
Oropharyngeal Candidiasis in HIV Infection: Analysis of Impaired Mucosal Immune Response to Candida albicans in Mice Expressing the HIV-1 Transgene

{kind=link}
{kind=link}
Abstract
:1. Introduction
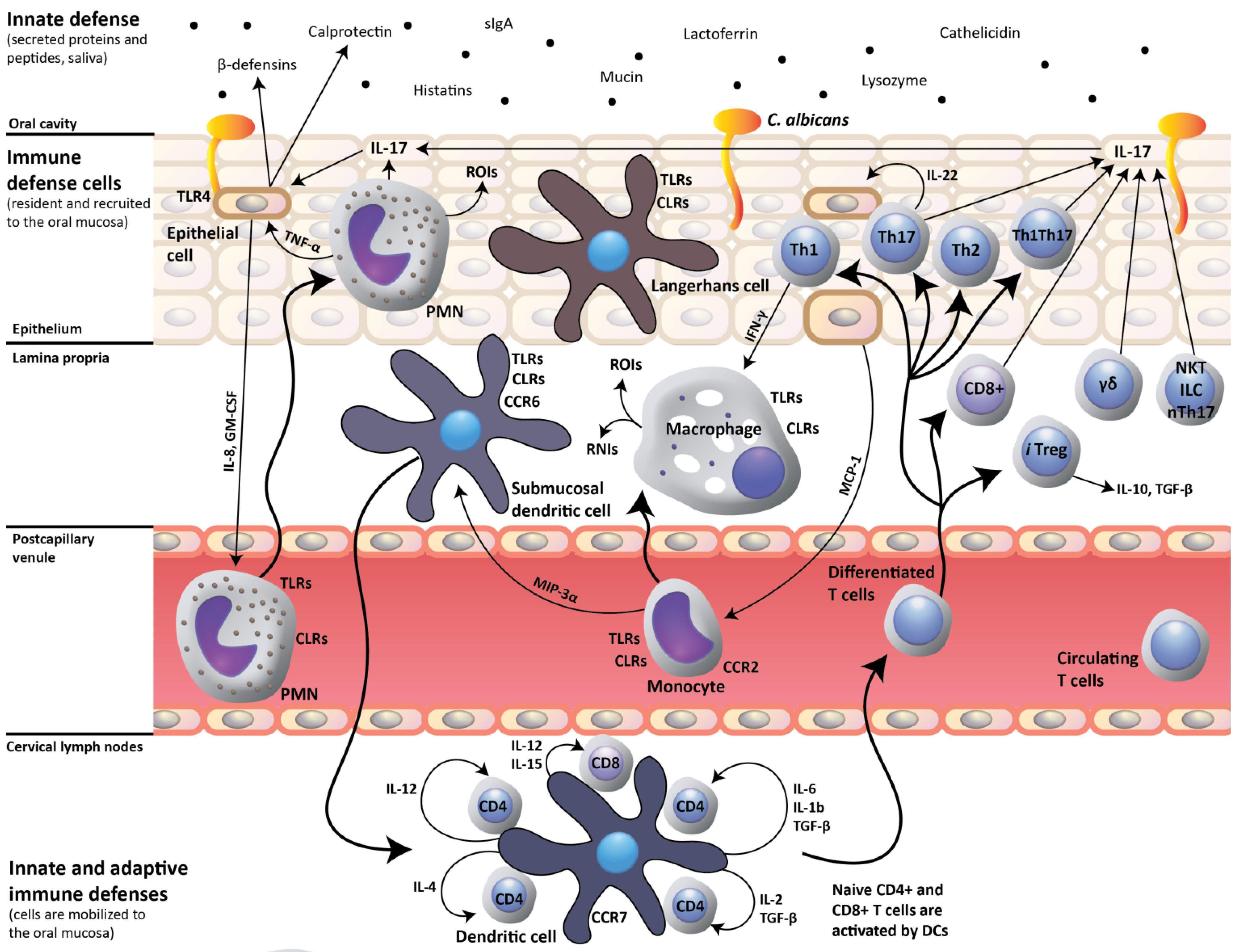
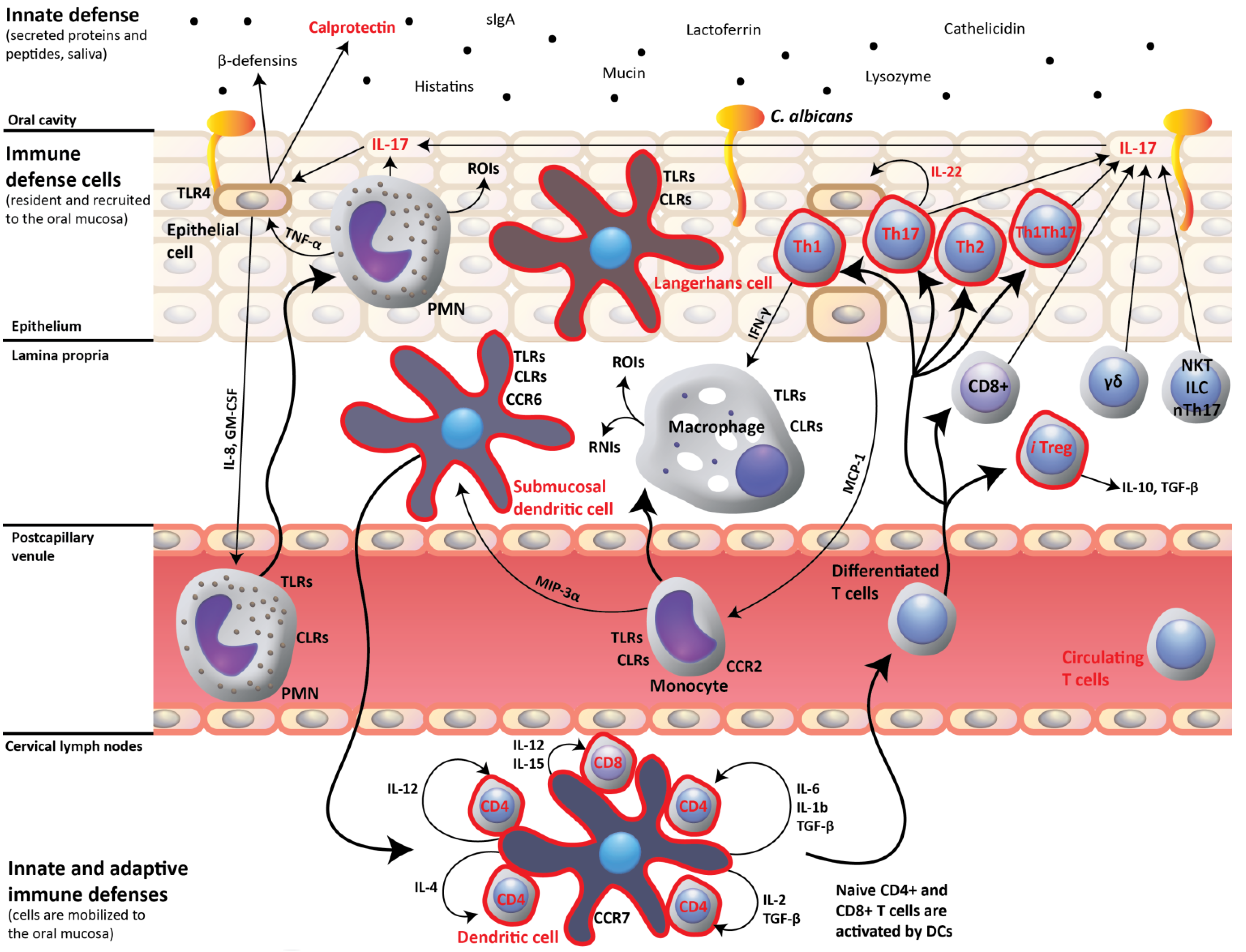
2. IL-17- and IL-22-Dependent Mucosal Host Response to Candida albicans

3. Impact of HIV Infection on Oral Mucosal Immunity to C. albicans
4. Oral C. albicans Infection in CD4C/HIV Transgenic Mice: Evidence for Defective IL-17- and IL-22-Mediated Mucosal Host Response

5. Oral C. albicans Infection in CD4C/HIV Transgenic Mice: Evidence for Protective CD8+ T-Cell Response
6. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCarthy, G.M. Host factors associated with HIV-related oral candidiasis. A review. Oral Surg. Oral Med. Oral Pathol. 1992, 73, 181–186. [Google Scholar] [CrossRef]
- McCarthy, G.M.; Mackie, I.D.; Koval, J.; Sandhu, H.S.; Daley, T.D. Factors associated with increased frequency of HIV-related oral candidiasis. J. Oral Pathol. Med. 1991, 20, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Bentsen, K.D.; Hojtved, L.; Willemoes, E.H.; Scheutz, F.; Schiodt, M.; Stoltze, K.; Pindborg, J.J. Oral candidiasis and immune status of HIV-infected patients. J. Oral Pathol. Med. 1994, 23, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Mercante, D.E.; Leigh, J.E.; Lilly, E.A.; McNulty, K.; Fidel, P.L., Jr. Assessment of the association between HIV viral load and CD4 cell count on the occurrence of oropharyngeal candidiasis in HIV-infected patients. J. Acquir. Immune Defic. Syndr. 2006, 42, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Campo, J.; Del Romero, J.; Castilla, J.; Garcia, S.; Rodriguez, C.; Bascones, A. Oral candidiasis as a clinical marker related to viral load, CD4 lymphocyte count and CD4 lymphocyte percentage in HIV-infected patients. J. Oral Pathol. Med. 2002, 31, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, P.; Pimpinelli, N.; Mori, M.; Reichart, P.A.; Eversole, L.R.; Ficarra, G. Immunocompetent cells in oral candidiasis of HIV-infected patients: An immunohistochemical and electron microscopical study. Oral Dis. 1997, 3, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Reichart, P.A.; Philipsen, H.P.; Schmidt-Westhausen, A.; Samaranayake, L.P. Pseudomembranous oral candidiasis in HIV infection: Ultrastructural findings. J. Oral Pathol. Med. 1995, 24, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Myers, T.A.; Leigh, J.E.; Arribas, A.R.; Hager, S.; Clark, R.; Lilly, E.; Fidel, P.L., Jr. Immunohistochemical evaluation of T cells in oral lesions from human immunodeficiency virus-positive persons with oropharyngeal candidiasis. Infect. Immun. 2003, 71, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J. Immunol. 2005, 175, 5–14. [Google Scholar] [PubMed]
