
Comparative De Novo and Pan-Genome Analysis of MDR Nosocomial Bacteria Isolated from Hospitals in Jeddah, Saudi Arabia

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approvement
2.2. Clinical Specimens
2.3. Isolation and Identification of MDR Bacteria
2.4. Amplification of 16S rRNA Gene, Sequencing, and Phylogenetic Analysis
2.5. Whole Genome Sequencing and Analysis Workflow
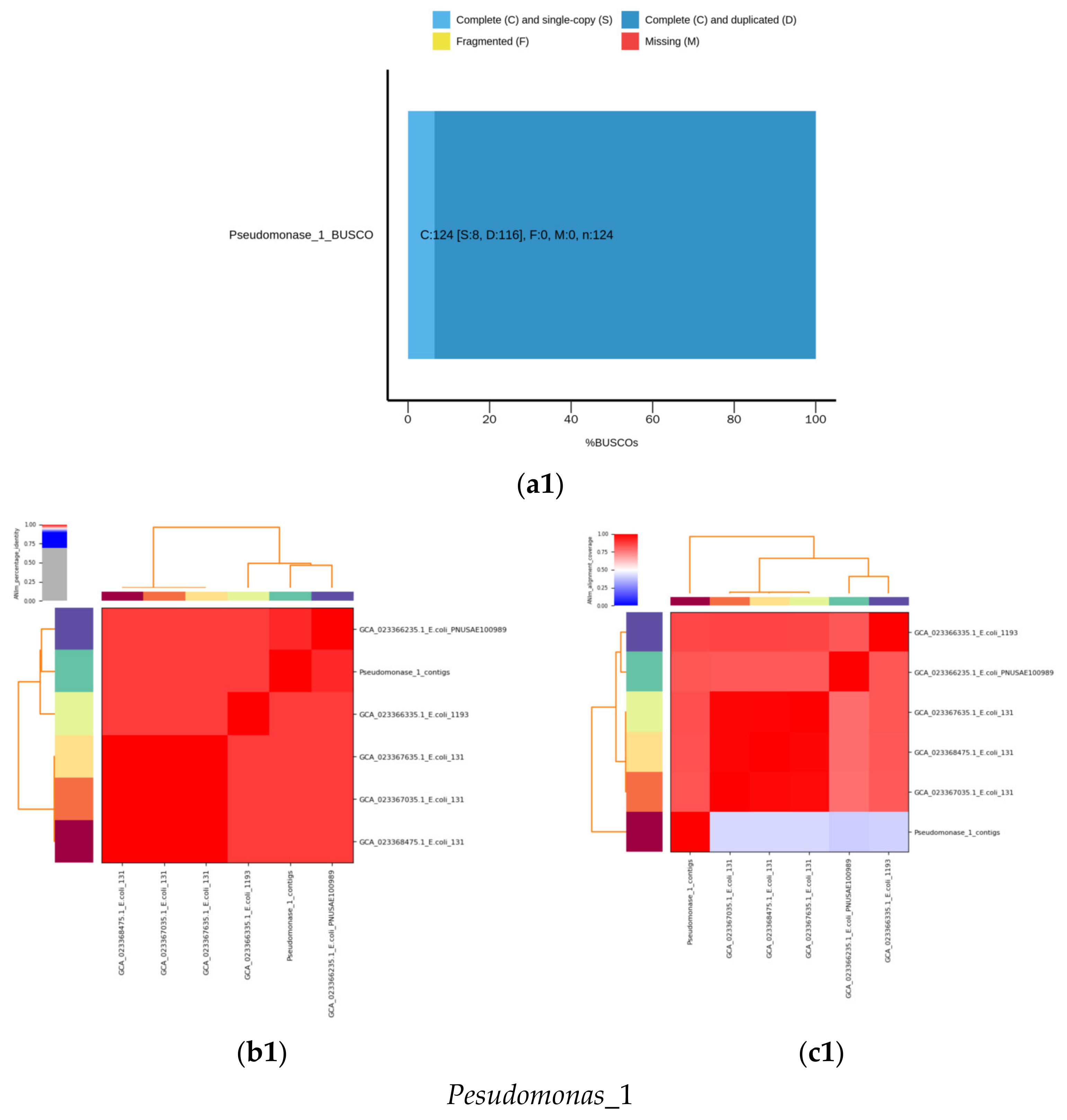
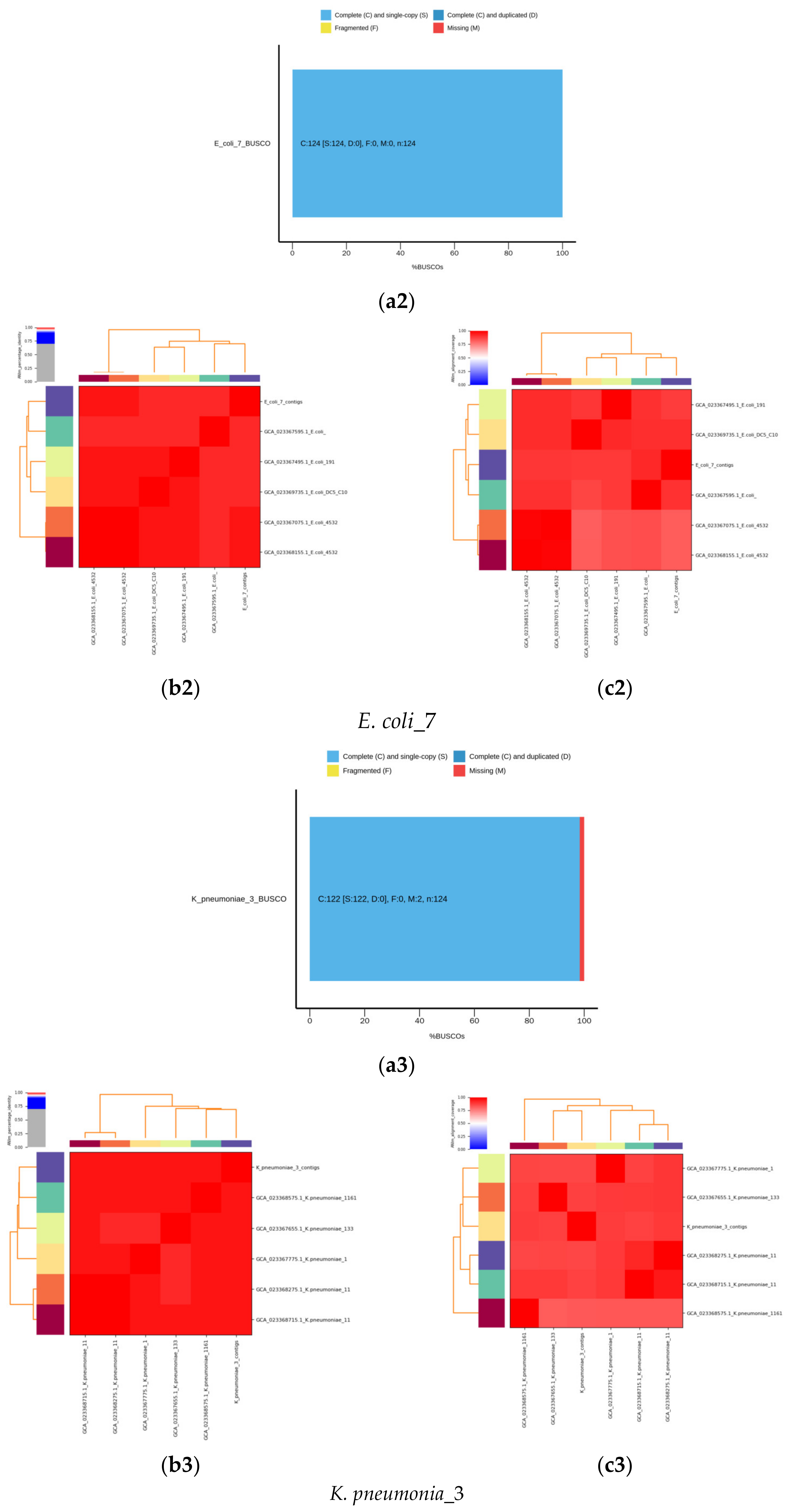
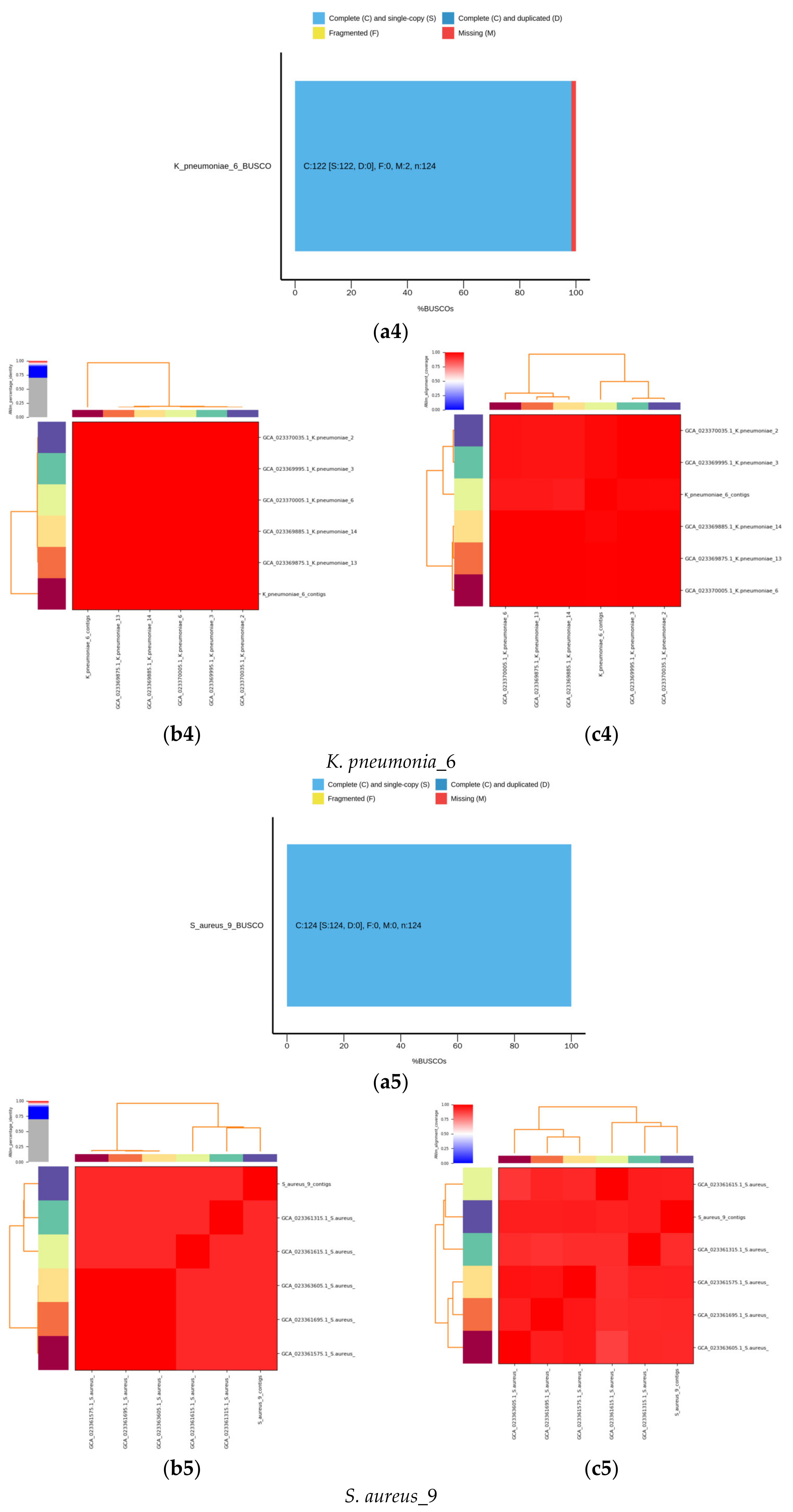
2.6. De Novo Assembly of MDR Bacterial Sequences
