
pH and Nitrate Drive Bacterial Diversity in Oil Reservoirs at a Localized Geographic Scale

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Geochemical Analysis
2.2. DNA Extraction and 16S rRNA Gene Sequencing
2.3. 16S rRNA Gene Sequence Processing and Statistical Analysis
3. Results
3.1. Physicochemical Characteristics of the Produced Water Samples
3.2. Microbial Compositions of the Produced Water Samples
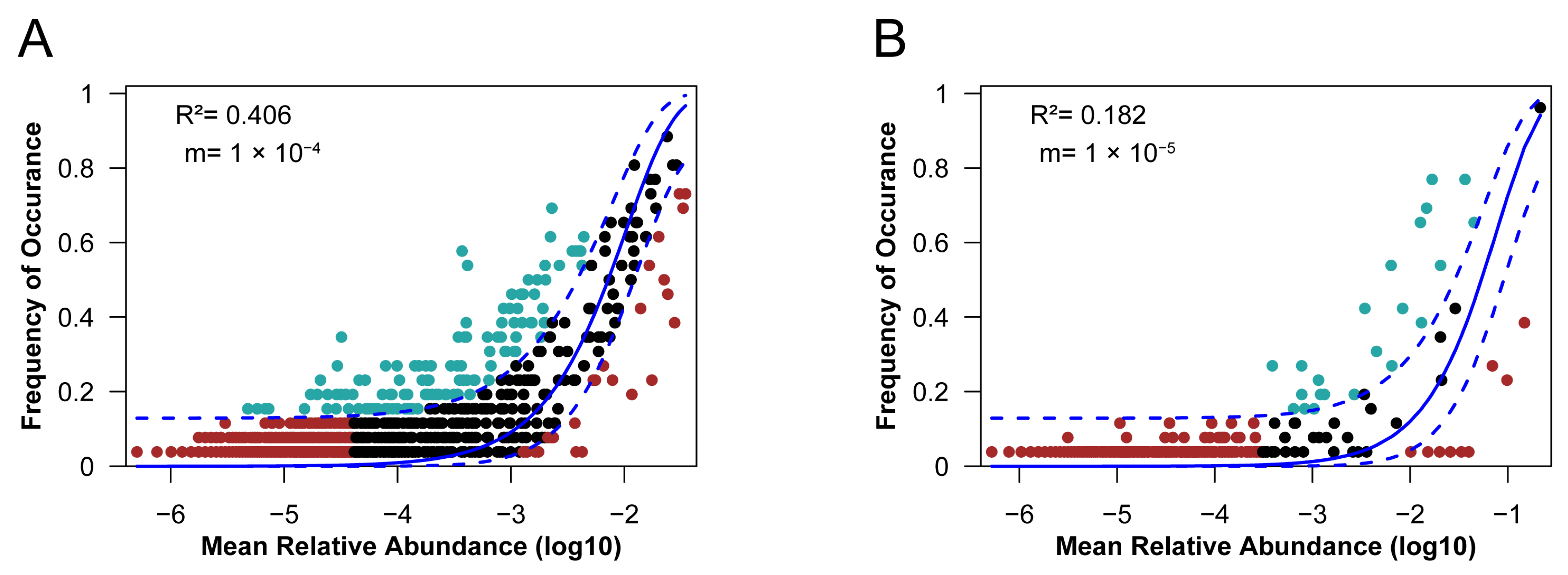
3.3. Assembly Mechanisms of Oil Reservoir Communities at a Local Geographic Scale
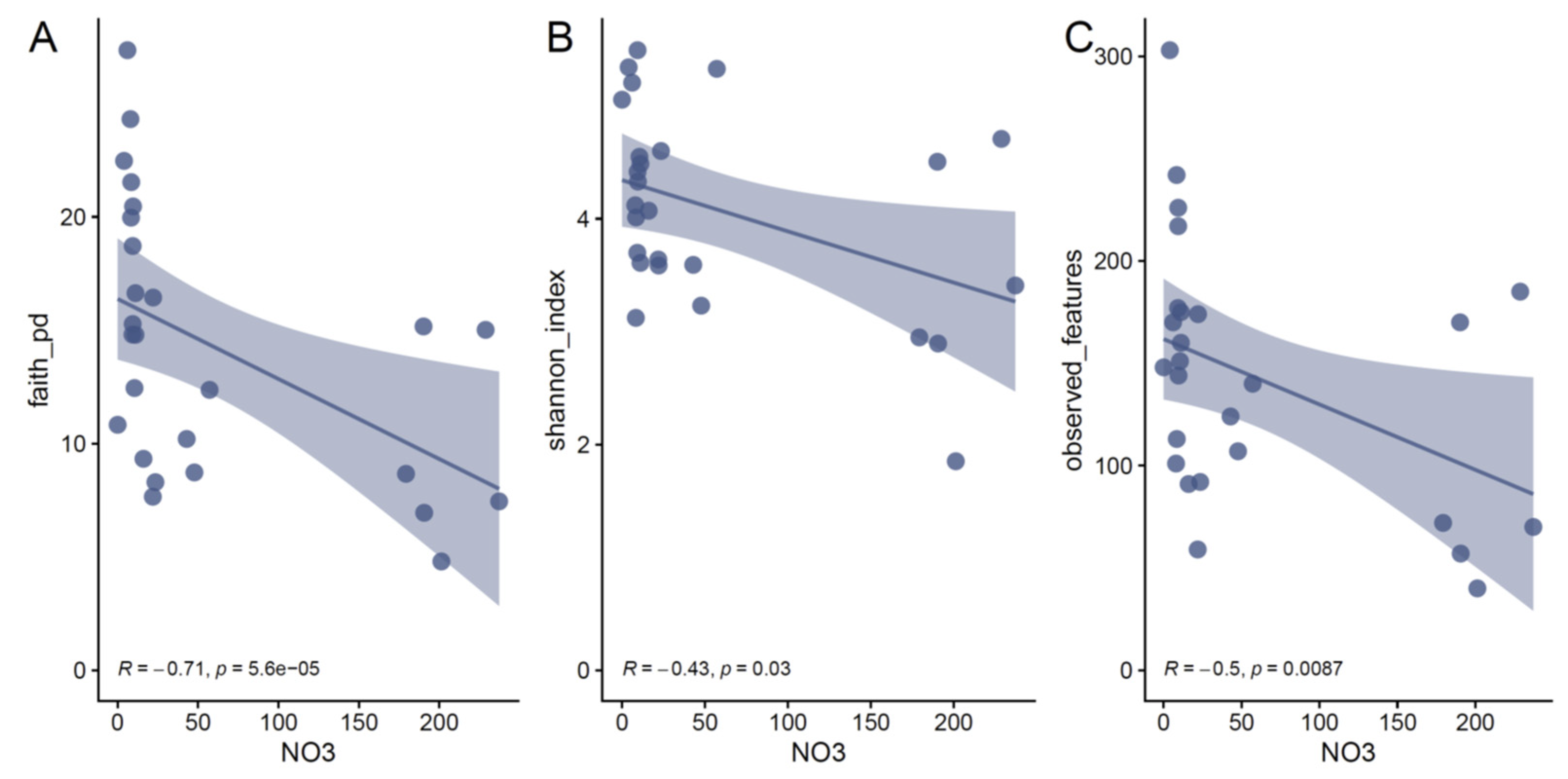
3.4. The Concentration of Nitrate Determines the Diversity of the Communities
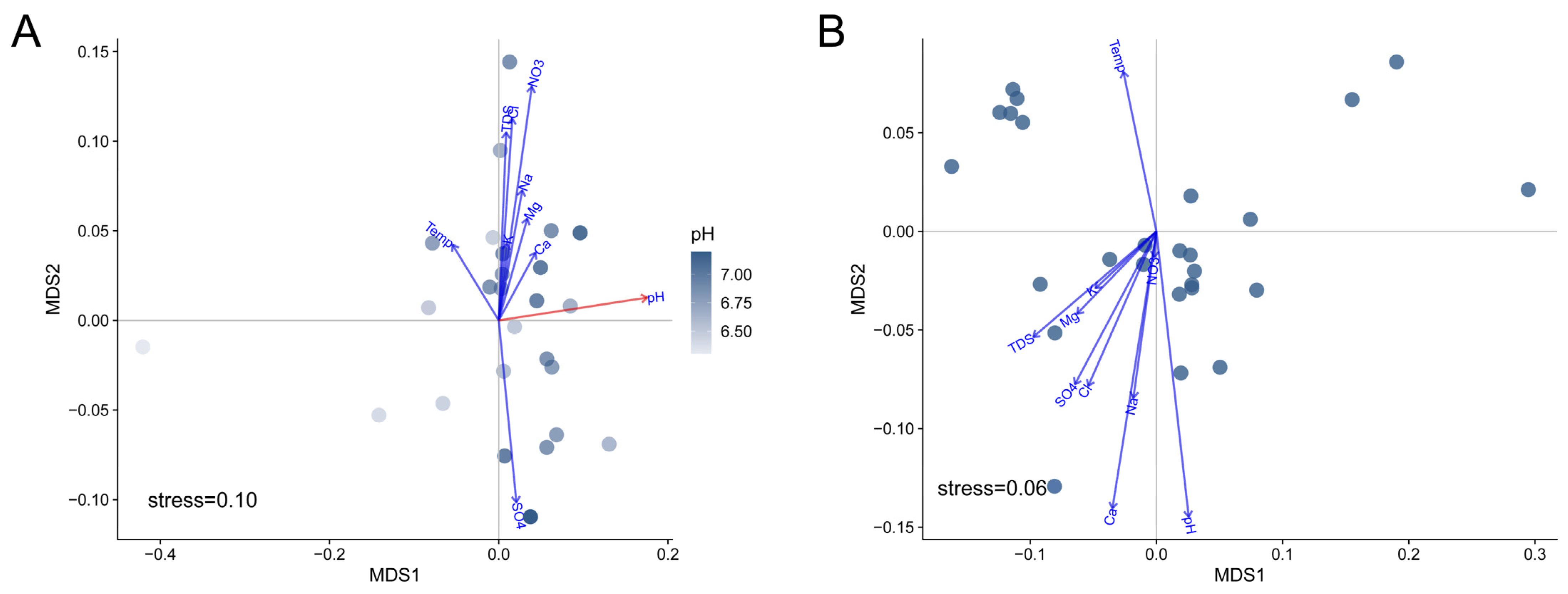
3.5. pH Drives the Microbial Compositions of the Communities
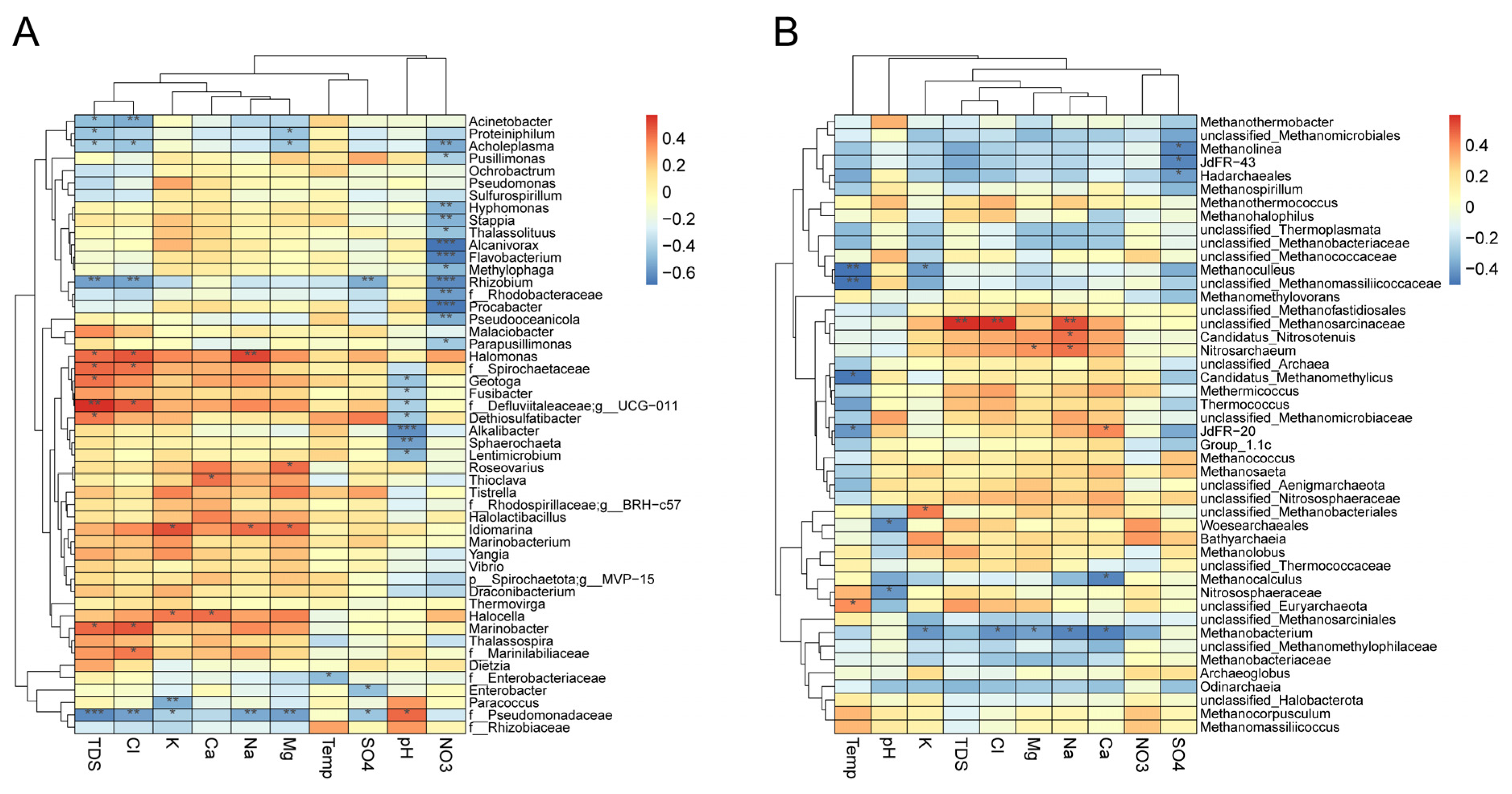
3.6. Taxonomic Association with Physicochemical Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Head, I.M.; Jones, D.M.; Larter, S.R. Biological activity in the deep subsurface and the origin of heavy oil. Nature 2003, 426, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.; Alsop, E.B.; Lomans, B.P.; Kyrpides, N.C.; Head, I.M.; Tsesmetzis, N. Succession in the petroleum reservoir microbiome through an oil field production lifecycle. ISME J. 2017, 11, 2141–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannekens, M.; Kroll, L.; Muller, H.; Mbow, F.T.; Meckenstock, R.U. Oil reservoirs, an exceptional habitat for microorganisms. New Biotechnol. 2019, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dahle, H.; Garshol, F.; Madsen, M.; Birkeland, N.K. Microbial community structure analysis of produced water from a high-temperature North Sea oil-field. Antonie Leeuwenhoek 2008, 93, 37–49. [Google Scholar] [CrossRef]