- Fidel, P.L., Jr. Candida-host interactions in HIV disease: Implications for oropharyngeal candidiasis. Adv. Dent. Res. 2011, 23, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L., Jr. Candida-host interactions in HIV disease: Relationships in oropharyngeal candidiasis. Adv. Dent. Res. 2006, 19, 80–84. [Google Scholar] [CrossRef] [PubMed]
- De Repentigny, L.; Lewandowski, D.; Jolicoeur, P. Immunopathogenesis of oropharyngeal candidiasis in human immunodeficiency virus infection. Clin. Microbiol. Rev. 2004, 17, 729–759. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Gaffen, S.L. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect. 2010, 12, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.A.; Casadevall, A. Rethinking T cell immunity in oropharyngeal candidiasis. J. Exp. Med. 2009, 206, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.S.; Hu, Y.; Riminton, S.; Ashman, R.B. Distinct roles for interleukin-12p40 and tumour necrosis factor in resistance to oral candidiasis defined by gene-targeting. Oral Microbiol. Immunol. 2006, 21, 252–255. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Zelante, T.; D’Angelo, C.; Zagarella, S.; Fallarino, F.; Spreca, A.; Iannitti, R.G.; Bonifazi, P.; Renauld, J.C.; Bistoni, F.; et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010, 3, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Wagener, J.; Wenzel, V.; Scarponi, C.; Pennino, D.; Albanesi, C.; Schaller, M.; Behrendt, H.; Ring, J.; Schmidt-Weber, C.B.; et al. IL-22 and TNF-alpha represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur. J. Immunol. 2011, 41, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Witte, K.; Warszawska, K.; Sabat, R. Biology of interleukin-22. Semin. Immunopathol. 2010, 32, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; McCray, P.B., Jr.; Chan, Y.R. Cytokine-mediated regulation of antimicrobial proteins. Nat. Rev. Immunol. 2008, 8, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Peck, A.; Mellins, E.D. Precarious balance: Th17 cells in host defense. Infect. Immun. 2010, 78, 32–38. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.L.; Roark, C.L.; Born, W.K. IL-17-producing gammadelta T cells. Eur. J. Immunol. 2009, 39, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Rachitskaya, A.V.; Hansen, A.M.; Horai, R.; Li, Z.; Villasmil, R.; Luger, D.; Nussenblatt, R.B.; Caspi, R.R. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J. Immunol. 2008, 180, 5167–5171. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Garcia-Hernandez Mde, L.; Reome, J.B.; Misra, S.K.; Strutt, T.M.; McKinstry, K.K.; Cooper, A.M.; Swain, S.L.; Dutton, R.W. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J. Immunol. 2009, 182, 3469–3481. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; Barlow, J.L.; McKenzie, A.N. Innate lymphoid cells--how did we miss them? Nat. Rev. Immunol. 2013, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernandez-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal. Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; LeibundGut-Landmann, S. Innate lymphoid cells: New players in IL-17-mediated antifungal immunity. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Luci, C.; Reynders, A.; Ivanov, II; Cognet, C.; Chiche, L.; Chasson, L.; Hardwigsen, J.; Anguiano, E.; Banchereau, J.; Chaussabel, D.; et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 2009, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Boisson-Dupuis, S.; Kong, X.F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.L. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Whibley, N.; Gaffen, S.L. Brothers in arms: Th17 and Treg responses in Candida albicans immunity. PLoS Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chou, L.L.; Epstein, J.; Cassol, S.A.; West, D.M.; He, W.; Firth, J.D. Oral mucosal Langerhans’ cells as target, effector and vector in HIV infection. J. Oral Pathol. Med. 2000, 29, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Charton-Bain, M.C.; Terris, B.; Dauge, M.C.; Marche, C.; Walker, F.; Bouchaud, O.; Xerri, L.; Potet, F. Reduced number of Langerhans cells in oesophageal mucosa from AIDS patients. Histopathology 1999, 34, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Rosini, S.; Caltagirone, S.; Tallini, G.; Lattanzio, G.; Demopoulos, R.; Piantelli, M.; Musiani, P. Depletion of stromal and intraepithelial antigen-presenting cells in cervical neoplasia in human immunodeficiency virus infection. Hum. Pathol. 1996, 27, 834–838. [Google Scholar] [CrossRef]
- McIlroy, D.; Autran, B.; Clauvel, J.P.; Oksenhendler, E.; Debre, P.; Hosmalin, A. Low CD83, but normal MHC class II and costimulatory molecule expression, on spleen dendritic cells from HIV+ patients. AIDS Res. Hum. Retrovir. 1998, 14, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.A.; Blyveis, N.; Palmer, B.E.; MaWhinney, S.; Wilson, C.C. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals. J. Infect. Dis. 2003, 187, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Pozniak, A.; Gazzard, B.; Qazi, N.; Gilmour, J.; Gotch, F.; Patterson, S. Loss of blood CD11c(+) myeloid and CD11c(−) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 2001, 98, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Hosmalin, A.; McIlroy, D.; Calvez, V.; Debre, P.; Autran, B. Depletion in blood CD11c-positive dendritic cells from HIV-infected patients. AIDS 1999, 13, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Lau, R.; Patterson, S.; Pinching, A.J.; Knight, S.C. Dendritic cell infection, depletion and dysfunction in HIV-infected individuals. Immunology 1990, 71, 38–45. [Google Scholar] [PubMed]
