Evaluation of Automated Ribosomal Intergenic Spacer Analysis for Bacterial Fingerprinting of Rumen Microbiome Compared to Pyrosequencing Technology

Abstract
:1. Introduction
2. Comparison of Local Richness Obtained from ARISA vs. Pyrosequencing
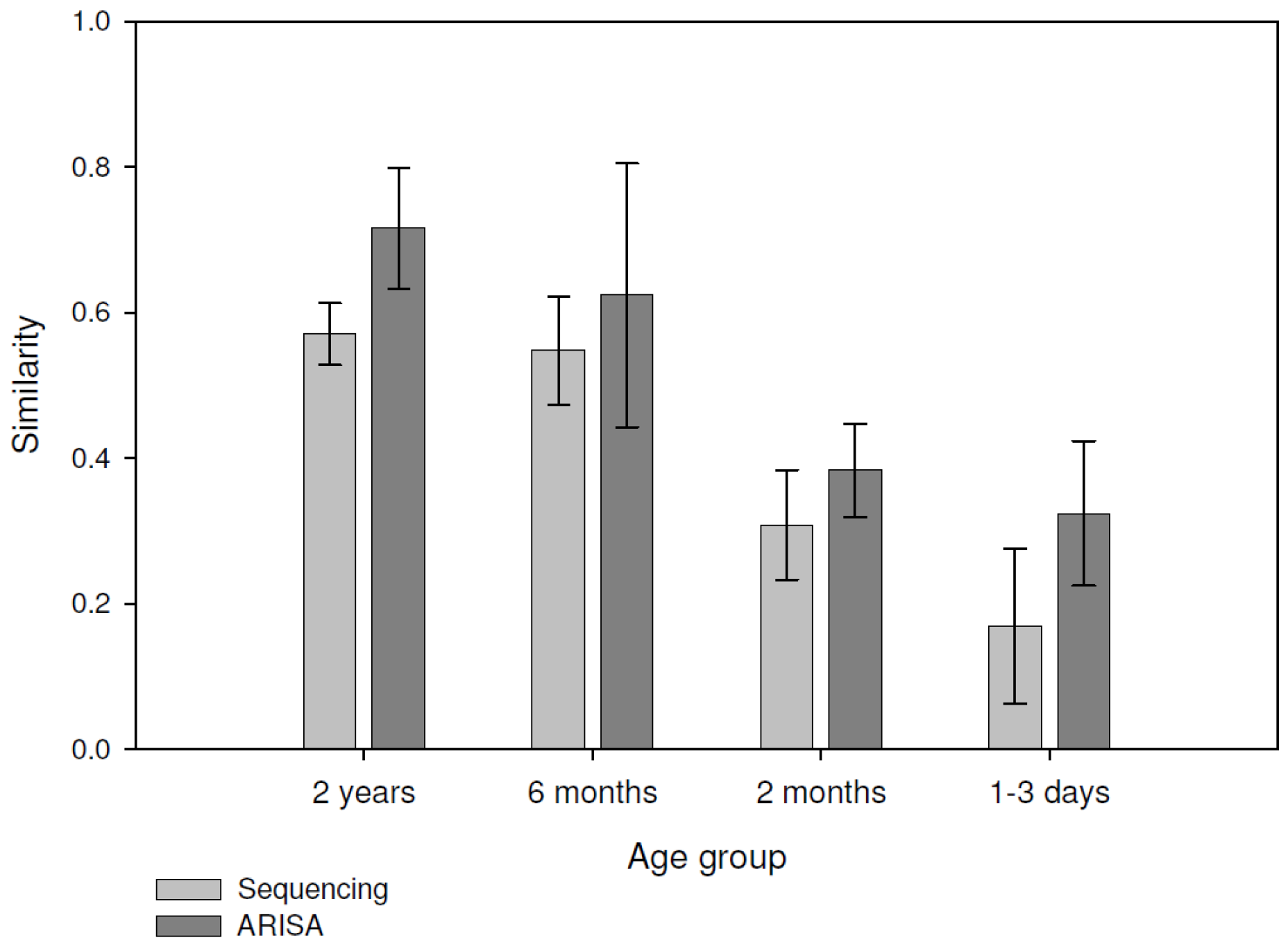
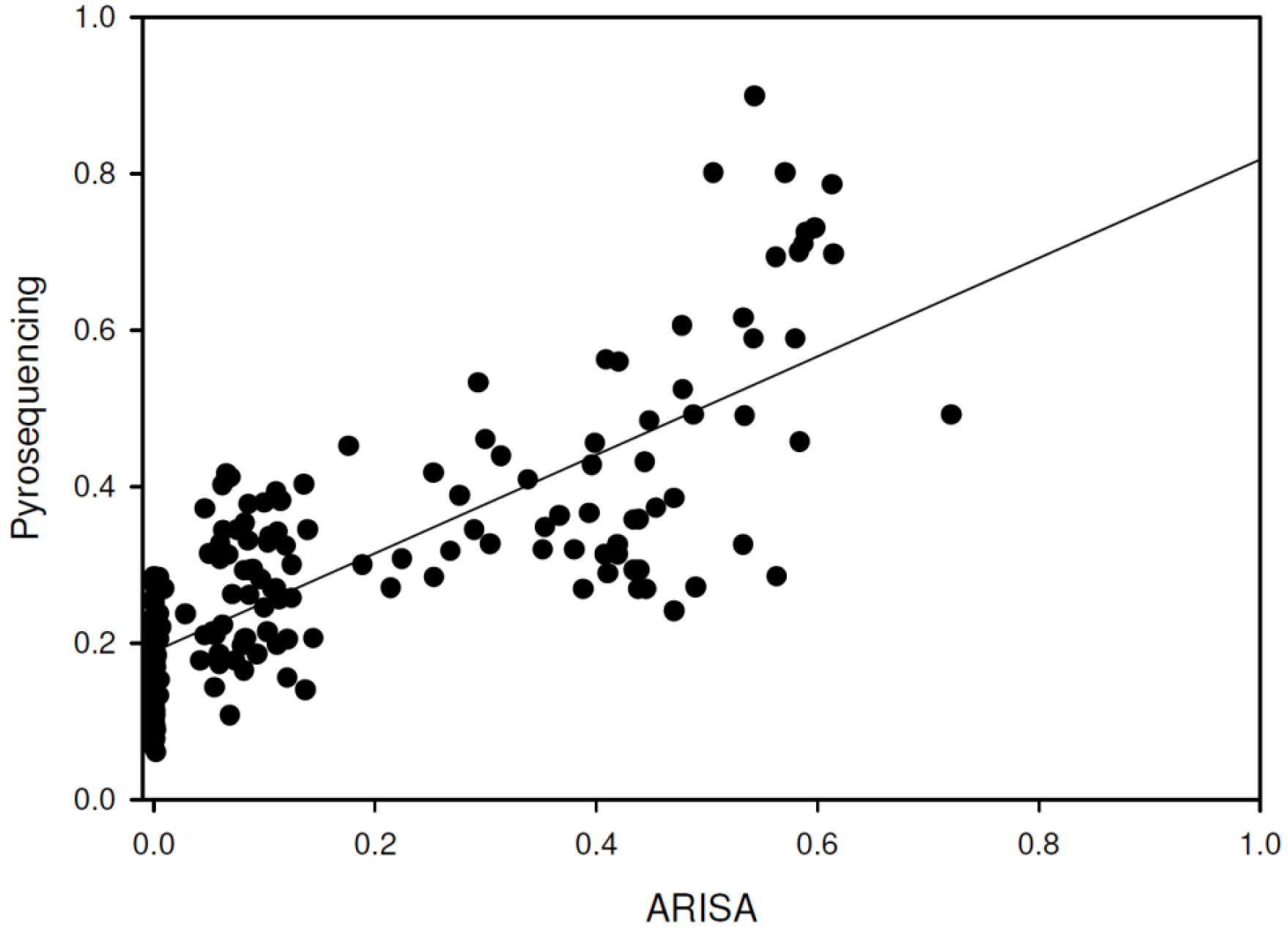
3. β-Diversity Calculation

4. Discussion



{kind=link}
{kind=link}
{kind=link}
| Number of OTUs identified (number of OTUs per sample) | Shannon-Wiener (H') diversity (SD) | |||
|---|---|---|---|---|
| Pyrosequencing | ARISA | Pyrosequencing | ARISA | |
| 1–3 days old | 380 (208 ± 47 a) | 206 (90 ± 18 a) | 2.8 (0.49) a | 3.7 (0.31) a |
| 2 months old | 1441 (620 ± 100 b) | 204 (116± 8 b) | 3.7 (0.36) b | 4.2 (0.15) b |
| 6 months old | 4074 (2051 ± 210 c) | 235 (141 ± 20 c) | 6.2 (0.3) c | 4.2 (0.23) b |
| 2 years old | 4885 (2382 ± 263 d) | 214 (148 ± 23 c) | 6.5 (0.08) d | 4.4 (0.35) b |
| Pyrosequencing | 2 years | 6 months | 2 months | 1–3 days |
|---|---|---|---|---|
| 2 years | 0 | 0.043 | 0.042 | 0.01 |
| 6 months | 0.916 | 0 | 0.034 | 0.012 |
| 2 months | 1 | 1 | 0 | 0.015 |
| 1–3 days | 1 | 1 | 1 | 0 |
| ARISA | 2 years | 6 months | 2 months | 1–3 days |
|---|---|---|---|---|
| 2 years | 0 | 0. 047 | 0. 047 | 0. 012 |
| 6 months | 0.96 | 0 | 0. 047 | 0. 013 |
| 2 months | 0.684 | 0.948 | 0 | 0. 012 |
| 1–3 days | 0.9387 | 0.7893 | 0.984 | 0 |
5. Experimental Section
5.1. Animal Handling and Sampling
5.2. Isolation of Microbial Fraction from the Rumen
5.3. DNA Extraction
5.4. Automated Ribosomal Intergenic Spacer Analysis (ARISA)
5.5. ARISA Resolution and Analysis
5.6. 454 Tag Amplicon Pyrosequencing and Data Analyses
5.7. Statistical Analyses
6. Conclusion
Conflicts of Interest
References
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16s rRNA and 16s rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Fisher, M.M.; Triplett, E.W. Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 1999, 65, 4630–4636. [Google Scholar]
- Kovacs, A.; Yacoby, K.; Gophna, U. A systematic assessment of automated ribosomal intergenic spacer analysis (arisa) as a tool for estimating bacterial richness. Res. Microbiol. 2010, 161, 192–197. [Google Scholar] [CrossRef]
