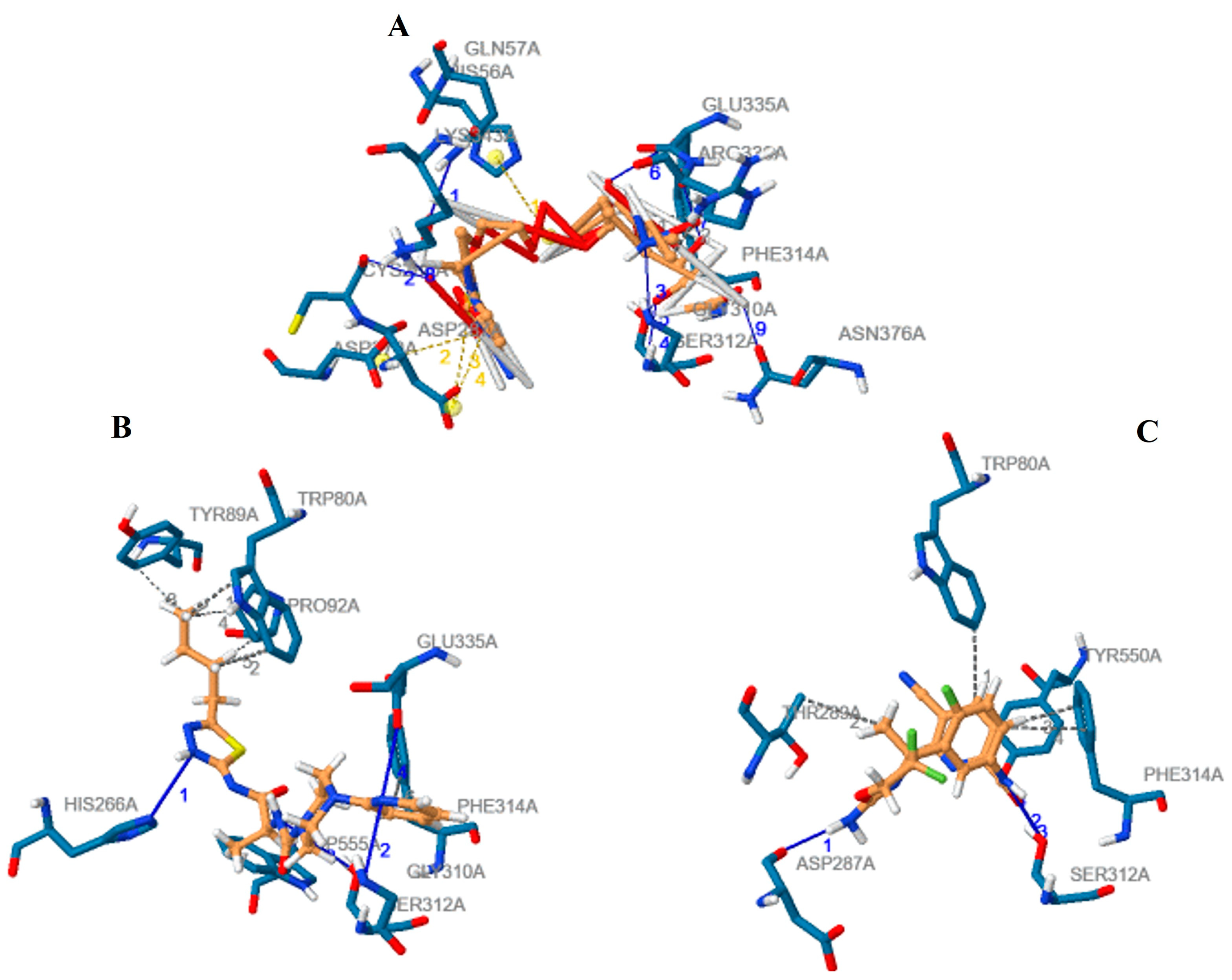
3.2. Analysis and Visualisation of the Docked Complex of CMAH and CMP-Neu5Ac
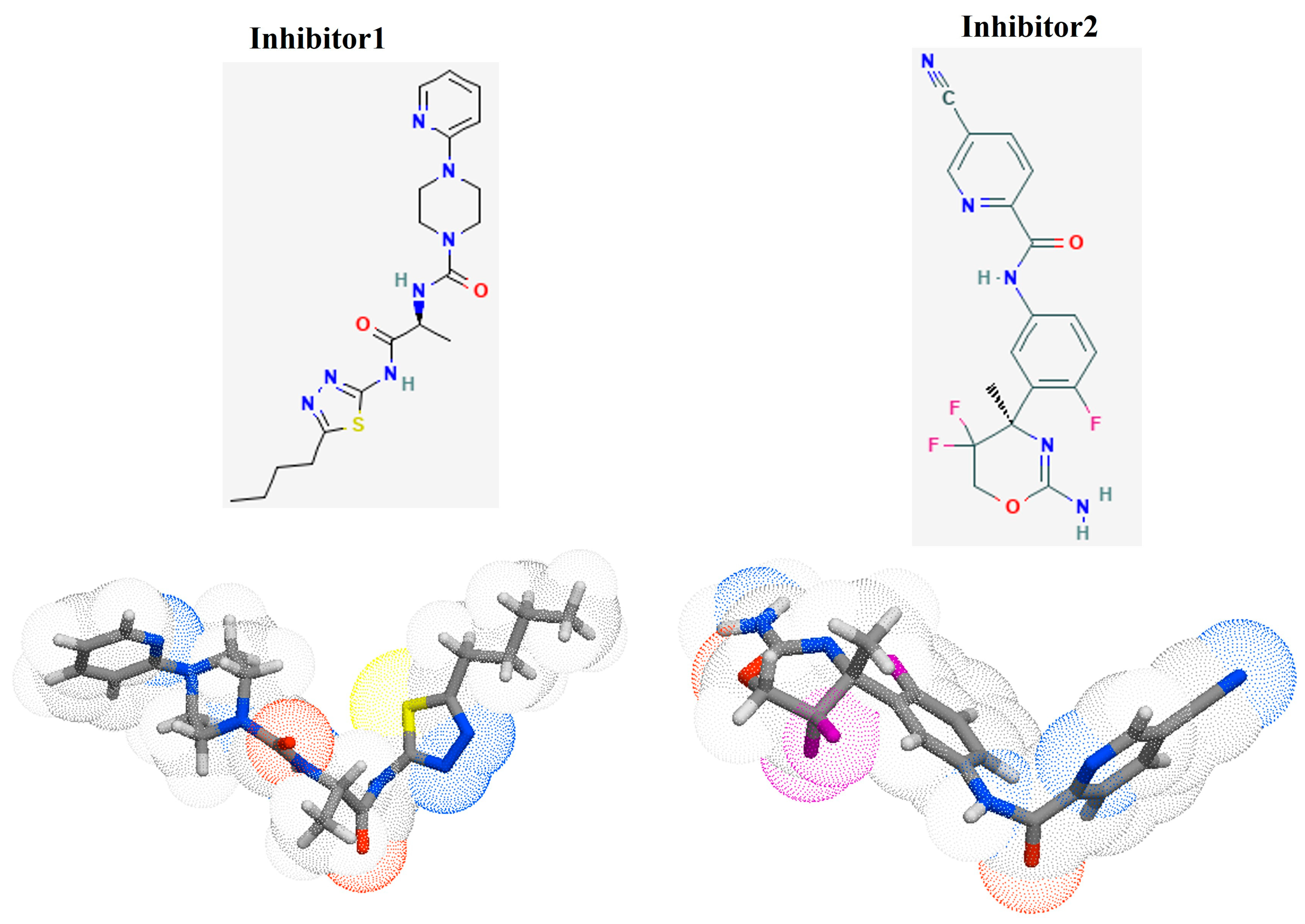