- Pacanowski, J.; Kahi, S.; Baillet, M.; Lebon, P.; Deveau, C.; Goujard, C.; Meyer, L.; Oksenhendler, E.; Sinet, M.; Hosmalin, A. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood 2001, 98, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Pimpinelli, N.; Borgognoni, L.; Riccardi, R.; Ficarra, G.; Mori, M.; Gaglioti, D.; Romagnoli, P. CD36(OKM5)+ dendritic cells in the oral mucosa of HIV− and HIV+ subjects. J. Investig. Dermatol. 1991, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Golebiowska, A.; Trumpfheller, C.; Siegal, F.P.; Steinman, R.M. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl. Acad Sci. USA 2004, 101, 7669–7674. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Patrick, C.N.; Cragg, M.S.; Honeychurch, J.; Mann, D.A.; Harris, M. Functional analysis of HIV type 1 Nef reveals a role for PAK2 as a regulator of cell phenotype and function in the murine dendritic cell line, DC2.4. J. Immunol. 2005, 175, 6560–6569. [Google Scholar] [CrossRef] [PubMed]
- Poudrier, J.; Weng, X.; Kay, D.G.; Hanna, Z.; Jolicoeur, P. The AIDS-like disease of CD4C/human immunodeficiency virus transgenic mice is associated with accumulation of immature CD11bHi dendritic cells. J. Virol. 2003, 77, 11733–11744. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Wurfl, S.; Benaroch, P.; Greenough, T.C.; Daniels, R.; Easterbrook, P.; Brenner, M.; Munch, J.; Kirchhoff, F. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 2003, 77, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Stumptner-Cuvelette, P.; Morchoisne, S.; Dugast, M.; Le Gall, S.; Raposo, G.; Schwartz, O.; Benaroch, P. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc. Natl. Acad Sci. USA 2001, 98, 12144–12149. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Jacque, J.M.; Santisteban, C.; Bristow, C.; Han, S.Y.; Villamide-Herrera, L.; Mehlhop, E.; Marx, P.A.; Steinman, R.M.; Gettie, A.; et al. Endogenously expressed nef uncouples cytokine and chemokine production from membrane phenotypic maturation in dendritic cells. J. Immunol. 2002, 169, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Vigerust, D.J.; Egan, B.S.; Shepherd, V.L. HIV-1 Nef mediates post-translational down-regulation and redistribution of the mannose receptor. J. Leukoc. Biol. 2005, 77, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Latorre, D.; Mele, F.; Foglierini, M.; De Gregorio, C.; Cassotta, A.; Fernandez, B.; Kelderman, S.; Schumacher, T.N.; Corti, D.; et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 2015, 347, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Surh, C.D.; Sprent, J. Homeostasis of naive and memory T cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, E.M.; Sprent, J.; Surh, C.D. Generation and maintenance of memory CD4(+) T Cells. Curr. Opin. Immunol. 2009, 21, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Udey, M.C. Mushrooming insights into skin dendritic cell physiology. Immunity 2015, 42, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, B.; Zhou, M.; Li, L.; Zhou, H.; Zhang, J.; Chen, H.; Wu, C. Memory IL-22-producing CD4+ T cells specific for Candida albicans are present in humans. Eur. J. Immunol. 2009, 39, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Elhed, A.; Unutmaz, D. Th17 cells and HIV infection. Curr. Opin. HIV AIDS 2010, 5, 146–150. [Google Scholar] [CrossRef] [PubMed]
