
Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease



Abstract
:1. Introduction
2. Herpesviruses’ dUTPases and Innate Immunity
3. Stress and Herpesvirus Reactivation
4. Herpesviruses dUTPases and Human Disease
4.1. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
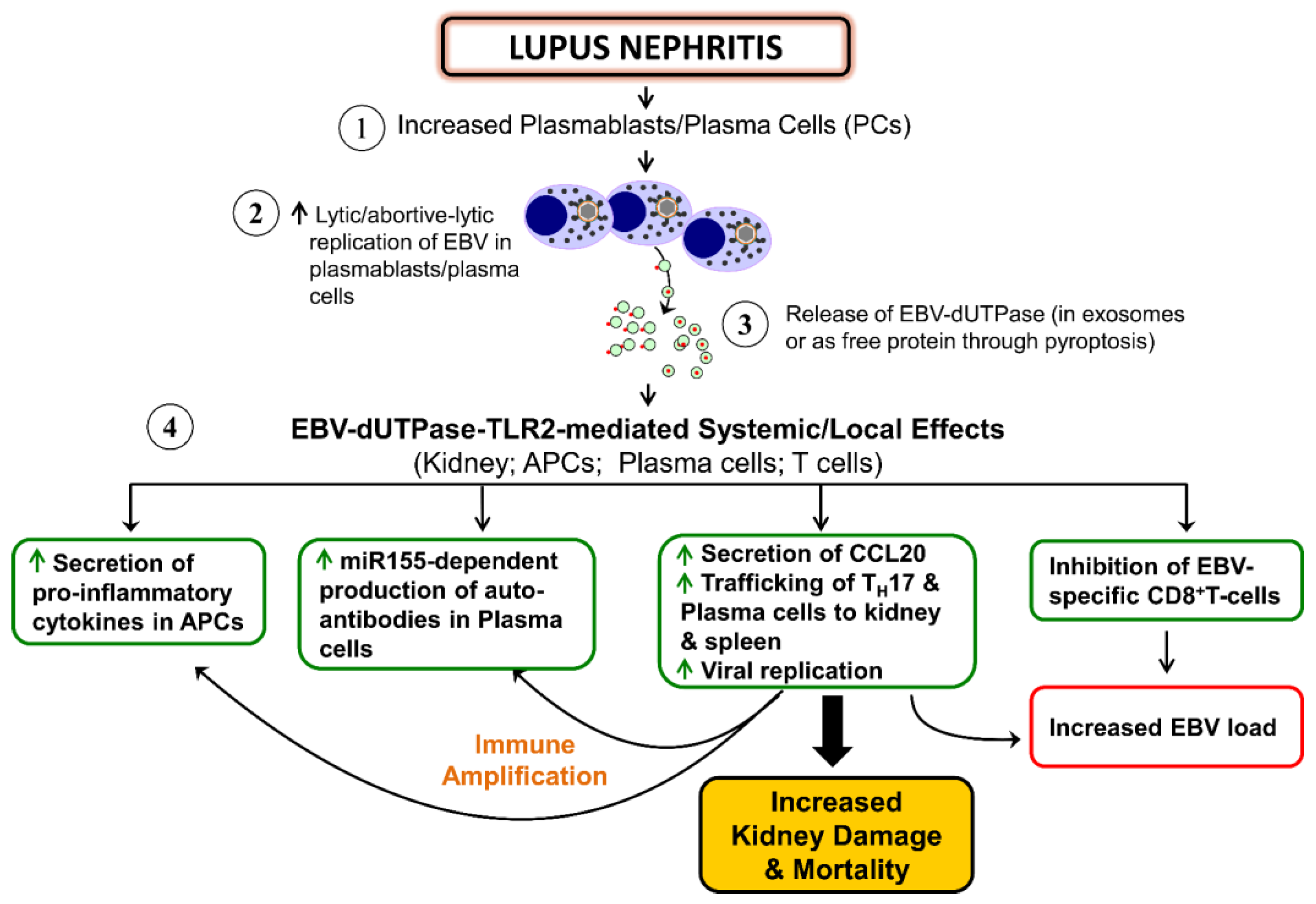
4.2. Autoimmune Diseases
4.3. Carcinogenesis
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nyman, P.O. Introduction. dUTPases. Curr. Protein Pept. Sci. 2001, 2, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Baldo, A.M.; McClure, M. Evolution and horizontal transfer of dUTPase encoding genes in viruses and their hosts. J. Virol. 1999, 73, 7710–7721. [Google Scholar] [PubMed]
- McGeoch, D.J.; Cook, S.; Dolan, A.; Jamieson, F.E.; Telford, E.A.R. Molecular phylogeny and evolutionary timescales for the family of mammalian herpesviruses. J. Mol. Biol. 1995, 247, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Tarbouriech, N.; Buisson, M.; Seigneurin, J.M.; Cusack, S.; Burmeiste, W.P. The monomeric dUTPase from Epstein-Barr virus mimic trimeric dUTPases. Structure 2005, 13, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.J.; Stow, N.D. New genes from old: Redeployment of dUTPase by herpesviruses. J. Virol. 2005, 79, 12880–12892. [Google Scholar] [CrossRef] [PubMed]
- Caposio, P.; Riera, L.; Hahn, G.; Landolfo, S.; Gribaudo, G. Evidence that the human cytomegalovirus 46-kDa UL72 protein is not an active dUTPase but a late protein dispensible for replication in fibroblasts. Virology 2004, 325, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Glaser, R.; Williams, M.V. Human herpesviruses encoded dUTPases: A family of proteins that modulate dendritic cells function and innate immunity. Front. Microbiol. 2014, 5, 504. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Arrebola, R.; Pena-Diaz, J.; Ruiz-Perez, L.M.; Gonzalez-Pacanowska, D. Description of a novel eukaryotic deoxyuridine-5’-triphosphate nucleotidohydrolase in Leishmania major. Biochem. J. 1997, 325, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Hidalgo-Zarco, F.; Bernier-Villamor, V.; Ruiz-Rerez, L.M. Properties of Leshmania major dUTP nucleotidohydrolase, a distinct nucleotide-hydrolyzing enzyme in kinetoplasts. Biochem. J. 2000, 346, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bernier-Villamor, V.; Camacho, A.; Hidalgo-Zarco, F.; Perez, J.; Ruiz-Perez, L.M.; Gonzalez-Pacanowska, D. Characterization of deoxyuridine triphosphate nucleotidohydrolase from Trypanosoma cruzi. FEBS Lett. 2002, 526, 147–150. [Google Scholar] [CrossRef]
- Hill, F.; Loakes, D.; Brown, D.M. Polymerase recognition of synthetic oligodeoxyribonucleotides incorporating degenerate pyrimidine and purine bases. Proc. Natl. Acad. Sci. USA 1998, 95, 4258–4263. [Google Scholar] [CrossRef] [PubMed]
- Parkhill, J.; Wren, B.W.; Mungall, K.; Ketley, J.M.; Churcher, C.; Basham, D.; Chillingworth, T.; Davies, R.M.; Feltwell, T.; Holroyd, S.; et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 2000, 40, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.B.; Preston, V.G. Isolation and characterization of herpes simplex virus type 1 mutants which fail to induce dUTPase activity. Virology 1986, 148, 190–197. [Google Scholar] [CrossRef]
- Ross, J.; Williams, M.; Cohen, J.I. Disruption of the varicella-zoster virus dUTPase and adjacent ORF9A gene results in impaired growth and reduced syncytia formation. Virology 1997, 234, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Lirette, R.; Caradonna, S. Inhibition of phosphorylation of cellular dUTP nucleotidohydrolase as a consequence of herpes simplex virus infection. J. Cell. Biochem. 1990, 43, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Tsuda, S.; Liu, Z.; Kozuka-Hata, H.; Oyama, M.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J. Virol. 2014, 88, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Hirohata, Y.; Arii, J.; Kawaguchi, Y. Phosphorylation of herpes simplex virus 1 dUTPase upregulated viral dUTPase activity to compensate for low cellular dUTPase activity for efficient viral replication. J. Virol. 2014, 88, 7776–7785. [Google Scholar] [CrossRef] [PubMed]
- Pyles, R.B.; Sawtell, N.M.; Thompson, R.L. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness and reactivation from latency. J. Virol. 1992, 66, 6706–6713. [Google Scholar] [PubMed]
- Song, M.J.; Hwang, S.; Wong, W.H.; Wu, T.T.; Lee, S.; Liao, H.I.; Sun, R. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Preston, V.G.; Fisher, F.B. Identification of the Herpes Simplex Virus Type 1 Gene Encoding the dUTPase. Virology 1984, 133, 58–68. [Google Scholar] [CrossRef]
- Williams, M.V. Deoxyuridine Triphosphate Nucleotidohydrolase Induced by Herpes Simplex Virus Type1. Purification and characterization of induced enzyme. J. Biol. Chem. 1984, 259, 10080–10084. [Google Scholar] [PubMed]