- Kaster, K.M.; Bonaunet, K.; Berland, H.; Kjeilen-Eilertsen, G.; Brakstad, O.G. Characterisation of culture-independent and -dependent microbial communities in a high-temperature offshore chalk petroleum reservoir. Antonie Leeuwenhoek 2009, 96, 423–439. [Google Scholar] [CrossRef]
- Nie, Y.; Zhao, J.Y.; Tang, Y.Q.; Guo, P.; Yang, Y.F.; Wu, X.L.; Zhao, F.Q. Species Divergence vs. Functional Convergence Characterizes Crude Oil Microbial Community Assembly. Front. Microbiol. 2016, 7, 1254. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.M.; Head, I.M.; Gray, N.D.; Adams, J.J.; Rowan, A.K.; Aitken, C.M.; Bennett, B.; Huang, H.; Brown, A.; Bowler, B.F.J.; et al. Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 2008, 451, 176–180. [Google Scholar] [CrossRef]
- Brown, L.R. Microbial enhanced oil recovery (MEOR). Curr. Opin. Microbiol. 2010, 13, 316–320. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Li, Y.; Zhao, J.Y.; Chi, C.Q.; Huang, L.X.; Dong, H.P.; Wu, X.L. Microbial Communities in Long-Term, Water-Flooded Petroleum Reservoirs with Different in situ Temperatures in the Huabei Oilfield, China. PLoS ONE 2012, 7, e33535. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, S.Z.; Mu, B.Z.; Rong, Z.F.; Zhang, J. Molecular analysis of the bacterial community in a continental high-temperature and water-flooded petroleum reservoir. FEMS Microbiol. Lett. 2006, 257, 92–98. [Google Scholar] [CrossRef]
- Lin, J.Z.; Hao, B.; Cao, G.Z.; Wang, J.; Feng, Y.; Tan, X.M.; Wang, W.D. A study on the microbial community structure in oil reservoirs developed by water flooding. J. Petrol. Sci. Eng. 2014, 122, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Li, G.Q.; Gao, P.K.; Wu, Y.Q.; Tian, H.M.; Dai, X.C.; Wang, Y.S.; Cui, Q.F.; Zhang, H.Z.; Pan, X.X.; Dong, H.P.; et al. Microbial Abundance and Community Composition Influence Production Performance in a Low-Temperature Petroleum Reservoir. Environ. Sci. Technol. 2014, 48, 5336–5344. [Google Scholar] [CrossRef] [PubMed]
