2.1. Building a Core Gene-Based Reference Database
Bacterial genomic sequences on public databases have a diverse range of genomic statistics, such as reference size, number of contigs, assembly status (complete, chromosome, scaffold, contig), N50 values among others. For some species, a high-quality assembly may not be available. When using these references for metagenomic shotgun profiling, this variation may provide a bias for higher quality and complete genomes, making abundance quantification unreliable, particularly for genomes that may be considered incomplete. To avoid this, we extracted UBCG sequences [
2] (92 core genes) from full genomic references from the EzBioCloud database [
8]. Regardless of the assembly status or genome length, extracting these core genes will remove any bias based on genome quality.
Figure 1a shows three streptococcal references with variable genome size, number of contigs, and N50 value. When extracting the UBCG sequences, all the references end up being represented by the same number of genes (92) and their sequence size is near identical. This shows that regardless of having a complete genome with one contig, or a contig assembly with 500 contigs, UBCG will provide an unbiased representation for any bacterial reference regardless of their assembly status, making detection and abundance estimation more reliable.
Figure 1b shows how genomic sequences from a variable length size for the genus
streptococcus can be translated into 92 UBCG sequences with a narrower difference in sequence length. After extracting all 92 UBCG sequences from the bacterial references, we built two Kraken [
12] based pipelines containing these sequences as references, one with just validly named bacterial species (KrakenUBCG-VNS) and another one containing also genomospecies references (KrakenUBCG).
Table 1 shows the difference between these two pipelines, highlighting their core algorithm and the number of bacterial and streptococcal species that their databases represent.
2.2. Evaluation of Pipelines Using Synthetic Metagenomes
To compare the classification accuracies by bacterial core genes as reference sequences, we created four different synthetic metagenomic datasets containing only species from the genus