- El Hed, A.; Khaitan, A.; Kozhaya, L.; Manel, N.; Daskalakis, D.; Borkowsky, W.; Valentine, F.; Littman, D.R.; Unutmaz, D. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J. Infect. Dis. 2010, 201, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.R.; Routy, J.P.; et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, A.; Prado, J.G.; Kang, Y.H.; Chen, F.; Riddell, L.A.; Luzzi, G.; Goulder, P.; Klenerman, P. HIV-1 infection is characterized by profound depletion of CD161+ Th17 cells and gradual decline in regulatory T cells. AIDS 2010, 24, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Nau, M.; Ehrenberg, P.; Chenine, A.L.; Macedo, C.; Zhou, Y.; Daye, Z.J.; Wei, Z.; Vahey, M.; Michael, N.L.; et al. Distinct gene-expression profiles associated with the susceptibility of pathogen-specific CD4 T cells to HIV-1 infection. Blood 2013, 121, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Bernier, A.; Cleret-Buhot, A.; Zhang, Y.; Goulet, J.P.; Monteiro, P.; Gosselin, A.; DaFonseca, S.; Wacleche, V.S.; Jenabian, M.A.; Routy, J.P.; et al. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology 2013, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Mattapallil, J.J. Loss and dysregulation of Th17 cells during HIV infection. Clin. Dev. Immunol. 2013, 2013, 852418. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; McKinnon, L.R.; Kovacs, C.; Kandel, G.; Huibner, S.; Chege, D.; Shahabi, K.; Benko, E.; Loutfy, M.; Ostrowski, M.; et al. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J. Immunol. 2013, 191, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Santos, N.; Gaffen, S.L. Th17 cells in immunity to Candida albicans. Cell Host Microbe 2012, 11, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Wagener, J.; Schaller, M. Epithelial cells and innate antifungal defense. J. Dent. Res. 2010, 89, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Gaffen, S.L.; Kolls, J.K. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009, 2, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; Erlandsen, J.E.; Kirkpatrick, W.R.; Berg, D.K.; Westbrook, S.D.; Louden, C.; Cornell, J.E.; Thompson, G.R.; Vallor, A.C.; Wickes, B.L.; et al. The Changing Epidemiology of Oropharyngeal Candidiasis in Patients with HIV/AIDS in the Era of Antiretroviral Therapy. AIDS Res. Treat. 2012. [Google Scholar] [CrossRef]
- Hanna, Z.; Kay, D.G.; Rebai, N.; Guimond, A.; Jothy, S.; Jolicoeur, P. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 1998, 95, 163–175. [Google Scholar] [CrossRef]
- Chrobak, P.; Simard, M.C.; Bouchard, N.; Ndolo, T.M.; Guertin, J.; Hanna, Z.; Dave, V.; Jolicoeur, P. HIV-1 Nef disrupts maturation of CD4+ T cells through CD4/Lck modulation. J. Immunol. 2010, 185, 3948–3959. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Priceputu, E.; Chrobak, P.; Poudrier, J.; Kay, D.G.; Hanna, Z.; Mak, T.W.; Jolicoeur, P. CD4+ T cells from CD4C/HIVNef transgenic mice show enhanced activation in vivo with impaired proliferation in vitro but are dispensable for the development of a severe AIDS-like organ disease. J. Virol. 2004, 78, 5244–5257. [Google Scholar] [CrossRef] [PubMed]