- Kotlar, H.K.; Lewin, A.; Johansen, J.; Throne-Holst, M.; Haverkamp, T.; Markussen, S.; Winnberg, A.; Ringrose, P.; Aakvik, T.; Ryeng, E.; et al. High coverage sequencing of DNA from microorganisms living in an oil reservoir 2.5 kilometres subsurface. Environ. Microbiol. Rep. 2011, 3, 674–681. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Hu, B.; Dolfing, J.; Li, Y.; Tang, Y.Q.; Jiang, Y.M.; Chi, C.Q.; Xing, J.M.; Nie, Y.; Wu, X.L. Thermodynamically favorable reactions shape the archaeal community affecting bacterial community assembly in oil reservoirs. Sci. Total Environ. 2021, 781, 146506. [Google Scholar] [CrossRef]
- Lewin, A.; Johansen, J.; Wentzel, A.; Kotlar, H.K.; Drablos, F.; Valla, S. The microbial communities in two apparently physically separated deep subsurface oil reservoirs show extensive DNA sequence similarities. Environ. Microbiol. 2014, 16, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; She, Y.H.; Chai, L.J.; Banat, I.M.; Zhang, X.T.; Shu, F.C.; Wang, Z.L.; Yu, L.J.; Hou, D.J. Microbial diversity in long-term water-flooded oil reservoirs with different in situ temperatures in China. Sci. Rep. 2012, 2, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.D.; O‘Farrell, C.; Toth, C.R.A.; Montoya, O.; Gieg, L.M.; Kwon, T.H.; Yoon, S. Microbial community analyses of produced waters from high-temperature oil reservoirs reveal unexpected similarity between geographically distant oil reservoirs. Microbial. Biotechnol. 2018, 11, 788–796. [Google Scholar] [CrossRef]
- Lenchi, N.; Inceoglu, O.; Kebbouche-Gana, S.; Gana, M.L.; Lliros, M.; Servais, P.; Garcia-Armisen, T. Diversity of Microbial Communities in Production and Injection Waters of Algerian Oilfields Revealed by 16S rRNA Gene Amplicon 454 Pyrosequencing. PLoS ONE 2013, 8, e66588. [Google Scholar] [CrossRef]