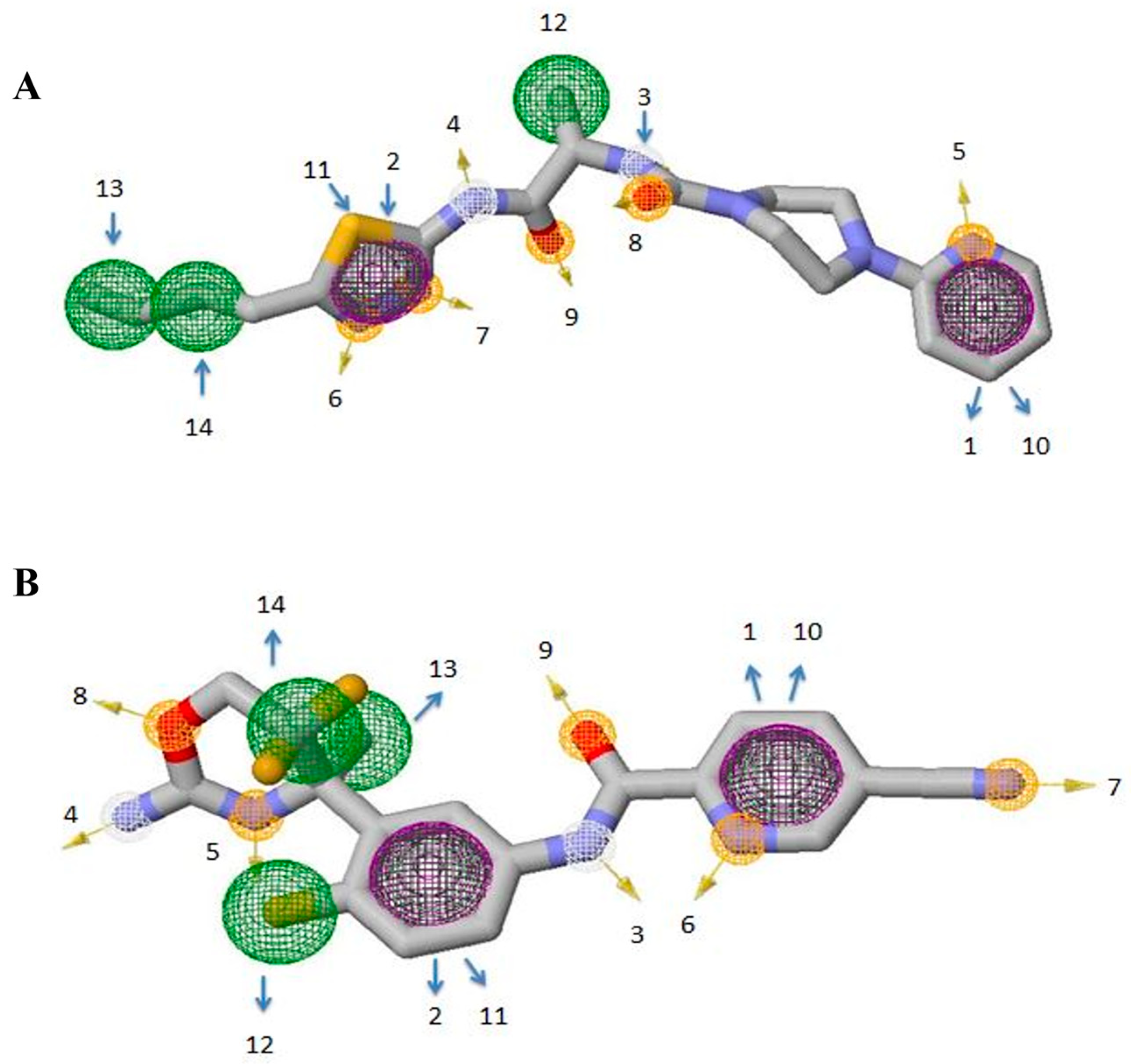
3.3. Identification of Potential Inhibitors through Structure-Based Virtual Screening