Streptococcus using the InSilicoSeq pipeline [
13] and proceeded to compare the results with MetaPhlAn2.
Supplementary Table S2 contains the description and accession number of the genomes used on these datasets along with their true abundances.
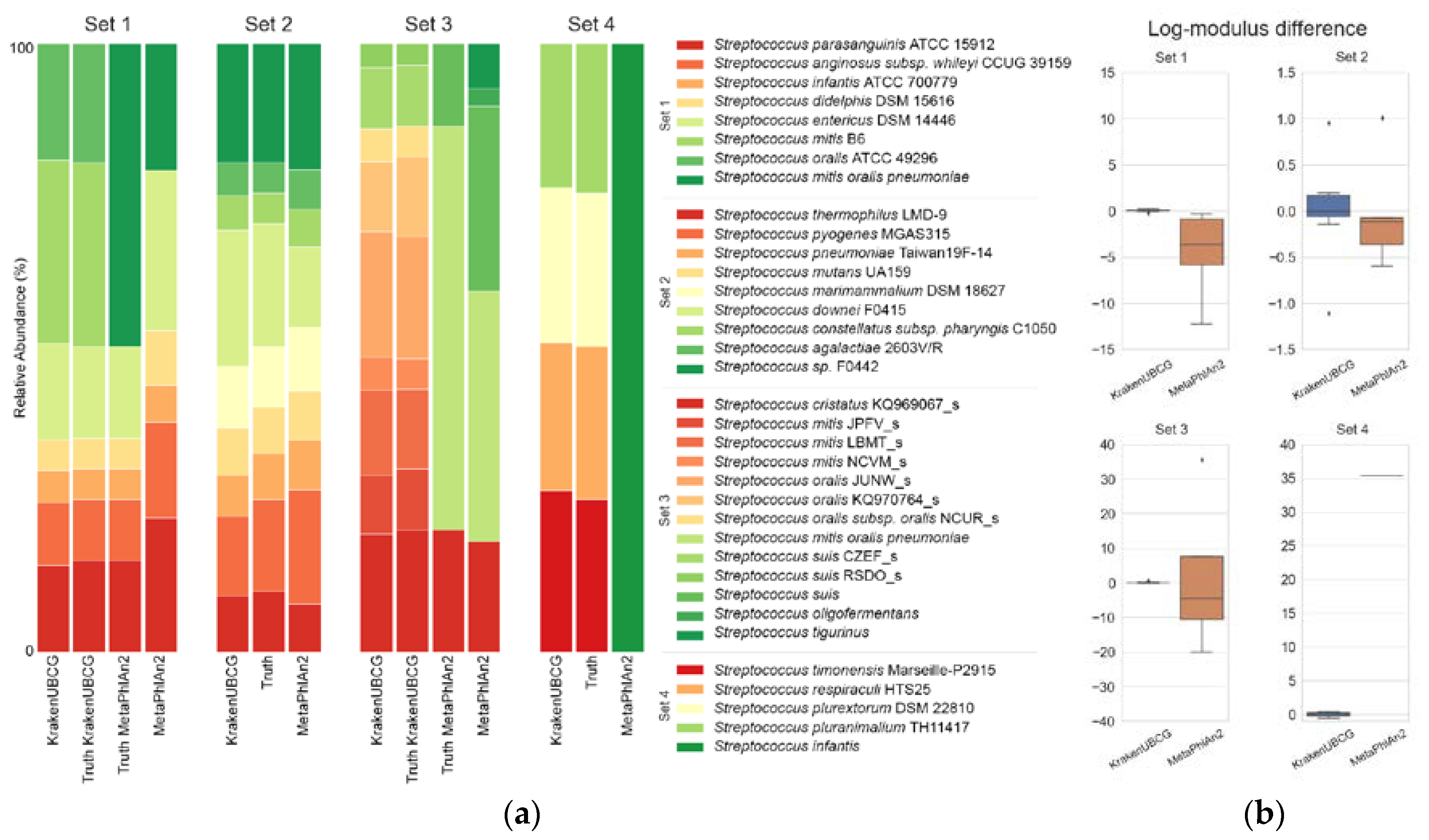
Figure 2a shows the predicted abundances for all four datasets using the KrakenUBCG pipeline and MetaPhlAn2, respectively, along with the true abundance of the synthetic sets. In order to compare some of MetaPhlAn2’s predictions to our pipeline KrakenUBCG, we included an alternative truth for some sets, since MetaPhlAn2 clusters several
Streptococcus species into a single taxon group.
Figure 2b shows the log-modulus difference of abundance prediction made by MetaPhlAn2 and the KrakenUBCG pipeline. The first synthetic set consists of seven different species of public assembled genomes from the NCBI database, which were included in both pipelines. MetaPhlAn2’s prediction was compared against a grouped truth, where all the genomes that belong to
S. mitis and
S. oralis are clustered into a single group (
S. mitis,
S. oralis, and
S. pneumoniae; referred as mitis group hereafter). Both MetaPhlAn2 and KrakenUBCG were able to identify correctly the presence of all species of
Streptococcus contained in the sample, however MetaPhlAn2’s marker gene database cannot distinguish between the species that are contained in the mitis group. Relative abundance predictions were less consistent for MetaPhlAn2, as it seems to be unable to predict correct abundances for the mitis group. While the expected abundance for
S. mitis and
S. oralis was expected at 30% and 20% respectively, MetaPhlAn2 only assigned a total abundance of 21.2% (mitis group), much lower than the expected 50%. KrakenUBCG, on the other hand, accurately predicted 29.9% and 19.3% relative abundance, respectively. Lower abundance prediction by MetaPhlAn2 for the mitis group indicates its overprediction of abundance for the remaining species, most notably
Streptococcus entericus with a predicted abundance of 26.3% instead of an expected abundance of 15%, while KrakenUBCG accurately predicted its abundance at 15.8%.