- Green, J.L.; Bohannan, B.J.M.; Whitaker, R.J. Microbial biogeography: From taxonomy to traits. Science 2008, 320, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Ning, D.L.; Deng, Y.; Tiedje, J.M.; Zhou, J.Z. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [Green Version]
- Stegen, J.C.; Lin, X.J.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.Z.; Ning, D.L. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcard, D.; Legendre, P.; Drapeau, P. Partialling out the Spatial Component of Ecological Variation. Ecology 1992, 73, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Yannarell, A.C.; Triplett, E.W. Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 2005, 71, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.; Gui, Z.Y.; Chen, Y.; Tian, X.F.; Gao, P.K.; Li, G.Q.; Ma, T. Disentangling the distinct mechanisms shaping the subsurface oil reservoir bacterial and archaeal communities across northern China. Sci. Total Environ. 2021, 789, 148074. [Google Scholar] [CrossRef]
- Bell, T. Experimental tests of the bacterial distance-decay relationship. ISME J. 2010, 4, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Atekwana, E.A.; Atekwana, E.A.; Rowe, R.S.; Werkema, D.D.; Legall, F.D. The relationship of total dissolved solids measurements to bulk electrical conductivity in an aquifer contaminated with hydrocarbon. J. Appl. Geophys. 2004, 56, 281–294. [Google Scholar] [CrossRef]
- Pires, A.C.C.; Cleary, D.F.R.; Almeida, A.; Cunha, A.; Dealtry, S.; Mendonca-Hagler, L.C.S.; Smalla, K.; Gomes, N.C.M. Denaturing Gradient Gel Electrophoresis and Barcoded Pyrosequencing Reveal Unprecedented Archaeal Diversity in Mangrove Sediment and Rhizosphere Samples. Appl. Environ. Microbiol. 2012, 78, 5520–5528. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Liu, Y.P.; Pan, B.Z.; Zhu, X.Z.; Zhao, X.H.; Sun, H.; He, H.R.; Jiang, W.X. Patterns of microbial communities and their relationships with water quality in a large-scale water transfer system. J. Environ. Manag. 2022, 319, 115678. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Yang, Y.F.; Xu, Y.Q.; Zhang, J.; Lu, Y.H. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020, 14, 202–216. [Google Scholar] [CrossRef]
- Chen, W.D.; Ren, K.X.; Isabwe, A.; Chen, H.H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Gittel, A.; Sorensen, K.B.; Skovhus, T.L.; Ingvorsen, K.; Schramm, A. Prokaryotic Community Structure and Sulfate Reducer Activity in Water from High-Temperature Oil Reservoirs with and without Nitrate Treatment. Appl. Environ. Microbiol. 2009, 75, 7086–7096. [Google Scholar] [CrossRef] [Green Version]
- Schwermer, C.U.; Lavik, G.; Abed, R.M.M.; Dunsmore, B.; Ferdelman, T.G.; Stoodley, P.; Gieseke, A.; de Beer, D. Impact of nitrate on the structure and function of bacterial biofilm communities in pipelines used for injection of seawater into oil fields. Appl. Environ. Microbiol. 2008, 74, 2841–2851. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Type * | Depth (m) | Temperature (°C) | pH | TDS (mg/L) | Cl− (mg/L) | NO3− (mg/L) | SO42− (mg/L) | Na+ (mg/L) | K+ (mg/L) | Mg2+ (mg/L) | Ca2+ (mg/L) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z1 | IW | / | / | 7.42 | 800 | 204 | 31 | 146 | 9 | <1 | 4 | 5 |
| Z3 | IW | / | / | 7.74 | 533 | 1069 | 6 | 128 | 40 | <1 | 4 | 10 |
| Z4 | IW | / | / | 7.80 | 567 | 594 | 12 | 75 | 23 | <1 | 3 | 4 |
| L68-1 | PW | 1773 | 61 | 6.76 | 15,867 | 7400 | 11 | 3 | 1800 | 43 | 60 | 752 |
| L77-1 | PW | 1570 | 56 | 6.38 | 8800 | 5025 | 11 | 2 | 2684 | 30 | 68 | 522 |
| L78-1 | PW | 1707 | 60 | 6.98 | 28,467 | 9900 | 22 | 13 | 2211 | 40 | 104 | 639 |