- Williams, M.V.; Parris, D.S. Characterization of herpes simplex virus type 2 induced deoxyuridine triphosphate nucleotidohydrolase and mapping of a type specific gene for the enzyme. Virology 1987, 156, 282–292. [Google Scholar] [CrossRef]
- Williams, M.V.; Holliday, J.; Glaser, R. Induction of deoxyuridine triphosphatase activity in Epstein-Barr virus infected cells. Virology 1985, 142, 326–333. [Google Scholar] [CrossRef]
- Pyles, R.B.; Thompson, R.L. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus Type 1 strains in cultured murine cells. J. Virol. 1994, 68, 4514–4524. [Google Scholar] [PubMed]
- Glaser, R.; Litsky, M.L.; Padget, D.A.; Baiocchi, R.A.; Yang, E.V.; Chin, M.; Yeh, P.E.; Green-Church, K.B.; Caligiuri, M.; Williams, M.V. The EBV-encoded dUTPase induces immune dysregulation: Implications for the pathophysiology of EBV-associated disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Waldman, W.J.; Williams, M.V.; Lemeshow, S.A.; Binkley, P.; Guttridge, D.; Kiecolt-Glaser, J.; Knight, D.A.; Ladner, K.J.; Glaser, R. Epstein-Barr virus-encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: Evidence for depression-induced atherosclerotic risk. Brain Behav. Immun 2008, 22, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M. The Epstein-Barr Virus (EBV)-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling Pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Binkley, P.F.; Cooke, G.E.; Lesinski, A.; Taylor, M.; Chen, M.; Laskowski, B.; Waldman, W.J.; Ariza, M.E.; Williams, M.V.; Knight, D.A.; et al. Evidence for the role of Epstein Barr virus infections in the pathogenesis of acute coronary events. PLoS ONE 2013, 8, e54008. [Google Scholar] [CrossRef]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2014, 8, e69827. [Google Scholar] [CrossRef] [PubMed]
- Padgett, D.A.; Hotchkiss, A.K.; Pyter, L.M.; Nelson, R.J.; Yang, E.; Yeh, P.; Litsky, M.; Williams, M.; Glaser, R. Epstein-Barr virus-encoded dUTPase modulates immune function and induces sickness behavior in mice. J. Med. Virol. 2004, 74, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Aubrecht, T.G.; Salloum, B.A.; Ariza, M.E.; Williams, M.; Reader, B.; Glaser, R.; Sheridan, J.; Nelson, R. Restraint induces sickness responses independent of injection with Epstein-Barr virus (EBV)-encoded dUTPase. J. Behav. Brain Sci. 2014, 4, 491–505. [Google Scholar] [CrossRef]
- Young, N.A.; Williams, M.V.; Jarjour, W.N.; Bruss, M.J.; Bolon, B.; Parikh, S.; Satoskar, A.; Ariza, M.E. Epstein-Barr virus (EBV) encoded dUTPase exacerbates the pathology of lupus nephritis in vivo. Int. J. Immunol. Immunother. 2016, in press. [Google Scholar]
- Kremmer, E.; Sommer, P.; Holzer, D.; Galetsky, S.A.; Molochkov, V.A.; Gurtsevitch, V.; Winkelmann, C.; Lisner, R.; Niedobitek, G.; Grasser, F.A. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) ORF54 encodes a functional dUTPase expressed in the lytic replication cycle. J. Gen. Virol. 1999, 80 Pt 5, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Madrid, A.S.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus ORF54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J. Virol. 2012, 86, 8693–8704. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hanto, H.; Sowinsk, S.; Monoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Chan, J.; Clement, M.V.; Pervaiz, S. Functional proteomics of reveratrol-induced colon cancer cell apoptosis: Caspase-6-mediated cleavage of lamin A is a major signaling loop. Proteomics 2006, 6, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shen, J.; Zhan, J.; Yu, Y. dUTP pyrophosphatase, its appearance in extracellular compartment may serve as a potential biomarker for N-methyl-N’-nitro-N-nitrosoguanidine exposure in mammalian cells. Proteomics 2006, 6, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Buschow, S.I.; van Balkom, B.W.M.; Alberts, M.; Heck, A.J.R.; Wauben, M.; Stoorvogel, M. MHC class-II associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 2010, 88, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.S.; Lee, S.S.; Prasad, B.V.; Javier, R.T. Human adenovirus early region 4 open reading frame 1 genes encode growth-transforming proteins that may be distantly related to dUTP pyrophosphate enzymes. J. Virol. 1997, 71, 1857–1870. [Google Scholar] [PubMed]
- Chung, A.H.; Frese, K.; Weiss, R.S.; Prasad, B.V.V.; Javier, R.T. A new crucial protein interaction element that targets the adenovirus E4-ORF1 oncoprotein to membrane vesicles. J. Virol. 2007, 81, 4787–4797. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Interferences, questions and possibilities in Toll-like receptor signaling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Guggemoos, S.; Hangel, D.; Hamm, S.; Heit, A.; Bauer, S.; Adler, H. TLR9 contributes to the antiviral immunity during gamma herpesvirus infection. J. Immnunol. 2008, 80, 438–443. [Google Scholar] [CrossRef]