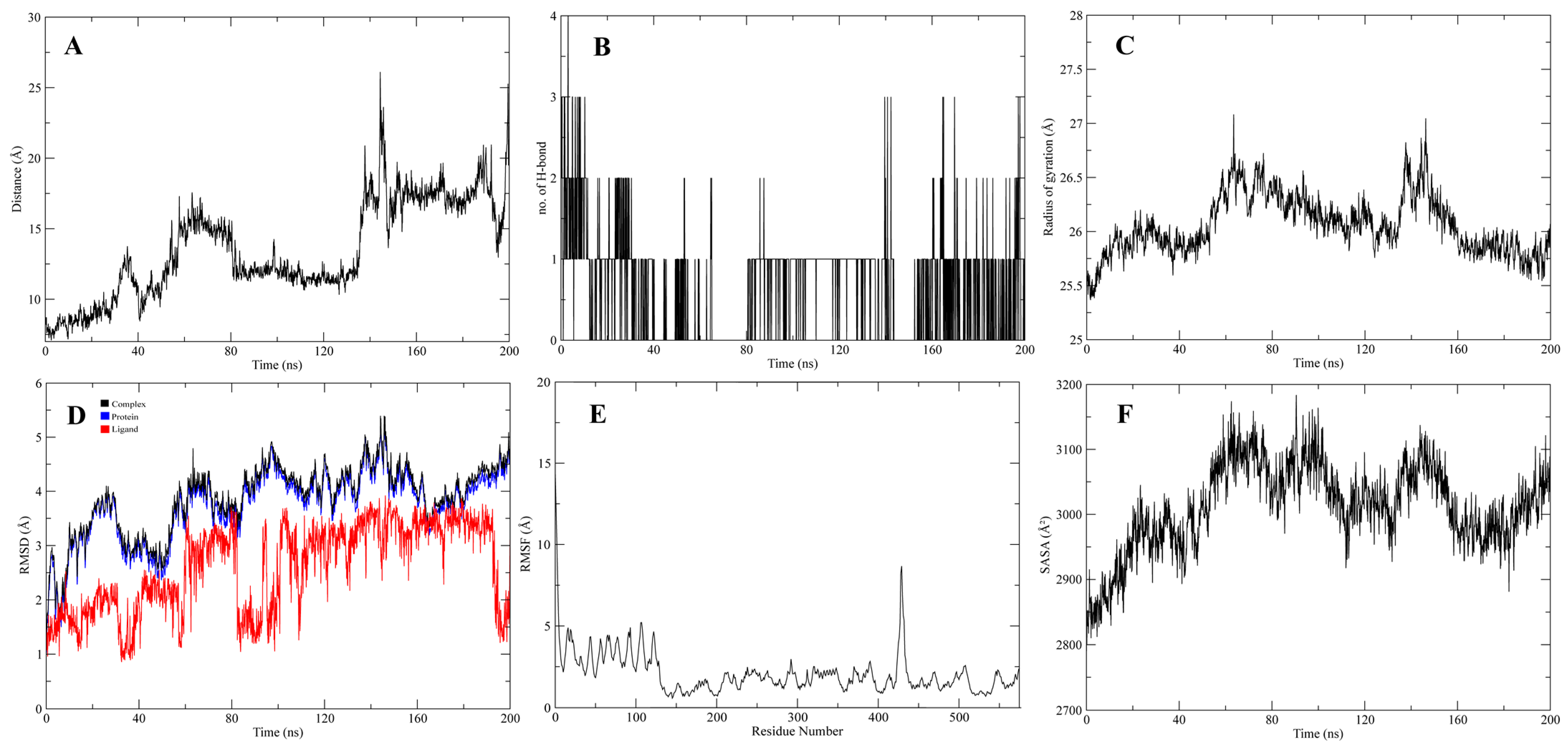
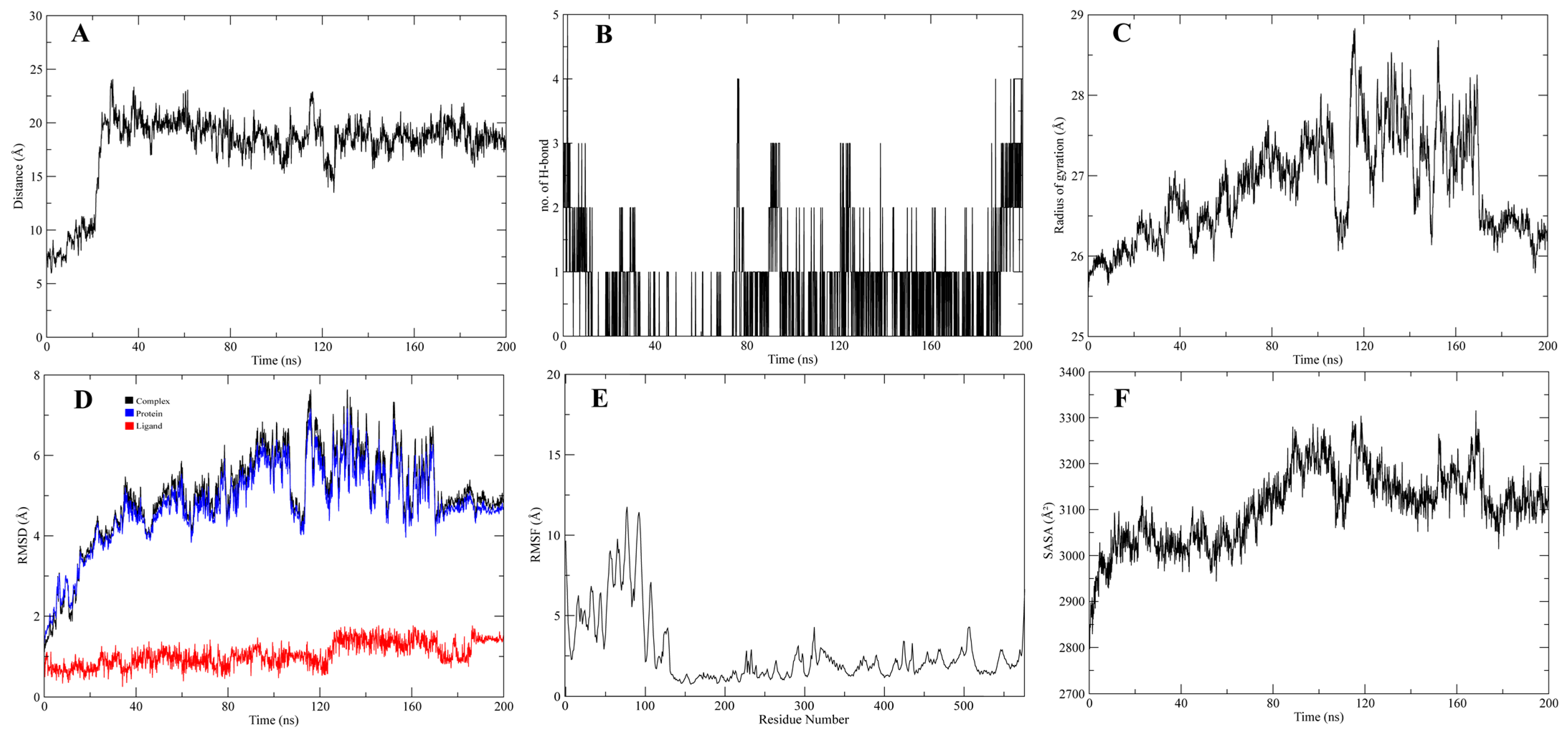
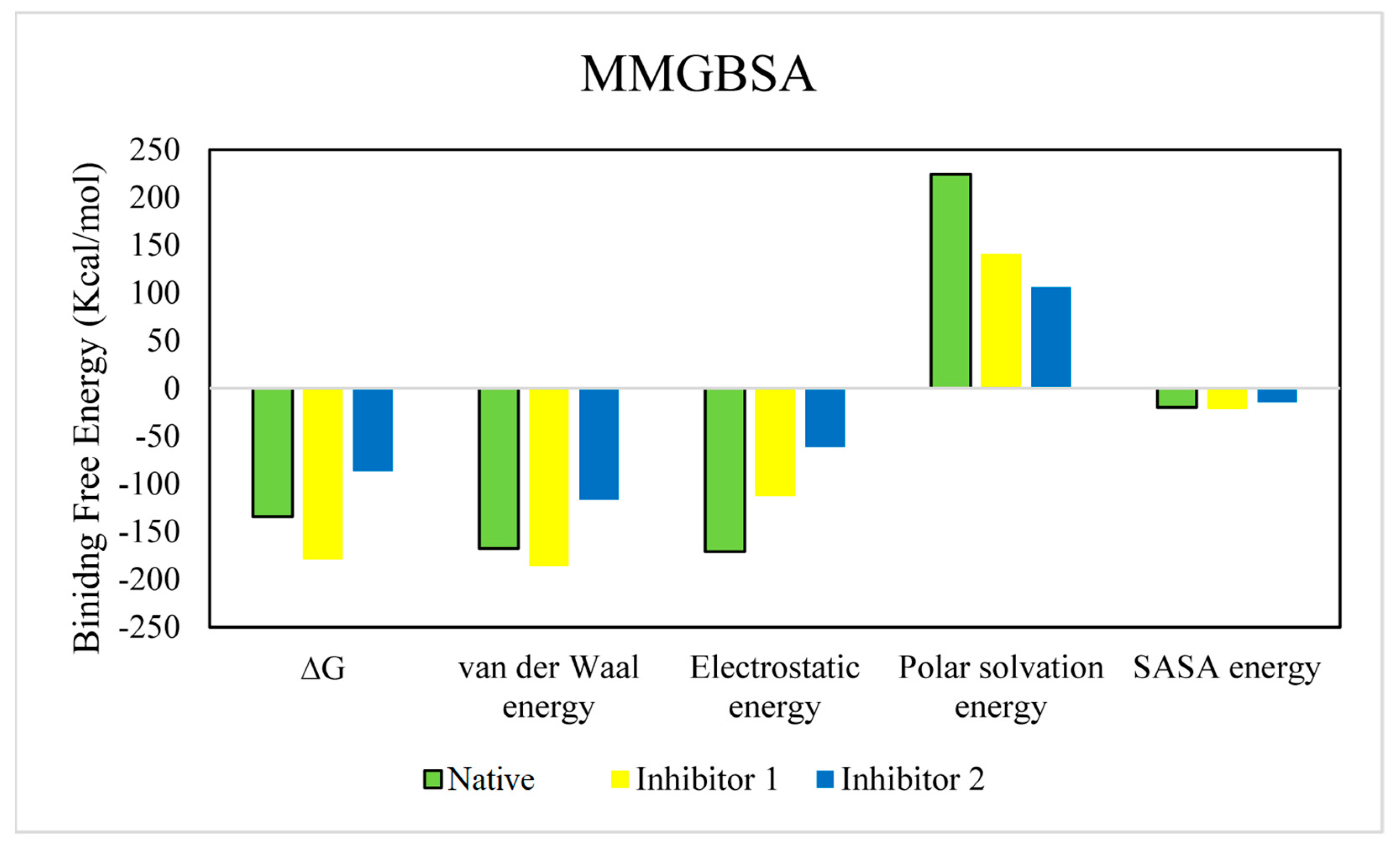
3.4. Molecular Dynamic Simulations of the Complexes
4. Discussion
5. Conclusions and Recommendations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agricultural Output—Meat Consumption—OECD Data, The OECD. Available online: https://data.oecd.org/agroutput/meat-consumption.htm (accessed on 21 April 2020).