- Koopman, M.M.; Fuselier, D.M.; Hird, S.; Carstens, B.C. The carnivorous pale pitcher plant harbors diverse, distinct, and time-dependent bacterial communities. Appl. Environ. Microbiol. 2010, 76, 1851–1860. [Google Scholar] [CrossRef]
- Bent, S.J.; Forney, L.J. The tragedy of the uncommon: Understanding limitations in the analysis of microbial diversity. ISME J. 2008, 2, 689–695. [Google Scholar] [CrossRef]
- Bent, S.J.; Pierson, J.D.; Forney, L.J.; Danovaro, R.; Luna, G.M.; Dell’sanno, A.; Pietrangeli, B. Measuring species richness based on microbial community fingerprints: The emperor has no clothes. Appl. Environ. Microbiol. 2007, 73, 2399–2401. [Google Scholar] [CrossRef]
- Gobet, A.l.; Boetius, A.; Ramette, A. Ecological coherence of diversity patterns derived from classical fingerprinting and next generation sequencing techniques. Environ. Microbiol. 2013. [Google Scholar] [CrossRef]
- Brusetti, L.; Borin, S.; Rizzi, A.; Mora, D.; Sorlini, C.; Daffonchio, D. Exploration of methods used to describe bacterial communities in silage of maize (zea mays) cultivars. Environ. Biosafety. Res. 2008, 7, 25–33. [Google Scholar] [CrossRef]
- Danovaro, R.; Luna, G.M.; Dell’anno, A.; Pietrangeli, B. Comparison of two fingerprinting techniques, terminal restriction fragment length polymorphism and automated ribosomal intergenic spacer analysis, for determination of bacterial diversity in aquatic environments. Appl. Environ. Microbiol. 2006, 72, 5982–5989. [Google Scholar] [CrossRef]
- Gillevet, P.M.; Sikaroodi, M.; Torzilli, A.P. Analyzing salt-marsh fungal diversity: Comparing arisa fingerprinting with clone sequencing and pyrosequencing. Fungal Ecology 2009, 2, 160–167. [Google Scholar] [CrossRef]
- Mills, D.K.; Fitzgerald, K.; Litchfield, C.D.; Gillevet, P.M. A comparison of DNA profiling techniques for monitoring nutrient impact on microbial community composition during bioremediation of petroleum-contaminated soils. J. Microbiol. Methods 2003, 54, 57–74. [Google Scholar] [CrossRef]
- Sikaroodi, M.; Gillevet, P.M. Quality control in multi-tag pyrosequencing of microbial communities. Biotechniques 2012, 53, 381–383. [Google Scholar]