- Lopes, J.A.G.; Borges-Canha, M.; Pimentel-Nunes, P. Innate immunity and hepatocellular carcinoma: Can toll-like receptors open the door to oncogenesis? World J. Hepatol. 2016, 8, 162–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Luz, A.; Fainstein, N.; Einstein, O.; Ben-Hur, T. The role of CNS TLR2 activation in mediating innate versus adaptive neuroinflammation. Exp. Neurol. 2015, 273, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, B.M.; Yao, X.D.; Rosenthal, K.L.; INFANT Study Team. HIV-1 structural proteins serve as PAMPs for TLR2 heterodimers significantly increasing infection and innate immune activation. Front. Immunol. 2015, 6, 426. [Google Scholar]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Kurt-Jones, E.A.; Shin, O.S.; Manchak, M.D.; Lewin, M.J.; Finberg, R.W. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J. Virol. 2005, 79, 12658–12666. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Goldenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed]
- Szomololanyi-Tsuda, E.; Liang, X.; Welsh, R.M.; Kurt-Jones, E.A.; Finberg, R.W. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 2006, 80, 4286–4291. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.D.; Kronenberger, W.G.; Edwards, J.F.; Marshall, G.S.; Schikler, K.N.; Causey, D.L. Psychological symptoms of chronic fatigue and juvenile rheumatoid arthritis. Pediatrics 1999, 103, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Theorell, T.; Blomkvist, V.; Lindh, G.; Evengard, B. Critical life events, infections, and symptoms during the year preceding chronic fatigue syndrome (CFS): An examination of CFS patients and subjects with a nonspecific life crisis. Psychosom. Med. 1999, 61, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Jason, L.A.; Zinn, M.L.; Zinn, M.A. Myalgic encephalomyelitis: Symptoms and biomarkers. Curr. Neurophamacol. 2015, 13, 701–731. [Google Scholar] [CrossRef]
- Glaser, R.; Padgett, D.A.; Litsky, M.L.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.E.; Klimas, N.G.; Gailen, D.; Marshall, G.D.; et al. Stress-associated changes in the steady-state expression of latent Epstein-Barr virus: Implications for chronic fatigue syndrome and cancer. Brain Behav. Immun. 2005, 19, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Stanton, A.L. Psychosocial concerns and interventions for cancer survivors. J. Clin. Oncol. 2006, 24, 5132–5137. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Chan, M.; Bhatti, H.; Halton, M.; Grassi, L.; Johansen, C.; Meader, N. Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: A meta-analysis of 94 interview-based studies. Lancet Oncol. 2011, 12, 160–174. [Google Scholar] [CrossRef]
- Cameron, B.; Bharadwaj, M.; Burrows, J.; Fazou, C.; Wakefield, D.; Hickie, I.; French, R.; Khanna, R.; Lloyd, A. Prolonged illness after infectious mononucleosis is associated with altered immunity but not with increased viral load. J. Infect. Dis. 2006, 193, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Bayat, A.; Wagner, J.; Nutman, T.B.; Baraniuk, J.N.; Iadarol, M.J. No serological evidence for a role of HHV-6 infection in chronic fatigue syndrome. Am. J. Transl. 2012, 4, 443–451. [Google Scholar]
- Castera, M.T.; McDermott, M.P.; Dewhurst, S.; Schnable, K.; Carnahan, J.A.; Gilbert, L.; Latham, G.; Lofthus, G.K.; Hall, C.B. Human herpesvirus 6 (HHV6) DNA persistence and reactivation in healthy children. J. Pediatr. 2004, 145, 478–484. [Google Scholar]
- Kutok, J.L.; Wang, F. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, S.D.; Chen, T.; Allison, P.; Tyring, S.K.; Heath, A. Herpesvirus prevalence and viral load in healthy blood donors by quantitative real-time polymerase chain reaction. Transfusion 2008, 48, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Biganzoli, P.; Ferreyra, L.; Sicilia, P.; Carabajal, C.; Frattari, S.; Littvik, A.; Nates, S.; Pavan, J. IgG subclasses and DNA detection of HHV-6 and HHV-7 in healthy individuals. J. Med. Virol. 2010, 82, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Papaevangelou, V.; Quinlivan, M.; Lockwood, J.; Papaloukas, O.; Sideri, G.; Critselis, E.; Papassotiriou, I.; Papadatos, J.; Breuer, J. Subclinical VZV reactivation in immunocompetent children hospitalized in the ICU associated with prolonged fever duration. Clin. Microbiol. Infect. 2013, 19, E245–E251. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Pearson, G.R.; Jones, J.F.; Hillhouse, J.; Kennedy, S.; Mao, H.; Kiecot-Glaser, J.K. Stress-related activation of Epstein-Barr virus. Brain Behav. Immun. 1991, 5, 219–232. [Google Scholar] [CrossRef]