Synthetic dataset 2 consists of nine species of Streptococcus that were contained in both databases while the genome assemblies representing each species were different. Both pipelines predicted correctly the presence of all the species contained in the sample, and while the KrakenUBCG pipeline contains different reference assemblies for all nine species, it was able to detect the presence of all nine species correctly. KrakenUBCG’s abundance predictions were more accurate than MetaPhlAn2’s, for the 20% expected abundance for Streptococcus downei, KrakenUBCG and MetaPhlAn2 predicted 22.2% and 13.19%, respectively, making KrakenUBCG overpredicting the abundance by 2.2% while MetaPhlAn2 underpredicted the abundance by 6.81%. Streptococcus pyogenes had an expected abundance of 15% with a predicted abundance of 13% and 18.64% for KrakenUBCG and MetaPhlAn2, respectively. For the remaining species, KrakenUBCG’s worst prediction was for Streptococcus pneumoniae, with a prediction of 6.72% against an expected abundance of 7.5%, while MetaPhlAn2’s worst prediction was Streptococcus agalactiae, with a predicted abundance of 6.5% against an expected abundance of 5%. Overall, both pipelines performed better than expected, however KrakenUBCG performed favorably over MetaPhlAn’s abundance prediction even though it did not contain the assemblies contained in the metagenomic sample.
The third set contains the assemblies of nine different genomes from the genus Streptococcus. Those genomes are annotated as genomospecies in the KrakenUBCG pipeline. MetaPhlAn2 contains the same assemblies, however, some of them are annotated as the same species under the NCBI database, so we expect MetaPhlAn2’s prediction to follow that annotation. Those annotations are as follows: genomospecies KQ969067_s is annotated at the NCBI database as Streptococcus cristatus; JPFV_s, LBMT_s, and NCVM_s as Streptococcus mitis; JUNW_s, KQ970764_s, and NCUR_s as Streptococcus oralis; and finally, CZEF_s and RSDO_s as Streptococcus suis. MetaPhlAn2 is also unable to distinguish between Streptococcus oralis and Streptococcus mitis, so while the KrakenUBCG prediction is expected to predict nine different genomospecies (KQ969067_s, JPFV_s, LBMT_s, NCVM_s, JUNW_s, KQ970764_s, NCUR_s, CZEF_s, and RSDO_s), MetaPhlAn2 is only expected to predict three distinct species (Streptococcus suis, Streptococcus cristatus, and mitis group), and while it predicted the presence of those three taxa correctly, it also predicted the presence of two other Streptococcus species (false positives). KrakenUBCG’s prediction, however, predicted correctly the presence of all nine genomospecies present in the sample. Abundance predictions for MetaPhlAn2 particularly for the mitis group were low, with an abundance prediction of 40.93% against an expected abundance of 66%. For Streptococcus suis, MetaPhlAn2 predicted 30.21% against an expected 14% abundance. KrakenUBCG’s worst abundance prediction was for the genomospecies KQ970764_s with 11.5% against an expected abundance of 13%.
Finally, set 4 consists of species present on the KrakenUBCG pipeline but absent on the MetaPhlAn2 database. MetaPhlAn2 predicted a single species (Streptococcus infantis) which is not present in the sample, and while it was expected that MetaPhlAn2 would not be able to predict correctly any of the species due to their absence in their database, it was not unable to predict the presence of more than one species. KrakenUBCG predicted all four species present, as for abundance predictions, each species had an expected abundance of 25% each, and KrakenUBCG predicted 26.4%, 24.2%, 25.3%, 23.9% abundance for Streptococcus timonensis, Streptococcus respiraculi, Streptococcus plurextorum, and Streptococcus pluranimalium, respectively.
Using four synthetic metagenomic samples containing full genome reads from Streptococcus, we demonstrated that using core genes as references not only predict accurate species composition, but also showed little difference when comparing predicted relative abundance versus the absolute abundance. We also showed that MetaPhlAn2 suffers from lower accuracy in predicting relative abundance, probably because its marker gene references are uneven in coverage, especially for the mitis group.