- Shade, A.; Read, J.S.; Youngblut, N.D.; Fierer, N.; Knight, R.; Kratz, T.K.; Lottig, N.R.; Roden, E.E.; Stanley, E.H.; Stombaugh, J.; et al. Lake microbial communities are resilient after a whole-ecosystem disturbance. ISME J. 2012, 6, 2153–2167. [Google Scholar] [CrossRef]
- Jami, E.; Shabtay, A.; Nikbachat, M.; Yosef, E.; Miron, J.; Mizrahi, I. Effects of adding a concentrated pomegranate-residue extract to the ration of lactating cows on in vivo digestibility and profile of rumen bacterial population. J. Dairy Sci. 2012, 95, 5996–6005. [Google Scholar] [CrossRef]
- Welkie, D.G.; Stevenson, D.M.; Weimer, P.J. Arisa analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe 2009, 16, 94–100. [Google Scholar]
- Weimer, P.J.; Stevenson, D.M.; Mertens, D.R.; Thomas, E.E. Effect of monensin feeding and withdrawal on populations of individual bacterial species in the rumen of lactating dairy cows fed high-starch rations. Appl. Microbiol. Biotechnol. 2008, 80, 135–145. [Google Scholar] [CrossRef]
- Jami, E.; Mizrahi, I. Similarity of the ruminal bacteria across individual lactating cows. Anaerobe 2012, 18, 338–343. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16s rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar]
- Gillevet, P.; Sikaroodi, M.; Keshavarzian, A.; Mutlu, E.A. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem. Biodivers. 2010, 7, 1065–1075. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef]
- Stevenson, D.M.; Weimer, P.J. Dominance of prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time pcr. Appl. Microbiol. Biotechnol. 2007, 75, 165–174. [Google Scholar] [CrossRef]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One 2012, 7, e33306. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughputcommunity sequencing data. Nat Methods 2011, 7, 335–336. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; Gonzalez, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef]
- Dunbar, J.; Ticknor, L.O.; Kuske, C.R. Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16s rrna genes from bacterial communities. Appl. Environ. Microbiol. 2001, 67, 190–197. [Google Scholar] [CrossRef]