- Coskun, O.; Sener, K.; Kilic, S.; Erdem, H.; Yaman, H.; Besirbellioglu, A.B.; Gul, H.C.; Eyigun, C.P. Stress-related Epstein-Barr virus reactivation. Clin. Exp. Med. 2010, 10, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, C.P.; Jaremka, L.M.; Glaser, R.; Alfano, C.M.; Povoski, S.P.; Lipari, A.M.; Agnese, D.M.; Yee, L.D.; Carson, W.E., III; Farrar, W.B.; et al. Attachment anxiety is related to Epstein-Barr virus latency. Brain Behav. Immun. 2014, 41, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.V.; Marketson, J.I.W.; Chen, M.; Lo, K.W.; Kim, S.J.; Glaser, R. Glucocorticoids activate Epstein Barr virus lytic replication through the upregulation of immediate early BZLF1 gene expression. Brain Behav. Immun. 2010, 24, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Al Tabaa, Y.; Tuaillon, E.; Bollore, K.; Foulongne, V.; Petitjean, G.; Seigneurin, J.M.; Duperray, C.; Desgranges, C.; Vendrell, J.P. Functional Epstein-Barr virus reservoir in plasma cells derived from infected peripheral blood memory B cells. Blood 2009, 113, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Al Tabaa, Y.; Tuaillon, E.; Jeziorski, E.; Ouedraogo, D.E.; Bolloré, K.; Rubbo, P.A.; Flulongne, V.; Rodiere, M.; Vendrell, J.P. B-cell polyclonal activation and Epstein-Barr viral abortive lytic cycle are two key features in acute infectious mononucleosis. J. Clin. Virol. 2011, 52, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine of the National Academies. Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; The National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Klimas, N.G.; Ironson, G.; Carter, A.; Balbin, E.; Bateman, L.; Felsenstein, D.; Levine, D.; Peterson, K.; Chiu, A.; Allen, K.; et al. Findings from a clinical and laboratory database developed for discovery of pathogenic mechanisms in myalgic encephalomyelitis/chronic fatigue syndrome. Fatigue Biomed. Health Discov. 2015, 3, 75–96. [Google Scholar] [CrossRef]
- Yalcin, S.; Kuratsune, H.; Yamaguchi, K.; Kitani, T.; Yamanishi, K. Prevalence of human herpesvirus 6 variants A and B in patients with chronic fatigue syndrome. Microbiol. Immunol. 1994, 38, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.L.; Natelson, B.; Gause, W.; Hay, J. Human herpesviruses in chronic fatigue syndrome. Clin. Diagn. Lab. Immunol. 1999, 6, 216–223. [Google Scholar] [PubMed]
- Ablashi, D.V.; Eastman, H.B.; Owen, C.B.; Roman, M.M.; Friedman, J.; Zabriskie, J.B.; Peterson, D.L.; Pearson, G.R.; Whitman, J.E. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J. Clin. Virol. 2000, 16, 179–191. [Google Scholar] [CrossRef]
- Komaroff, A.L. Is human herpesvirus 6 a trigger for chronic fatigue syndrome? J. Clin. Virol. 2006, 37, S39–S46. [Google Scholar] [CrossRef]
- Chapenko, S.; Krumina, A.; Kozireva, S.; Nora, Z.; Sultanova, A.; Viksna, L.; Murovska, M. Activation of human herpesviruses 6 and 7 in patients with chronic fatigue syndrome. J. Clin. Virol. 2006, 37, S47–S51. [Google Scholar] [CrossRef]
- Chapenko, S.; Krumina, A.; Logina, I.; Rasa, S.; Chistjakovs, M.; Sultanova, A.; Viksna, L.; Murovska, M. Association of active human herpesvirus-6, -7 and parvovirus B19 infection with clinical outcomes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Adv. Virol. 2012, 2012, 205085. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z.; Shiraishi, Y.; Mears, C.J.; Binns, H.J.; Taylor, R. Chronic fatigue syndrome after infectious mononucleosis in adolescents. Pediatrics 2009, 124, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L.; Cho, T.A. Role of infection and neurologic dysfunction in chronic fatigue syndrome. Semin. Neurol. 2011, 31, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Loebel, M.; Strohschein, K.; Giannini, C.; Koelsch, U.; Bauer, S.; Doebis, C.; Thomas, S.; Unterwalder, N.; von Baehr, V.; Reinke, P.; et al. Deficient EBV-specific B-and T-cell response in patients with chronic fatigue syndrome. PLoS ONE 2014, 9, e85387. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.; Flamand, L.; Juwana, H.; Middeldorp, J.; Naing, Z.; Rawlinson, W.; Ablashi, D.; Lloyd, A. Serological and virological investigation of the role of the herpesviruses EBV, CMV and HHV-6 in post-infective fatigue syndrome. J. Med. Virol. 2010, 82, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Ariza, M.E.; Williams, M.; Jason, L.; Beqaj, S.; Fitzgerald, J.T.; Lemeshow, S.; Glaser, G. Antibody to Epstein-Barr virus deoxyuridine triphosphate nucleotidohydrolase and deoxyribonucleotide polymerase in a chronic fatigue syndrome subset. PLoS ONE 2012, 7, e47891. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Z.; Russell, T.A.; Spelman, T.; Carbone, F.R.; Tscharke, D.C. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell- intrinsic transcriptional response. PLoS Pathog. 2014, 10, e1004237. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.A.; Harth-Hertle, M.L.; Malterer, G.; Haas, J.; Ellwart, J.; Schulz, T.F.; Kempkes, B. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog. 2013, 9, e1003336. [Google Scholar] [CrossRef] [PubMed]