| L79-1 | PW | 1739 | 60 | 6.30 | 36,000 | 13,920 | 22 | 64 | 2942 | 72 | 71 | 716 |
| L79-2 | PW | 1711 | 60 | 6.75 | 30,667 | 9055 | 9 | 9 | 3569 | 29 | 70 | 1164 |
| L79-3 | PW | 1505 | 54 | 6.84 | 30,167 | 14,195 | 9 | 2 | 5485 | 85 | 178 | 1767 |
| L79-4 | PW | 1700 | 60 | 6.94 | 27,333 | 10,605 | 8 | 8 | 5360 | 58 | 107 | 1146 |
| L83-1 | PW | 1519 | 54 | 6.82 | 33,733 | 17,185 | 9 | 3 | 8224 | 85 | 194 | 2206 |
| L83-2 | PW | 1654 | 58 | 6.77 | 38,967 | 19,745 | 8 | 22 | 5653 | 294 | 80 | 1037 |
| L84-1 | PW | 1563 | 55 | 6.60 | 42,567 | 19,855 | 48 | 31 | 7915 | 88 | 198 | 1739.5 |
| L84-2 | PW | 1483 | 53 | 6.86 | 42,200 | 21,325 | 10 | 21 | 10,138 | 220 | 315 | 2748.5 |
| L85-1 | PW | 1549 | 55 | 6.96 | 33,467 | 12,340 | 8 | 14 | 4693 | 70 | 145 | 1235 |
| L87-1 | PW | 1700 | 60 | 6.64 | 37,000 | 15,980 | 237 | 276 | 10,380 | 146 | 286 | 2112 |
| L87-2 | PW | 1742 | 60 | 6.66 | 50,867 | 24,250 | 179 | 36 | 9050 | 46 | 128 | 1149 |
| L87-3 | PW | 1646 | 58 | 7.07 | 40,167 | 17,470 | 191 | 18 | 6970 | 43 | 109 | 1534 |
| L90-1 | PW | 1666 | 58 | 6.73 | 33,433 | 12,950 | 190 | 54 | 6505 | 114 | 180 | 1258 |
| L90-2 | PW | 1645 | 58 | 6.45 | 89,467 | 40,960 | 229 | 53 | 20,450 | 199 | 690 | 3469 |
| L90-3 | PW | 1807 | 62 | 6.84 | 32,667 | 14,980 | 201 | 30 | 6975 | 105 | 196 | 1345 |
| L1 | PW | 1773 | 62 | 6.48 | 58,967 | 24,650 | 57. | 3 | 10,598 | 111 | 281 | 3436 |
| L2 | PW | 1581 | 55 | 6.83 | 57,000 | 25,050 | 43 | 254 | 12,580 | 113 | 333 | 4010 |
| L3 | PW | 1802 | 62 | 6.43 | 23,533 | 10,150 | 4 | 8 | 4544 | 114 | 118 | 1151 |
| L4 | PW | 1566 | 56 | 6.56 | 48,100 | 16,350 | 6 | 19 | 6650 | 92 | 189 | 1969 |
| L5 | PW | 1590 | 57 | 6.82 | 39,733 | 19,295 | 0 | 5 | 21,370 | 85 | 253 | 2007 |
| L6 | PW | 1699 | 59 | 7.20 | 12,833 | 4997.5 | 10 | 768 | 6798 | 74 | 211 | 1817 |