2.3. Chronic Obstructive Pulmonary Disease Samples
Chronic obstructive pulmonary disease (COPD) is an obstructive lung disease where the affected individual suffers from long-term breathing problems and airflow. Cameron et al. studied the association between the microbiome and COPD by comparing the metagenomic profiles of two different groups, patients with COPD and ‘healthy’ smoking controls [
14], and showed significant changes in abundance of bacterial species, particularly in the genus
Streptococcus. They also found the presence of
Streptococcus pneumoniae in all the samples. Using KrakenUBCG with 201 different
Streptococcus reference genomes and MetaPhlAn2’s database, we profiled all the samples and compared the prediction of
Streptococcus at genus and species level.
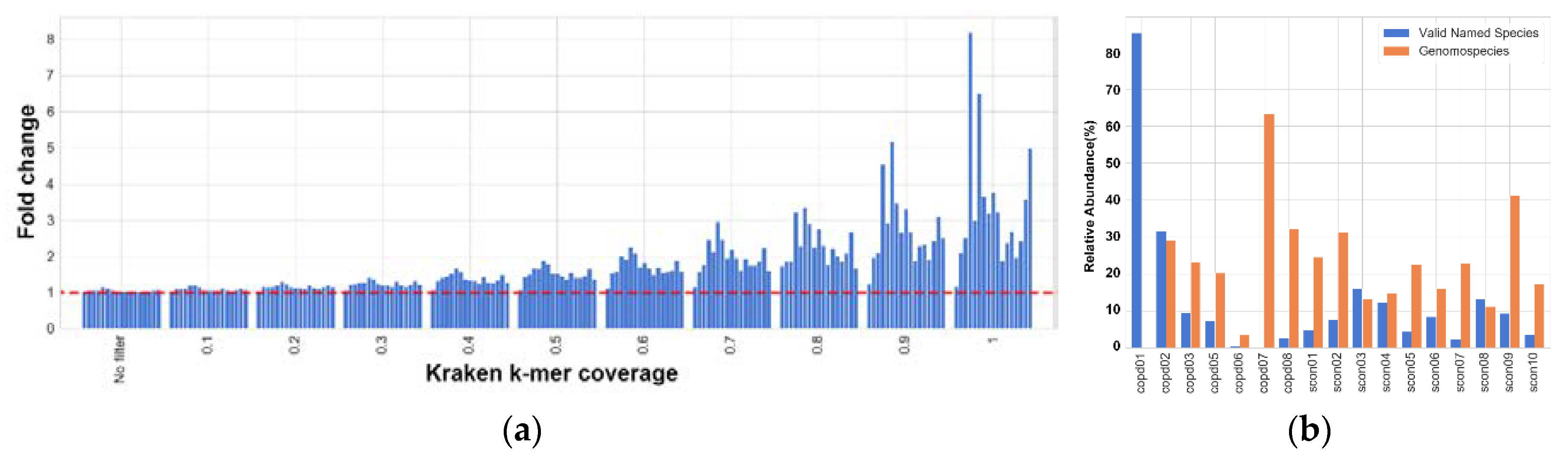
Figure 3 shows the abundance prediction at genus level by both KrakenUBCG and MetaPhlAn2. While the presence of other genera is variable among the databases, they are outside the scope of this study and will not be discussed in detail, however, it is important to mention that the number of classified reads for other genera will also have a direct impact on the predicted abundance of
Streptococcus. As an example, this can be observed on sample copd04, MetaPhlAn2 was unable to detect the presence of any bacteria, while KrakenUBCG detected the presence of three distinct genera.
Figure 4 shows heatmaps for predictions made by KrakenUBCG and MetaPhlAn2 along with their respective ANI dendrogram. Only samples that contained any
Streptococcus in either of the predictions were included. References from the MetaPhlAn2 database were matched with the KrakenUBCG pipeline and aligned on the heatmap accordingly. Predictions with a minimum of 0.5% abundance are shown, also only the species and genomospecies that were contained in at least one of the samples were included.