- Halpin, P.; Williams, M.V.; Klimas, N.G.; Fletcher, M.A.; Barnes, Z.; Ariza, M.E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Gulf War Illness patients exhibit increased humoral responses to the Herpesviruses-encoded dUTPase: Implications in disease pathophysiology. J. Med. Virol. 2016. Submitted. [Google Scholar]
- Aubrecht, T.G.; Weil, Z.M.; Ariza, M.E.; Williams, M.; Reader, B.; Glaser, R.; Sheridan, J.; Nelson, R. Epstein-Barr virus (EBV)-encoded dUTPase and chronic restraint induce anxiety-like behavior, impaired learning and memory and sickness responses. Physiol. Behav. 2014, 137, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, D.; Tai, J.; Levi-Schaffer, F.; Dovrat, S.; Mandelboim, O. Human herpesvirus 6 downregulates the expression of activating ligands during lytic infection to escape elimination by natural killer cells. J. Virol. 2016, 90, 9608–9617. [Google Scholar] [CrossRef] [PubMed]
- Kogelnik, A.M.; Loomis, K.; Hoegh-Petersen, M.; Rossoa, F.; Hischier, C.; Montoya, J.G. Use of valganciclovir in patients with elevated antibody titers against human herpesvirus-6 (HHV-6) and Epstein-Barr virus (EBV) who were experiencing central nervous system dysfunction including long-standing fatigue. J. Clin. Virol. 2006, 37, S33–S38. [Google Scholar] [CrossRef]
- Montoya, J.G.; Kogelnik, A.M.; Bhangoo, M.; Lunn, M.R.; Flamand, L.; Merrihew, L.E.; Watt, T.; Kubo, J.T.; Paik, J.; Desai, M. Randomized clinical trial to evaluate the efficacy and safety of valganciclovir in a subset of patient with chronic fatigue syndrome. J. Med. Virol. 2013, 85, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Beqaj, S. A paradigm linking herpesvirus immediate-early gene expression apoptosis and myalgic encephalomyelitis chronic fatigue syndrome. Virus Adapt. Treat. 2011, 3, 19–24. [Google Scholar] [CrossRef]
- Watt, T.; Oberfoell, S.; Balise, R.; Lunn, M.R.; Kar, A.K.; Merrihew, L.; Bhangoo, M.S.; Montoya, J.G. Response to valganciclovir in chronic fatigue syndrome patients with human herpesvirus 6 and Epstein–Barr virus IgG antibody titers. J. Med. Virol. 2012, 84, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Fluge, O.; Mella, O. Clinical impact of B-cell depletion with the anti-CD20 antibody rituximab in chronic fatigue syndrome: A preliminary case series. BMC Neurol. 2009, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Fluge, O.; Bruland, O.; Risa, K.; Storstein, A.; Kristoffersen, E.K.; Sapkota, D.; Næss, H.; Dahl, O.; Nyland, H.; Mella, O. Benefit from B-lymphocyte depletion using the anti-CD20 antibody Rituximab in chronic fatigue syndrome. A Double-Blind and Placebo-Controlled Study. PLoS ONE 2011, 6, e26358. [Google Scholar] [CrossRef] [PubMed]
- Fluge, O.; Risa, K.; Lunde, S.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Kristoffersen, E.K.; Sørland, K.; Bruland, O.; Dahl, O.; et al. B-lymphocyte depletion in Myalgic Encephalopathy/Chronic Fatigue Syndrome. An open-label Phase II study with Rituximab maintenance treatment. PLoS ONE 2015, 10, e0129898. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Hengel, H. Human cytomegalovirus and autoimmune disease. BioMed Res. Int. 2014, 2014, 472978. [Google Scholar] [CrossRef] [PubMed]
- Broccolo, F.; Fusetti, L.; Ceccherini-Nelli, L. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. Sci. World J. 2013, 2013, 867389. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.J.; Hochberg, D.; Rand, W.M.; Thorley-Lawson, D.A. EBV and systemic lupus erythematous: A new perspective. J. Immunol. 2005, 174, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, G.; Desani, J.; Anders, H.J. Lupus nephritis: Update on mechanisms of systemic autoimmunity and kidney immunopathology. Curr. Opin. Nephrol. Hypertens. 2014, 23, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.J. The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Fogo, A.R. Immunopathology of lupus nephritis. Semin. Immunopathol. 2014, 36, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Zickert, A.; Amoudruz, P.; Sundstrom, Y.; Ronnelid, J.; Malstrom, V.; Gunnarsson, I. IL-21 and IL-23 in lupus nephritis-association to histopathology and response to treatment. BMC Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Xu, L.; Chen, X.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J. Immunol. 2013, 190, 5411–5422. [Google Scholar] [CrossRef] [PubMed]