| L7 | PW | 1781 | 61 | 6.50 | 47,333 | 18,670 | 23 | 513 | 3526 | 84 | 204 | 444 |
| L8 | PW | 1513 | 53 | 6.76 | 36,000 | 12,540 | 16 | 11 | 2096 | 33 | 61 | 575 |
| Factors | Mantel r Statistic | p-Value | Tail Type |
|---|---|---|---|
| Bacteria | |||
| Temp | −0.0100 | 0.521 | two-sided |
| pH | 0.2876 | 0.005 | two-sided |
| TDS | 0.0351 | 0.364 | two-sided |
| Cl− | 0.0098 | 0.440 | two-sided |
| NO3− | −0.0564 | 0.648 | two-sided |
| SO42− | −0.0376 | 0.581 | two-sided |
| Na+ | −0.1168 | 0.846 | two-sided |
| K+ | −0.1374 | 0.889 | two-sided |
| Mg2+ | −0.1239 | 0.883 | two-sided |
| Ca2+ | −0.1316 | 0.888 | two-sided |
| Archaea | |||
| Temp | 0.0511 | 0.228 | two-sided |
| pH | 0.0629 | 0.239 | two-sided |
| TDS | 0.1313 | 0.133 | two-sided |
| Cl− | 0.0724 | 0.225 | two-sided |
| NO3− | 0.0974 | 0.179 | two-sided |
| SO42− | 0.0621 | 0.287 | two-sided |
| Na+ | −0.0272 | 0.560 | two-sided |
| K+ | −0.0885 | 0.792 | two-sided |
| Mg2+ | −0.0269 | 0.569 | two-sided |
| Ca2+ | −0.0098 | 0.487 | two-sided |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wang, J.; Liu, Q.; Zhang, Q.; Wu, J.; Zhou, M.; Nie, Y.; Wu, X.-L. pH and Nitrate Drive Bacterial Diversity in Oil Reservoirs at a Localized Geographic Scale. Microorganisms 2023, 11, 151. https://doi.org/10.3390/microorganisms11010151
Xu Y, Wang J, Liu Q, Zhang Q, Wu J, Zhou M, Nie Y, Wu X-L. pH and Nitrate Drive Bacterial Diversity in Oil Reservoirs at a Localized Geographic Scale. Microorganisms. 2023; 11(1):151. https://doi.org/10.3390/microorganisms11010151
Chicago/Turabian StyleXu, Ying, Jianwei Wang, Qingjie Liu, Qun Zhang, Jiazhong Wu, Minghui Zhou, Yong Nie, and Xiao-Lei Wu. 2023. "pH and Nitrate Drive Bacterial Diversity in Oil Reservoirs at a Localized Geographic Scale" Microorganisms 11, no. 1: 151. https://doi.org/10.3390/microorganisms11010151