- Poudrier, J.; Weng, X.; Kay, D.G.; Pare, G.; Calvo, E.L.; Hanna, Z.; Kosco-Vilbois, M.H.; Jolicoeur, P. The AIDS disease of CD4C/HIV transgenic mice shows impaired germinal centers and autoantibodies and develops in the absence of IFN-gamma and IL-6. Immunity 2001, 15, 173–185. [Google Scholar] [CrossRef]
- Priceputu, E.; Rodrigue, I.; Chrobak, P.; Poudrier, J.; Mak, T.W.; Hanna, Z.; Hu, C.; Kay, D.G.; Jolicoeur, P. The Nef-mediated AIDS-like disease of CD4C/human immunodeficiency virus transgenic mice is associated with increased Fas/FasL expression on T cells and T-cell death but is not prevented in Fas-, FasL-, tumor necrosis factor receptor 1-, or interleukin-1beta-converting enzyme-deficient or Bcl2-expressing transgenic mice. J. Virol. 2005, 79, 6377–6391. [Google Scholar] [PubMed]
- Kay, D.G.; Yue, P.; Hanna, Z.; Jothy, S.; Tremblay, E.; Jolicoeur, P. Cardiac disease in transgenic mice expressing human immunodeficiency virus-1 nef in cells of the immune system. Am. J. Pathol. 2002, 161, 321–335. [Google Scholar] [CrossRef]
- De Repentigny, L.; Aumont, F.; Ripeau, J.S.; Fiorillo, M.; Kay, D.G.; Hanna, Z.; Jolicoeur, P. Mucosal candidiasis in transgenic mice expressing human immunodeficiency virus type 1. J. Infect. Dis. 2002, 185, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, D.; Marquis, M.; Aumont, F.; Lussier-Morin, A.C.; Raymond, M.; Senechal, S.; Hanna, Z.; Jolicoeur, P.; de Repentigny, L. Altered CD4+ T cell phenotype and function determine the susceptibility to mucosal candidiasis in transgenic mice expressing HIV-1. J. Immunol. 2006, 177, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Hanna, Z.; Priceputu, E.; Chrobak, P.; Hu, C.; Dugas, V.; Goupil, M.; Marquis, M.; de Repentigny, L.; Jolicoeur, P. Selective expression of human immunodeficiency virus Nef in specific immune cell populations of transgenic mice is associated with distinct AIDS-like phenotypes. J. Virol. 2009, 83, 9743–9758. [Google Scholar] [CrossRef] [PubMed]
- Goupil, M.; Cousineau-Cote, V.; Aumont, F.; Senechal, S.; Gaboury, L.; Hanna, Z.; Jolicoeur, P.; de Repentigny, L. Defective IL-17- and IL-22-dependent mucosal host response to Candida albicans determines susceptibility to oral candidiasis in mice expressing the HIV-1 transgene. BMC Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Huppler, A.R.; Conti, H.R.; Hernandez-Santos, N.; Darville, T.; Biswas, P.S.; Gaffen, S.L. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J. Immunol. 