- Patole, P.S.; Pawar, R.D.; Lech, M.; Zecher, D.; Schmidt, H.; Segerer, S.; Ellwart, A.; Henger, A.; Kretzler, M.; Anders, H.J. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas (lpr) mice. Nephrol. Dial. Transplant. 2006, 21, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Crispin, J.C.; Tsokos, G.C. Interleukin-17 and systemic lupus erythematosus: Current concepts. Clin. Exp. Immunol. 2009, 157, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Amarilyo, G.; Lourenco, E.V.; Shi, F.D.; La Cava, A. Il-17 promotes murine lupus. J. Immunol. 2014, 193, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Baeten, D.L.; Josien, R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin. Immunol. 2014, 154, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liao, J.; Zhao, M.; Wu, H.; Yung, S.; Chan, T.M.; Yoshimura, A.; Lu, Q. Increased expression of TLR2 in CD4+ T cells from SLE patients enhances expression through histone modifications. Eur. J. Immunol. 2015, 45, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Galil, S.M.A.; Ezzeldin, M.; El-Boshy, M.E. The role of serum IL-17 and IL-6 biomarkers of disease activity and predictors of remission in patients with lupus nephritis. Cytokine 2015, 76, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Turpin, D.; Truchetet, M.E.; Faustin, B.; Augusto, J.F.; Contin-Bordes, C.; Brisson, A.; Blanco, P.; Duffau, P. Role of extracellular vesicles in autoimmune diseases. Autoimmun. Rev. 2016, 15, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Farre, B.; Rovira, J.; Martinez, D.; Valera, A.; Garcia-Herrera, A.; Marcos, M.A.; Sole, C.; Roue, G.; Colomer, D.; Gonzalvo, E.; et al. In vivo intratumoral Epstein-Barr virus replication is associated with XBP1 activation and early-onset post-transplant lymphoproliferative disorders with prognostic implications. Mod. Pathol. 2014, 27, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Cochet, C.; Martel-Renoir, D.; Greunewald, V.; Bosq, J.; Cochet, G.; Schwaab, G.; Bernaudin, J.F.; Joab, I. Expression of the Epstein-Barr virus immediate early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 1993, 197, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, A.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Montone, K.T.; Hodinka, R.L.; Salhany, K.E.; Lavi, E.; Rostami, A.; Tomaszewski, J.E. Identification of Epstein-Barr virus lytic activity in post-transplantion lymphoproliferative disease. Mod. Pathol. 1996, 9, 621–630. [Google Scholar] [PubMed]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; et al. Promiscuous expression of Epstein-Barr virus in Burkitt’s lymphoma from the central Africian country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Li, S.; Levi, M.; Weinberg, A. Lytic and latent EBV gene expression in transplant recipients with and without post-transplant lymphoproliferative disorder. J. Clin. Virol. 2011, 52, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Straon, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.B.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gartner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Munz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr Virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr Virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hedge, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowski-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef] [PubMed]
- Traylen, C.; Ramasubramanyan, S.; Zuo, J.; Rowe, M.; Almohammad, R.; Heesom, K.; Sweet, S.M.M.; Matthews, D.A.; Sinclair, A.J. Identification of Epstein-Barr virus replication proteins in Burkitt’s lymphoma cells. Pathogens 2015, 4, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yoshida, A.; Yamamoto, Y.; Katano, H.; Hagihara, K.; Oka, S.; Kimura, S.; Yoshizaki, K. Viral load of human herpesvirus-8 (HHV-8) in the circulatory blood cells correlates with clinical progression in a patient with HHV-8 associated solid lymphoma with aids-associated Kaposi’s sarcoma. Leuk. Lymphoma 2004, 45, 2343–2347. [Google Scholar] [CrossRef] [PubMed]
- Laney, A.S.; Cannon, M.J.; Jaffe, H.W.; Offermann, M.K.; Ou, C.Y.; Radford, K.W.; Patel, M.M.; Spira, T.J.; Gunthel, C.J.; Pellett, P.E.; et al. Human herpesvirus 8 presence and viral load are associated with progression of AIDS-associated Kaposi’s sarcoma. AIDS 2007, 21, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cell responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Bataille, R.; Pellat-Deceunynck, C. Interleukin-6 is a growth factor for nonmalignant plasmablasts. Blood 2001, 97, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Geginat, J.; Larghi, P.; Paroni, M.; Nizzoli, G.; Penatti, A.; Pagani, M.; Gagliani, N.; Meroni, P.; Abrignani, S.; Flavell, R.A. The light and dark sides of interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev. 2016, 30, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Kohlhass, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting Edge: The Foxp3 target mir-155 contributes to the development of regulatory T cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Toomey, N.L.; Diaz, L.A.; Walker, G.; Ramos, J.C.; Barber, G.N.; Ning, S. Oncogenic IRFs provide a survival advantage for Epstein-Barr virus- or human T-cell leukemia virus type 1-transformed cells through induction of BIC expression. J. Virol. 