One of the major differences between both pipelines is the mitis group on MetaPhlAn2, which is covered by 21 different species and genomospecies at KrakenUBCG, showing clear differences as expected. On KrakenUBCG, only 3 samples showed the presence of S. pneumoniae, and another 3 showed presence of S. mitis, but none of the samples showed the presence of both at the same time; genomospecies CP016207_s, KV802702_s, KB373321_s, JPFY_s, JPFT_s, NCVM_s, CP012646_s, JUQO_s, JPFU_s, and JUUO_s were predicted only once among the samples, while the remaining genomospecies were found in two or more of the samples. While MetaPhlAn2 found zero presence of bacteria in the mitis group for sample copd02, copd06, scon05, and scon10, KrakenUBCG found the presence of ASZZ_s and AFUU_s for copd02, and JPFV_s and JVWC_s for copd06. For samples scon05 and scon10, KrakenUBCG did not find the presence of any species or genomospecies closer to S. pneumoniae or S. mitis matching MetaPhlAn2’s prediction. Looking at some samples individually we can observe distinct predictions, for example, MetaPhlAn2 predicted only one species of Streptococcus (S. salivarius) with a 17.5% abundance, while KrakenUBCG found three distinct species (ALIF_s, JYGT_s and S. intermedius) with a total abundance of 20.7%. If we analyze each sample individually, we can observe more differences than coincidences. These differences are the result of the lack of references on MetaPhlAn2’s database, and the argument can be made that only formally named species should be added to a reference database. However, if MetaPhlAn2’s authors decide to add genomospecies references to their database, they would require to recalculate all gene markers to those genus affected with newly added genomospecies, and even if somehow, these recalculations can be done within reasonable computational time, we can already observe that these gene markers cannot differentiate between three already recognized species (S. mitis, S. oralis and S. pneumoniae), so adding any more references may result in more larger groups of species, just like the mitis group.
Figure 5a shows the taxonomic biomarkers found by LEfSe [
15] by KrakenUBCG and MetaPhlAn2. While the study was not aimed at finding associations between COPD and healthy controls, our analyses demonstrated that any kind of conclusion driven by the use of distinct databases may lead to different findings. Both databases found a great diversity of streptococcal species found in the control samples that were absent in the COPD samples. KrakenUBCG found 24 streptococcal biomarkers (8 validly named species and 16 genomospecies) contained in the control metagenomic samples while MetaPhlAn2 detected 6 streptococcal biomarkers, demonstrating the impact that a high species and genomospecies coverage would have on the biomarker discovery process.
Figure 5b shows four biomarkers found by both pipelines while the remaining biomarkers were exclusive to each pipeline. These common biomarker predictions done by both pipelines show that regardless of the core algorithm or database, when a species is predicted accurately, the process for taxonomic biomarker discovery can reliable. However, it also shows how the absence of streptococcal species on MetaPhlAn2’s database would miss several biomarkers that were detected by KrakenUBCG (
Figure 5a). Absence of species may also incur on false taxonomic predictions, particularly when a reference species has no genomically close species. For example, MetaPhlAn2 detected
Streptococcus infantis as a biomarker, however this may be explained by
Figure 4, where it can be seen that MetaPhlAn2’s database lacks reference species genomically close to its genome, while KrakenUBCG detected the presence of
Streptococcus infantis only in two control samples while the rest of the prediction was assigned to other genomically close genomospecies. This example showcases how an outdated database might impact metagenomic profiling and biomarker discovery, since the lack of genomic diversity around the reference of
Streptococcus infantis will potentially incur in taxonomic false positives and result in a false biomarker prediction.