- Brown, M.V.; Schwalbach, M.S.; Hewson, I.; Fuhrman, J.A. Coupling 16s-its rdna clone libraries and automated ribosomal intergenic spacer analysis to show marine microbial diversity: Development and application to a time series. Environ. Microbiol. 2005, 7, 1466–1479. [Google Scholar] [CrossRef]
- Crosby, L.D.; Criddle, C.S. Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. Biotechniques 2003, 34, 790–803. [Google Scholar]
- Roesch, L.F.; Lorca, G.L.; Casella, G.; Giongo, A.; Naranjo, A.; Pionzio, A.M.; Li, N.; Mai, V.; Wasserfall, C.H.; Schatz, D.; et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 2009, 3, 536–548. [Google Scholar] [CrossRef]
- Dehority, B.A.; Grubb, J.A. Effect of short-term chilling of rumen contents on viable bacterial numbers. Appl. Environ. Microbiol. 1980, 39, 376–381. [Google Scholar]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16s rRNA bacterial tag-encoded flx amplicon pyrosequencing (btefap). BMC Microbiol. 2008. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16s rRNA sequence formation and detection in sanger and 454-pyrosequenced pcr amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jami, E.; Shterzer, N.; Mizrahi, I. Evaluation of Automated Ribosomal Intergenic Spacer Analysis for Bacterial Fingerprinting of Rumen Microbiome Compared to Pyrosequencing Technology. Pathogens 2014, 3, 109-120. https://doi.org/10.3390/pathogens3010109
Jami E, Shterzer N, Mizrahi I. Evaluation of Automated Ribosomal Intergenic Spacer Analysis for Bacterial Fingerprinting of Rumen Microbiome Compared to Pyrosequencing Technology. Pathogens. 2014; 3(1):109-120. https://doi.org/10.3390/pathogens3010109
Chicago/Turabian StyleJami, Elie, Naama Shterzer, and Itzhak Mizrahi. 2014. "Evaluation of Automated Ribosomal Intergenic Spacer Analysis for Bacterial Fingerprinting of Rumen Microbiome Compared to Pyrosequencing Technology" Pathogens 3, no. 1: 109-120. https://doi.org/10.3390/pathogens3010109
APA StyleJami, E., Shterzer, N., & Mizrahi, I. (2014). Evaluation of Automated Ribosomal Intergenic Spacer Analysis for Bacterial Fingerprinting of Rumen Microbiome Compared to Pyrosequencing Technology. Pathogens, 3(1), 109-120. https://doi.org/10.3390/pathogens3010109