2011, 85, 8328–8337. [Google Scholar] [CrossRef] [PubMed]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen-1 in Hodgkin’s lymphoma cells mediates up-regulation of CCL20 and the migration of regulatory T cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Komai-Koma, M.; Jones, L.; Ogg, G.S.; Xu, D.; Liew, F.Y. TLR2 is expressed on activated T cells as a costimulatory molecule. Proc. Natl. Acad. Sci. USA 2004, 101, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Komai-Koma, M.; Xu, D.; Liew, F.Y. Toll-like receptor 2 signaling modulates the functions of CD4+CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7048–7053. [Google Scholar] [CrossRef] [PubMed]
- Sutmuller, R.P.M.; den Brok, M.H.G.M.; Kramer, M.; Bennink, E.J.; Toonen, L.W.J.; Kullberg, B.J.; Joosten, L.A.; Akira, S.; Netea, M.G.; Adema, G.J. Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin. Investig. 2006, 116, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, M.H.; Morandi, E.; Vinkemeier, U.; Constantin-Teodosiu, D.; Drinkwater, S.; Mee, M.; King, L.; Podda, G.; Zhang, G.X.; Ghaemmaghami, A.; et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: A novel mechanism of reduced regulatory T cell function in multiple sclerosis. J. Immunol. 2015, 194, 5761–5774. [Google Scholar] [CrossRef] [PubMed]
- Hoeppli, R.E.; Wu, D.; Cook, L.; Levings, M.K. The environment of regulatory T cell biology: Cytokines, metabolites and the microbiome. Front. Immunol. 2015, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Burrows, S.R. Role of cytotoxic T lymphocytes in Epstein-Barr virus-associated diseases. Annu. Rev. Microbiol. 2000, 54, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Purtilo, D.T. Simple assay for evaluation of Epstein-Barr virus specific cytotoxic T lymphocytes. J. Immunol. Methods 1995, 184, 149–152. [Google Scholar] [CrossRef]
- Ariza, M.E.; Williams, M.V. EBV-dUTPase modulates host immune responses potentially altering the tumor microenvironment in EBV-associated malignancies. J. Curr. Res. HIV/AIDS 2016, 2016, 1–9. [Google Scholar]
- Strauss, I.; Bergmann, G.; Szezepanski, M.J.; Lang, S.; Kirkwood, J.M.; Whiteside, T.L. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxps3+ T regulatory cells: Implications and impact on tumor-mediated immune suppression. J. Immunol. 2008, 180, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Le, K.S.; Thibult, M.L.; Just-Landi, S.; Pastor, S.; Gondois-Rey, F.; Granjeaud, S.; Broussais, F.; Bouabdallah, R.; Colisson, R.; Caux, C.; et al. Follicular B lymphomas generate regulatory T cells via the ICOS/ICOSL pathway and are susceptible to treatment by anti-ICOS/ICOSL therapy. Cancer Res. 2016, 76, 4648–4680. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E. The challenge of the microenvironment in B-cell lymphomas. Histopathology 2011, 58, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zirakzadeh, A.; Marits, P.; Sherif, A.; Winqvist, O. Multiplex B Cell characterization in blood, lymph nodes, and tumors from patients with malignancies. J. Immunol. 2013, 190, 5847–5855. [Google Scholar] [CrossRef] [PubMed]
- Marits, P.; Zirakzadeh, A.A.; Sherif, A.; Winqvist, O. The many flavors of tumor-associated B cells. Oncoimmunology 2013, 2, e25237. [Google Scholar] [CrossRef] [PubMed]
- Shiley, K.; Blumberg, E. Herpes Viruses in transplant recipients: HSV, VZV, Human Herpes Viruses, 6 and 7 and EBV. Infect. Dis. Clin. N. Am. 2010, 24, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, R.A.; Limaye, A.P. Varicella zoster virus (VZV) and herpes simplex virus (HSV) in solid organ transplant patients. Am. J. Transpl. 2013, 13, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Inazawa, N.; Hori, T.; Hatakeyama, N.; Yamamoto, M.; Yoto, Y.; Nojima, M.; Suzuki, N.; Shimizu, N.; Tsutsumi, H. Large-scale multiplex polymerase chain reaction assay for diagnosis of viral reactivations after allogeneic hematopoietic stem cell transplantation. J. Med. Virol. 2015, 87, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Walton, A.H.; Muenzer, J.T.; Rasche, D.; Boomer, J.S.; Sato, B.; Brownstein, B.H.; Pachot, A.; Brooks, T.L.; Deych, E.; Shannon, W.D.; et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE 2014, 9, e98819. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Hiraharas, K.; Sakuma, K.; Shiohara, T. Several herpesviruses can reactivate in a severe drug-induced multiorgan reaction in the same sequential order as in graft-versus-host disease. Br. J. Dermatol. 