2014, 192, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Lacasse, M.; Fortier, C.; Trudel, L.; Collet, A.J.; Deslauriers, N. Experimental oral candidosis in the mouse: Microbiologic and histologic aspects. J. Oral Pathol. Med. 1990, 19, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; Wagner, S.A.; Matias, M.A.; Hu, Y.; Zaini, Z.M.; Farah, C.S. Early activation of the interleukin-23–17 axis in a murine model of oropharyngeal candidiasis. Mol. Oral Microbiol. 2010, 25, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Eversole, L.R.; Reichart, P.A.; Ficarra, G.; Schmidt-Westhausen, A.; Romagnoli, P.; Pimpinelli, N. Oral keratinocyte immune responses in HIV-associated candidiasis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1997, 84, 372–380. [Google Scholar] [CrossRef]
- Marquis, M.; Lewandowski, D.; Dugas, V.; Aumont, F.; Senechal, S.; Jolicoeur, P.; Hanna, Z.; de Repentigny, L. CD8+ T cells but not polymorphonuclear leukocytes are required to limit chronic oral carriage of Candida albicans in transgenic mice expressing human immunodeficiency virus type 1. Infect. Immun. 2006, 74, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Goupil, M.; Trudelle, E.B.; Dugas, V.; Racicot-Bergeron, C.; Aumont, F.; Senechal, S.; Hanna, Z.; Jolicoeur, P.; de Repentigny, L. Macrophage-mediated responses to Candida albicans in mice expressing the human immunodeficiency virus type 1 transgene. Infect. Immun. 2009, 77, 4136–4149. [Google Scholar] [CrossRef] [PubMed]
- McNulty, K.M.; Plianrungsi, J.; Leigh, J.E.; Mercante, D.; Fidel, P.L., Jr. Characterization of CD8+ T cells and microenvironment in oral lesions of human immunodeficiency virus-infected persons with oropharyngeal candidiasis. Infect. Immun. 2005, 73, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [PubMed]
- Nurieva, R.; Yang, X.O.; Chung, Y.; Dong, C. Cutting edge: In vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J. Immunol. 2009, 182, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Repentigny, L.; Goupil, M.; Jolicoeur, P. Oropharyngeal Candidiasis in HIV Infection: Analysis of Impaired Mucosal Immune Response to Candida albicans in Mice Expressing the HIV-1 Transgene. Pathogens 2015, 4, 406-421. https://doi.org/10.3390/pathogens4020406
De Repentigny L, Goupil M, Jolicoeur P. Oropharyngeal Candidiasis in HIV Infection: Analysis of Impaired Mucosal Immune Response to Candida albicans in Mice Expressing the HIV-1 Transgene. Pathogens. 2015; 4(2):406-421. https://doi.org/10.3390/pathogens4020406
Chicago/Turabian StyleDe Repentigny, Louis, Mathieu Goupil, and Paul Jolicoeur. 2015. "Oropharyngeal Candidiasis in HIV Infection: Analysis of Impaired Mucosal Immune Response to Candida albicans in Mice Expressing the HIV-1 Transgene" Pathogens 4, no. 2: 406-421. https://doi.org/10.3390/pathogens4020406
APA StyleDe Repentigny, L., Goupil, M., & Jolicoeur, P. (2015). Oropharyngeal Candidiasis in HIV Infection: Analysis of Impaired Mucosal Immune Response to Candida albicans in Mice Expressing the HIV-1 Transgene. Pathogens, 4(2), 406-421. https://doi.org/10.3390/pathogens4020406