Here, by comparing classification results using samples from COPD patients with our genomospecies database KrakenUBCG, we found the presence of thirty-three different genomospecies and nineteen species across the samples. On the other hand, MetaPhlAn2 only found the presence of seventeen species of
Streptococcus, demonstrating the importance of keeping a database up to date (
Figure 4). This finding indicates that lack of all species and genomospecies in the database being used and their taxonomies inconsistencies can exert a dramatic impact on the discovery of reliable biomarkers and biodiversity estimates. While MetaPhlAn2’s algorithmic predictions may not be incorrect, their use of an outdated database with an incorrect taxonomy annotation may have an impact on their prediction performance, specifically since they rely on that taxonomy structure in order to generate their reference marker genes. Additionally, the use of marker genes as reference sequences makes the process of updating their database difficult. While generating those marker genes itself requires substantial time and efforts, the process is further complicated by the fact that every time when a new species is added, all the marker genes have to be recalculated. To this end, using UBCG as a reference appears to be more convenient, as once these core genes have been extracted, if a new species needed to be added to the database, only the core genes for that species have to be calculated.
2.4. Identifying Accurately a Streptococcal Infection Using Clinical Data
Tracing the origin of pathogenic bacteria in bloodstream infections can be a challenge. Tamburini et al. [
16] proposed that the source of infection on some patients is the human gut. They isolated and sequenced pathogenic bacteria present in the bloodstream of patients at Stanford University Hospital and performed shotgun metagenomics on stool samples for those same patients. For this research, we will focus on patient 22, whereby using his blood isolate and stool sample, the authors were able to match the genome assembled from the bloodstream isolate with the metagenomic assembly generated form the stool sample and classified it as
Streptococcus mitis.
We analyzed the streptococcal bacterial isolate from this patient (Sample id: SRR7407865) using the TrueBac ID system [
17], which utilizes the exact same species and genomospecies references from the EzBioCloud database included in our KrakenUBCG pipeline.
Table 2 shows the results of TrueBac ID when comparing the isolate with the EzBioCloud database. Our analysis indicates that the closest reference to the isolate is actually streptococcal genomospecies JPFV_s (not
Streptococcus mitis) with an ANI identity value of 94.61% with a coverage of 86.9%. Since the ANI identity value is below the species threshold (95%) [
18], we will assume that this isolate belongs to a tentative new genomospecies and does not belong to
Streptococcus mitis.
To further illustrates the utility of broader species coverage and need to update the reference databases frequently, we built a third metagenomic reference database (KrakenUBCG+ SRR7407865) with the addition of this tentative new genomospecies by assembling the genome and extracting the 92 core genes using the UBCG pipeline and simply adding them to the KrakenUBCG database. We then perform a metagenome classification analysis using all three databases.
Figure 8 shows the classification results for all the available pipelines. Relative abundance at the genus level for
Streptococcus yields an increase of 0.07% after classifying with KrakenUBCG, while it only showed an increase of 0.002% when adding the bloodstream isolate from the sample (KrakenUBCG+ SRR7407865). However, at the species level, the classifications for all three databases showed big differences. For the classification with only valid named species (KrakenUBCG-VNS), the highest, most abundant species of
Streptococcus is
Streptococcus mitis, however by using KrakenUBCG, the most abundant species is JPFV_s. By observing those two different classifications using the ANI tree shown in
Figure 9, we can observe that they are both correct, with the absence of references from streptococcal genomospecies, the classification should go to the closest reference, in this case,
Streptococcus mitis. This is also consistent with the initial classification of the isolate in the original paper as
Streptococcus mitis [
16].
Finally, the third classification using KrakenUBCG+ SRR7407865, showed that by comparing the stool sample with an updated database that contains a bloodstream isolate, will successfully assign the top streptococcal species as this tentative novel genomospecies.
While for this case, we had access to the bacterial blood isolate, the majority of the time, the user will have to rely on precompiled databases, and in this specific case, if we were to use our genomospecies database, the classification for this streptococcal species would be assigned to the closest reference available in the database, in this case, the genomospecies JPFV_s. We also show that in the absence of any genomospecies in the reference database, this streptococcal strain would be classified as Streptococcus mitis. This demonstrates the importance of updating reference databases frequently, even if those references have not been published or named formally.