2006, 155, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Shiohara, T.; Inaoka, M.; Kano, Y. Drug-induced HypersensitivitySyndrome (DIHS): A reaction induced by a complex interplay among herpesviruses and antiviral and antidrug immune responses. Allergol. Int. 2006, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seishima, M.; Yamanaka, S.; Fujisawa, T.; Tohyama, M.; Hashimoto, K. Reactivation of human herpesvirus (HHV) family members other than HHV-6 in drug-induced hypersensitivity syndrome. Br. J. Dermatol. 2006, 155, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Picard, D.; Janela, B.; Descamps, V.; D’Incan, M.; Courville, P.; Jacquot, S.; Rogez, S.; Mardivirin, L.; Moins-Teisseterenc, H.; Toubert, A.; et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): A multiorgan antiviral T cell response. Sci. Transl. Med. 2010, 2, 46ra62. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chiang, H.H.; Cho, Y.T.; Chang, C.Y.; Chen, K.L.; Yang, C.W.; Lee, Y.H.; Chu, C.Y. Human herpes virus reactivations and dynamic cytokine profiles in patients with cutaneous adverse drug reactions—A prospective comparative study. Allergy 2015, 70, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, J.; Abuabara, K.; Rerman, M.J.; Yan, A.C. HHV6 involvement in pediatric drug hypersensitivity. Br. J. Dermatol. 2015, 172, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Flamand, L.; Stefanescu, I.; Ablashi, D.V.; Menezes, J. Activation of Epstein-Barr virus replicative cycle by human herpesvirus 6. J. Virol. 1993, 61, 6768–6777. [Google Scholar]
- McCarthy, M.; Auger, D.; He, J.; Wood, C. Cytomegalovirus and human herpesvirus-6 trans-activate the HIV-1 long terminal repeat via multiple response regions in human fetal astrocytes. J. Neurovirol. 1998, 4, 495–511. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, P.; Carbone, A. Microenvironmental abnormalities induced by viral cooperation: Impact on lymphomagenesis. Semin. Cancer Biol. 2015, 34, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.-F.; Kenney, S.C.; Lambert, P.F. Human papilloma virus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Rezaie, F.; Moghim, S.; Mogharehabed, A.; Rezaei, M.; Mehraban, B. Periodontopathic bacteria and herpesviruses in chronic periodontitis. Mol. Oral Microbiol. 2010, 25, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Saygun, I.; Kubar, A.; Sahin, S.; Sener, K.; Slots, J. Quantitative analysis of association between herpesviruses and bacterial pathogens in periodontitis. J. Periodontal Res. 2008, 43, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: Future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Matar, C.G.; Jacobs, N.T.; Speck, S.H.; Lamb, T.J.; Moorman, A.M. Does EBV alter the pathogenesis of malaria? Parasite Immunol. 2015, 37, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Williams, M.V. A human endogenous retrovirus K dUTPase triggers a TH1, TH17 cytokine response: Does it have a role in Psoriasis? J. Investig. Dermatol. 2011, 131, 2419–2427. [Google Scholar] [CrossRef] [PubMed]
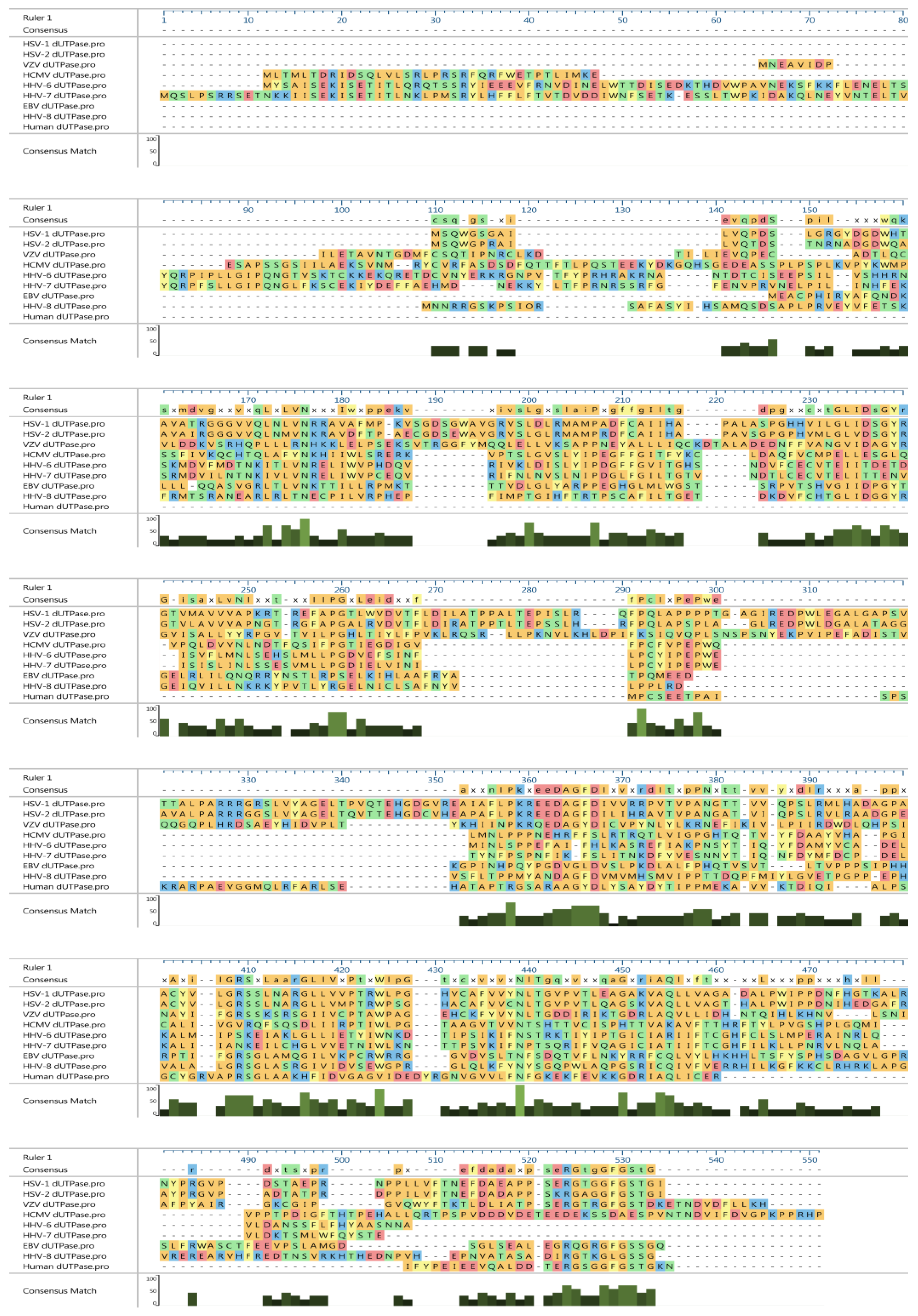

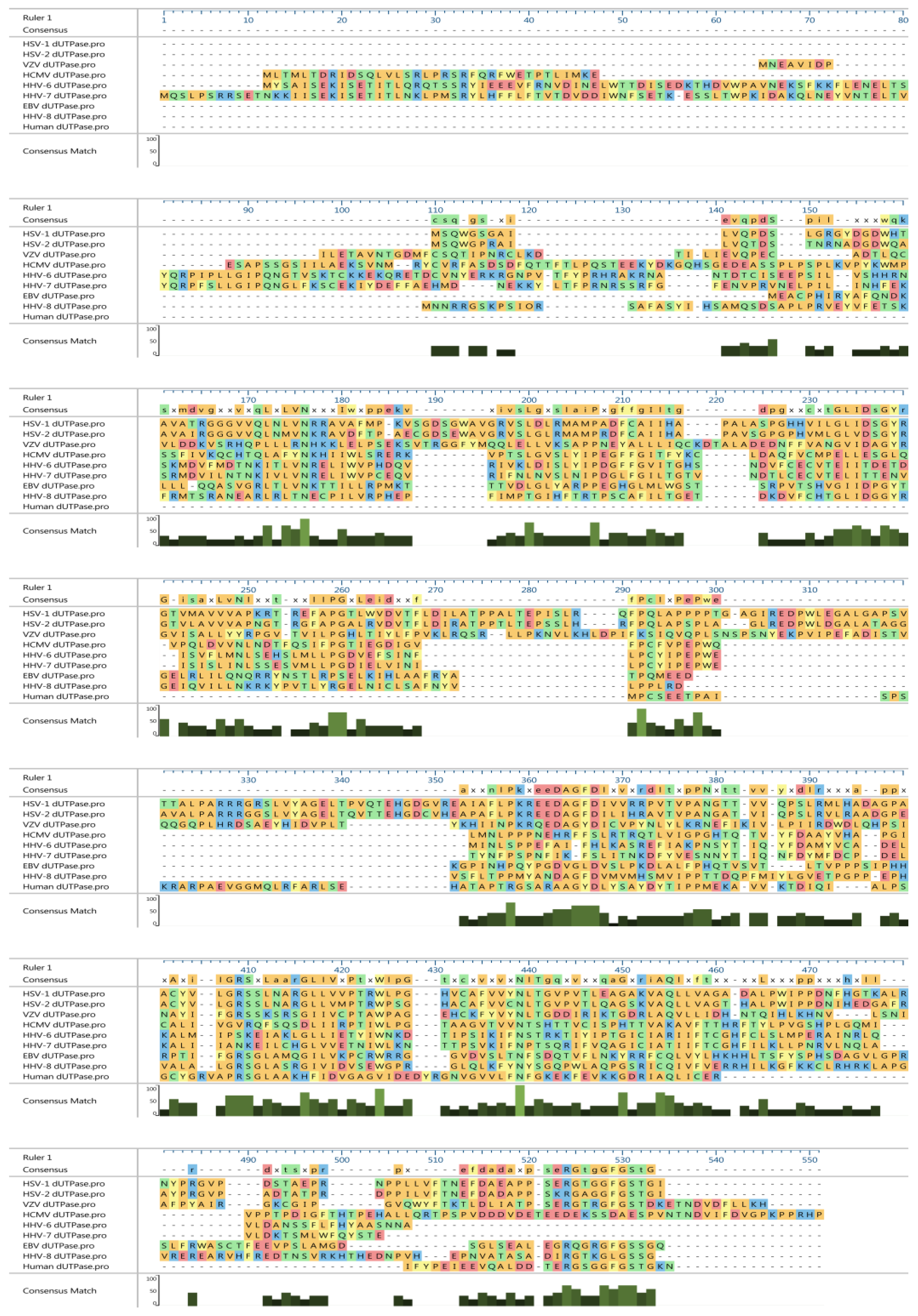{kind=link}
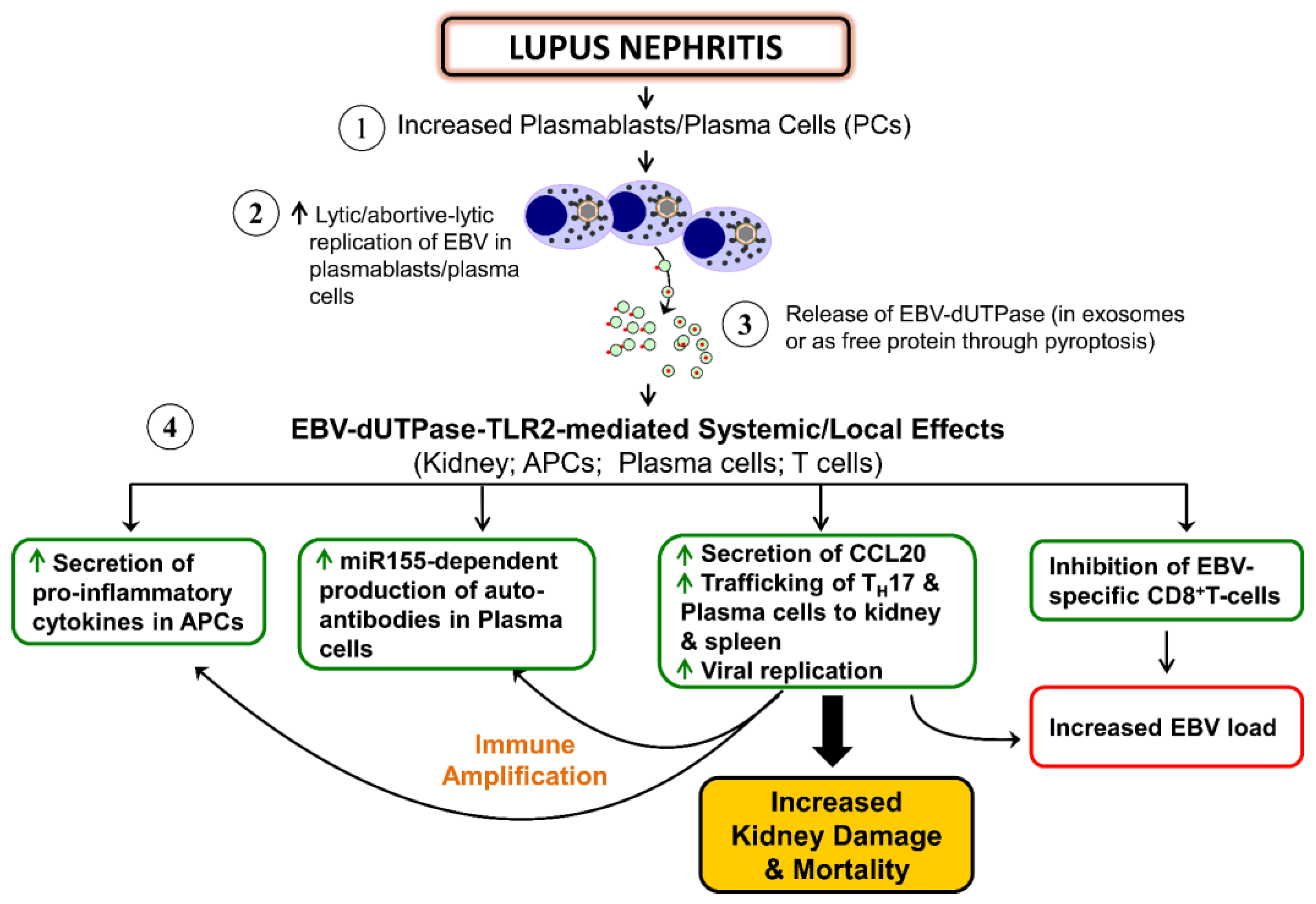{kind=link}
| Virus | Gene | Enzymatic Activity | Crystal Structure | Protein Homology b (%) | Required for In Vitro Replication | Immune Modulatory Function c |
|---|---|---|---|---|---|---|
| HSV-1/2 | UL50 | Yes [20,21,22,23] | ND | 29 | No [13,24] | Induces IL-10, IL-12p70, IL-1β, IL-6, IL-8, TNFα in human PBMC and human dendritic cells (hDC) [7] |
| VZV | ORF8 | Yes [14] | ND | 24 | No [14] | Induces IL-10, IL-12p70, IL-1β, IL-6, IL-8, TNFα in hPBMC and hDC [7] |
| HCMV | UL72 | No [6] | ND | 24 | No [6] | ND |
| HHV-6A | U45 | No [7] | ND | 21 | ND | Induces IL-10, IL-12p70, IL-1β, IL-6, IL-8, TNFα in hPBMC and hDC [7] |
| HHV-6B | U45 | ND a | ND | 21 | ND | ND |
| HHV-7 | U45 | ND | ND | 23 | ND | ND |
| EBV | BLLF3 | Yes [23] | Yes [4] | 100 | ND | Induces IL-10, IL-12p70, IL-1β, IL-6, IL-8, IL-17A, TNFα in hPBMC and hDC [25,26,27,28,29] as well as IL-1β, IL-6 and IL-17 in vivo [30,31,32] |
| HHV-8 | ORF54 | Yes [33] | ND | 31 | ND | Induces IL-10, IL-12p70, IL-1β, IL-6, IL-8, TNFα in hPBMC and hDC [7] Downregulates NKp44L [34] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, M.V.; Cox, B.; Ariza, M.E. Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease. Pathogens 2017, 6, 2. https://doi.org/10.3390/pathogens6010002
Williams MV, Cox B, Ariza ME. Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease. Pathogens. 2017; 6(1):2. https://doi.org/10.3390/pathogens6010002
Chicago/Turabian StyleWilliams, Marshall V., Brandon Cox, and Maria Eugenia Ariza. 2017. "Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease" Pathogens 6, no. 1: 2. https://doi.org/10.3390/pathogens6010002
APA StyleWilliams, M. V., Cox, B., & Ariza, M. E. (2017). Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease. Pathogens, 6(1), 2. https://doi.org/10.3390/pathogens6010002