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the porcine species in biomedical research. Front. Genet. 2015, 6, 293. [Google Scholar] [CrossRef] [PubMed]
- Ogun, O.J.; Thaller, G.; Becker, D. An Overview of the Importance and Value of Porcine Species in Sialic Acid Research. Biology 2022, 11, 903. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal, A.M.; Caicedo, L.A.; Martínez, J.M.; Moreno, M.; Echeverri, G.J. Clinical Xenotransplantation, a Closer Reality: Literature Review. Cirugía Española (Engl. Ed.) 2017, 95, 62–72. [Google Scholar] [CrossRef]
- Dhar, C.; Sasmal, A.; Varki, A. From “Serum Sickness” to “Xenosialitis”: Past, Present, and Future Significance of the Non-human Sialic Acid Neu5Gc. Front. Immunol. 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed]
- Samraj, A.N.; Läubli, H.; Varki, N.; Varki, A. Involvement of a Non-Human Sialic Acid in Human Cancer. Front. Oncol. 2014, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Angata, T.; Varki, A. Chemical Diversity in the Sialic Acids and Related α-Keto Acids: An Evolutionary Perspective. Chem. Rev. 2002, 102, 439–470. [Google Scholar] [CrossRef]
- Kooner, A.S.; Yu, H.; Chen, X. Synthesis of N-Glycolylneuraminic Acid (Neu5Gc) and Its Glycosides. Front. Immunol. 2019, 10, 2004. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Schauer, R. Sialic acids. In Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Delorme, C.; Brüssow, H.; Sidoti, J.; Roche, N.; Karlsson, K.-A.; Neeser, J.-R.; Teneberg, S. Glycosphingolipid Binding Specificities of Rotavirus: Identification of a Sialic Acid-Binding Epitope. J. Virol. 2001, 75, 2276–2287. [Google Scholar] [CrossRef]
- Kyogashima, M.; Ginsburg, V.; Krivan, H.C. Escherichia coli K99 binds to N-glycolylsialoparagloboside and N-glycolyl-GM3 found in piglet small intestine. Arch. Biochem. Biophys. 1989, 270, 391–397. [Google Scholar] [CrossRef]
- Schwegmann, C.; Zimmer, G.; Yoshino, T.; Enss, M.-L.; Herrler, G. Comparison of the sialic acid binding activity of transmissible gastroenteritis coronavirus and E. coli K99. Virus Res. 2001, 75, 69–73. [Google Scholar] [CrossRef]
- Scholtissek, C. Pigs as ‘mixing vessels’ for the creation of new pandemic influenza A viruses. Med. Princ. Pract. 1990, 2, 65–71. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef]
- Nelli, R.K.; Kuchipudi, S.V.; White, G.A.; Baquero Perez, B.; Dunham, S.P.; Chang, K.-C. Comparative distribution of human and avian type sialic acid influenza receptors in the pig. BMC Vet. Res. 2010, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G. Antigenic Variation in Influenza Viruses. In Origin and Evolution of Viruses; Domingo, E., Webster, R., Holland, J., Eds.; Academic Press: London, UK, 1999; pp. 377–390. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Loffredo, M.; Lucero, H.; Chen, D.-Y.; O’connell, A.; Bergqvist, S.; Munawar, A.; Bandara, A.; De Graef, S.; Weeks, S.D.; Douam, F.; et al. The in-vitro effect of famotidine on SARS-CoV-2 proteases and virus replication. Sci. Rep. 2021, 11, 5433. [Google Scholar] [CrossRef]
- Guruprasad, L. Human SARS-CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinform. 2021, 89, 569–576. [Google Scholar] [CrossRef]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef]
- Fischer, K.; Rieblinger, B.; Hein, R.; Sfriso, R.; Zuber, J.; Fischer, A.; Klinger, B.; Liang, W.; Flisikowski, K.; Kurome, M.; et al. Viable pigs after simultaneous inactivation of porcine MHC class I and three xenoreactive antigen genes GGTA1, CMAH and B4GALNT2. Xenotransplantation 2020, 27, e12560. [Google Scholar] [CrossRef]