2.5. Effects of Different ANI Thresholds on the Classificwtion of Genomospecies
The ANI threshold for genomospecies identification proposed by Chun et al. [
18] is 95%, and this threshold is applied at the EzBioCloud database. As the authors mention in their article, these are minimal standards, which means that in some cases, a different ANI threshold could be applied in order to set species boundaries for a specific taxon. Similarly, the Genome Taxonomic Database (GTDB) [
19] also proposes a 95% ANI threshold for these novel species using FastANI [
20], although using a different nomenclature for these genomospecies, supporting this ANI threshold as a consensus in the prokaryote taxonomic community. In the previous example shown in
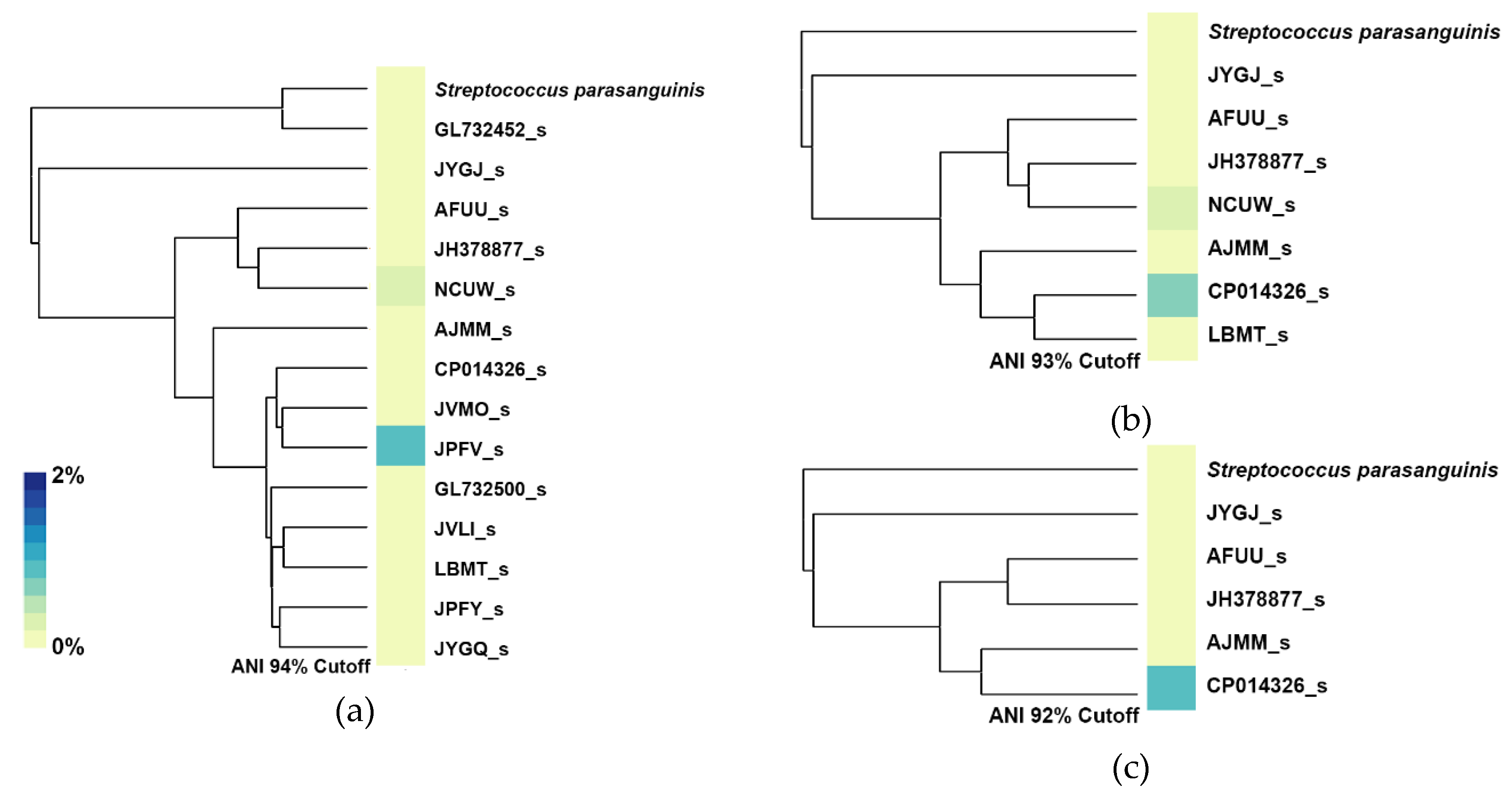
Figure 8, the blood isolate from patient 22 was recognized as a potential new species because the ANI value between the isolate and the closest genomospecies JPFV_s is 94.24%, which is below the proposed species boundary. However, if a different ANI threshold was set, for example 94%, this blood isolate would be considered as a part of the genomospecies JPFV. With this in mind, we decided to change the ANI threshold for genomospecies for the case of patient 22 in order to see the effects that a different ANI threshold would have on the classification of streptococcal bacteria present in the stool sample.
Figure 9 shows the ANI dendrogram marked with the different ANI thresholds used in this analysis.
For this experiment, we built three additional databases, each one containing a different number of streptococcal genomospecies depending on the ANI threshold set for each database. When setting a new ANI threshold, only one reference would be used as a reference for that genomospecies. For example, at an ANI threshold of 94%, the genomospecies JUQO_s and LBMT_s would be considered as a single genomospecies, so in order to select which reference would be used to represent this new genomospecies, we selected the genome with the highest N50 value. After building these three additional databases, we profiled the stool sample of patient 22 and generated heatmaps with their respective ANI dendrogram as shown in