- Burlak, C.; Paris, L.L.; Lutz, A.J.; Sidner, R.A.; Estrada, J.; Li, P.; Tector, M.; Tector, A.J. Tector, Reduced Binding of Human Antibodies to Cells from GGTA1/CMAH KO Pigs: Crossmatch Analysis of GGTA1/CMAH KO Pigs. Am. J. Transplant. 2014, 14, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Matsunari, H.; Watanabe, M.; Nakano, K.; Umeyama, K.; Sakai, R.; Takayanagi, S.; Takeishi, T.; Fukuda, T.; Yashima, S.; et al. Generation of α1,3-galactosyltransferase and cytidine monophospho-N-acetylneuraminic acid hydroxylase gene double-knockout pigs. J. Reprod. Dev. 2015, 61, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Martens, G.R.; Reyes, L.M.; Butler, J.R.; Ladowski, J.M.; Estrada, J.L.; Sidner, R.A.; Eckhoff, D.E.; Tector, M.; Tector, A.J. Humoral Reactivity of Renal Transplant-Waitlisted Patients to Cells from GGTA1/CMAH/B4GalNT2, and SLA Class I Knockout Pigs. Transplantation 2017, 101, e86–e92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Burlak, C.; Estrada, J.L.; Li, P.; Tector, M.F.; Tector, A.J. Erythrocytes from GGTA1/CMAH knockout pigs: Implications for xenotransfusion and testing in non-human primates. Xenotransplantation 2014, 21, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Shakil, S.; Rizvi, S.M.D.; Greig, N.H. High Throughput Virtual Screening and Molecular Dynamics Simulation for Identifying a Putative Inhibitor of Bacterial CTX-M-15. Antibiotics 2021, 10, 474. [Google Scholar] [CrossRef]
- Sehailia, M.; Chemat, S. Antimalarial-agent artemisinin and derivatives portray more potent binding to Lys353 and Lys31-binding hotspots of SARS-CoV-2 spike protein than hydroxychloroquine: Potential repurposing of artenimol for COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 6184–6194. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, X.; Russell, P.; Li, J.; Pan, W.; Fu, J.; Zhang, A. Evaluation of the binding performance of flavonoids to estrogen receptor alpha by Autodock, Autodock Vina and Surflex-Dock. Ecotoxicol. Environ. Saf. 2022, 233, 113323. [Google Scholar] [CrossRef]
- Viegas, D.J.; Edwards, T.G.; Bloom, D.C.; Abreu, P.A. Virtual screening identified compounds that bind to cyclin dependent kinase 2 and prevent herpes simplex virus type 1 replication and reactivation in neurons. Antivir. Res. 2019, 172, 104621. [Google Scholar] [CrossRef]
- Breznik, M.; Ge, Y.; Bluck, J.P.; Briem, H.; Hahn, D.F.; Christ, C.D.; Mortier, J.; Mobley, D.L.; Meier, K. Prioritizing Small Sets of Molecules for Synthesis through in-silico Tools: A Comparison of Common Ranking Methods. ChemMedChem 2023, 18, e202200425. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year 2021: Protein structure prediction. Nat. Methods 2022, 19, 1. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Dym, O.; Eisenberg, O.D.; Yeates, T. International Tables for Crystallography; Reidel: Dordrecht, The Netherlands, 2012; pp. 678–679. [Google Scholar]
- Zhao, J.; Cao, Y.; Zhang, L. Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 2020, 18, 417–426. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Li, Q.; Shah, S. Structure-Based Virtual Screening. In Protein Bioinformatics; Wu, C.H., Arighi, C.N., Ross, K.E., Eds.; Springer: New York, NY, USA, 2017; pp. 111–124. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Chemin 2012, 4, P17. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Colleton, C.; Brewster, D.; Chester, A.; Clarke, D.O.; Heining, P.; Olaharski, A.; Graziano, M. The Use of Minipigs for Preclinical Safety Assessment by the Pharmaceutical Industry. Toxicol. Pathol. 2016, 44, 458–466. [Google Scholar] [CrossRef]
- Bollen, P.; Ellegaard, L. The Göttingen Minipig in Pharmacology and Toxicology. Basic Clin. Pharmacol. Toxicol. 1997, 80, 3–4. [Google Scholar] [CrossRef]
- Henze, L.J.; Koehl, N.J.; O’Shea, J.P.; Kostewicz, E.S.; Holm, R.; Griffin, B.T. The pig as a preclinical model for predicting oral bioavailability and in vivo performance of pharmaceutical oral dosage forms: A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 581–602. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Qureshi, K.A.; Al Nasr, I.; Koko, W.S.; Khan, T.A.; Fatmi, M.Q.; Imtiaz, M.; Khan, R.A.; Mohammed, H.A.; Jaremko, M.; Emwas, A.-H.; et al. In Vitro and In Silico Approaches for the Antileishmanial Activity Evaluations of Actinomycins Isolated from Novel Streptomyces smyrnaeus Strain UKAQ_23. Antibiotics 2021, 10, 887. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- Pathak, R.K.; Seo, Y.-J.; Kim, J.-M. Structural insights into inhibition of PRRSV Nsp4 revealed by structure-based virtual screening, molecular dynamics, and MM-PBSA studies. J. Biol. Eng. 2022, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Scobie, L.; Padler-Karavani, V.; Le Bas-Bernardet, S.; Crossan, C.; Blaha, J.; Matouskova, M.; Hector, R.D.; Cozzi, E.; Vanhove, B.; Charreau, B.; et al. Long-Term IgG Response to Porcine Neu5Gc Antigens without Transmission of PERV in Burn Patients Treated with Porcine Skin Xenografts. J. Immunol. 2013, 191, 2907–2915. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Xu, Z.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. C−X··· H contacts in biomolecular systems: How they contribute to protein−ligand binding affinity. J. Phys. Chem. B 2009, 113, 12615–12621. [Google Scholar] [CrossRef]
- Kurczab, R.; Śliwa, P.; Rataj, K.; Kafel, R.; Bojarski, A.J. Salt Bridge in Ligand–Protein Complexes—Systematic Theoretical and Statistical Investigations. J. Chem. Inf. Model. 2018, 58, 2224–2238. [Google Scholar] [CrossRef] [PubMed]
- Donald, J.E.; Kulp, D.W.; DeGrado, W.F. Salt bridges: Geometrically specific, designable interactions. Proteins 2011, 79, 898–915. [Google Scholar] [CrossRef]
- Bastolla, U.; Demetrius, L. Stability constraints and protein evolution: The role of chain length, composition and disulfide bonds. Protein Eng. Des. Sel. 2005, 18, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Kuddus, M. Introduction to Food Enzymes. In Enzymes in Food Biotechnology; Kuddus, M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–18. [Google Scholar] [CrossRef]
- Ramakrishnan, J.; Rathore, S.S.; Raman, T. Review on fungal enzyme inhibitors—Potential drug targets to manage human fungal infections. RSC Adv. 2016, 6, 42387–42401. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Pharmacokinetics and Metabolism in Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Ogun, O.J.; Soremekun, O.S.; Thaller, G.; Becker, D. An In Silico Functional Analysis of Non-Synonymous Single-Nucleotide Polymorphisms of Bovine CMAH Gene and Potential Implication in Pathogenesis. Pathogens 2023, 12, 591. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Molecular Formula | Hydrophobic Interactions | Hydrogen Bonds | Salt Bridges | Vina Score (Kcal/mol) |
|---|---|---|---|---|---|
| Native | C20H31N4O16P | Phe314 & Arg336 | Gln57, Cys288, Gly310, Ser312, Glu335, Lys343, Asn376 | His56, Asp270, Asp287 | −8.7 |
| Inhibitor 1 | C19H27N7O2S | Trp80, Tyr89, Pro92, Phe314, Trp555 | His266, Gly310, Ser312, Glu335 | None | −9.9 |
| Inhibitor 2 | C18H14F3NSO2 | Trp80, Thr289, Phe314, Try550 | Asp287, Ser312, Tyr550 | None | −9.4 |
| Compound | ∆G | Van der Waal Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy |
|---|---|---|---|---|---|
| Native | −134.317 | −167.815 | −170.890 | 224.374 | −19.985 |
| ±89.180 | ±44.570 | ±162.416 | ±119.177 | ±3.904 | |
| Inhibitor 1 | −179.038 | −185.644 | −112.857 | 140.880 | −21.418 |
| ±12.127 | ±13.649 | ±34.330 | ±46.563 | ±0.989 | |
| Inhibitor 2 | −86.716 | −116.820 | −61.125 | 105.857 | −14.629 |
| ±22.650 | ±15.080 | ±48.771 | ±61.576 | ±2.232 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogun, O.J.; Thaller, G.; Becker, D. Molecular Structural Analysis of Porcine CMAH–Native Ligand Complex and High Throughput Virtual Screening to Identify Novel Inhibitors. Pathogens 2023, 12, 684. https://doi.org/10.3390/pathogens12050684
Ogun OJ, Thaller G, Becker D. Molecular Structural Analysis of Porcine CMAH–Native Ligand Complex and High Throughput Virtual Screening to Identify Novel Inhibitors. Pathogens. 2023; 12(5):684. https://doi.org/10.3390/pathogens12050684
Chicago/Turabian StyleOgun, Oluwamayowa Joshua, Georg Thaller, and Doreen Becker. 2023. "Molecular Structural Analysis of Porcine CMAH–Native Ligand Complex and High Throughput Virtual Screening to Identify Novel Inhibitors" Pathogens 12, no. 5: 684. https://doi.org/10.3390/pathogens12050684