Figure 10. With a 94% ANI cutoff, the blood isolate from patient 22 is now considered as the JPFV_s genomospecies, at the same time, JUPI_s now is considered a strain of GL732452_s; despite these changes, the main streptococcal species found on the sample is JPFV_s, which is the genomospecies that now represents the blood isolate from the patient. A small abundance is also detected for the genomospecies NCUW_s. Lowering the ANI threshold to 93%, decreases the number of genomospecies present in this tree, with CP014326_s now representing a vast majority of genomospecies from the 95% ANI tree, including ASZZ_s, JUPG_s, JVMO_s, JPFV_s, and the blood isolate SRR7407865. In this case, CP014326_s is classified as the main streptococcal species with NCUW_s also being found present with a low abundance. Lastly, with an ANI threshold of 92%, only six species remain, this time, the genomospecies CP014326_s maintains its predominant presence of streptococcal abundance, with a slight abundance increment, and this could be mainly due to the absence of NCUW_s, now being represented by JH378877_s.
The example shown in
Figure 10, demonstrated that regardless of the ANI threshold set for genomospecies, the detection of a specific species can be achieved if a representative species close to the one of interest is present in the database. With an ANI threshold of 92%, genomospecies CP014326_s represents our blood isolate SRR7407865, and with an ANI distance between these two of 93.591%, it can be seen that with a small sacrifice of abundance detection, the presence of either genomospecies can be achieved regardless of the ANI threshold set by the user.
Here, we showed that even with different ANI thresholds, the classification of the streptococcal strain was consistent with the location of the blood isolate within the original ANI dendrogram. However, a loss of abundance was seen mainly because of the higher ANI distance between the species present in the stool sample and the reference used to represent the genomospecies on each of the examples. While the ANI threshold for setting boundaries for new species can be debatable, when using a computational method that is based on sequence similarity, we believe that a fixed sequence similarity ANI threshold should be implemented, regardless of the taxonomic rank we set to given a taxon node (species, genomospecies, or even strain).

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}