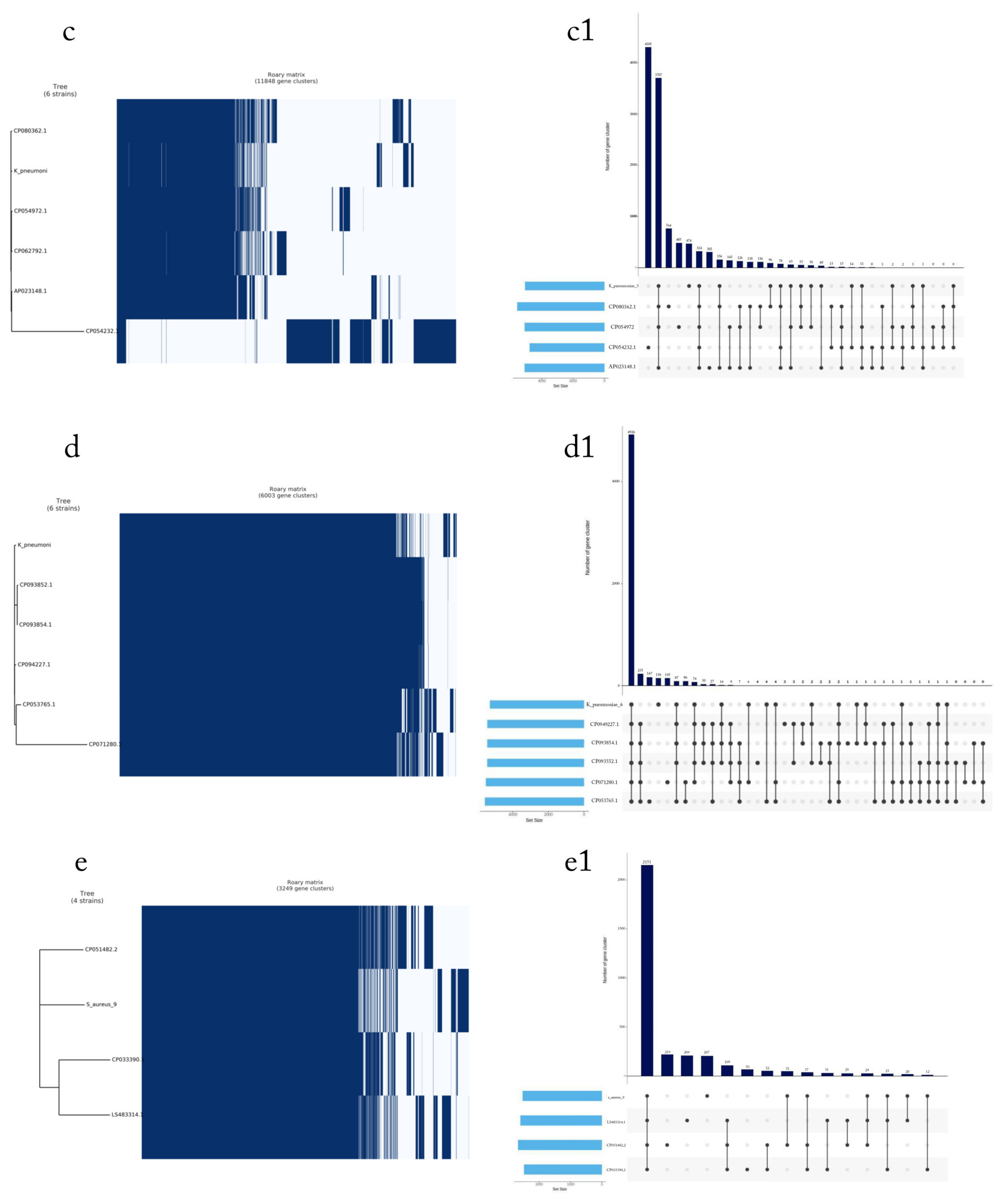
2.7. Pan-Genome Analysis
2.7.1. Calculation of Mash Distances and ANI
2.7.2. Profiling of Virulence and Antibiotic Resistance Genes
3. Statistical Analysis
4. Results
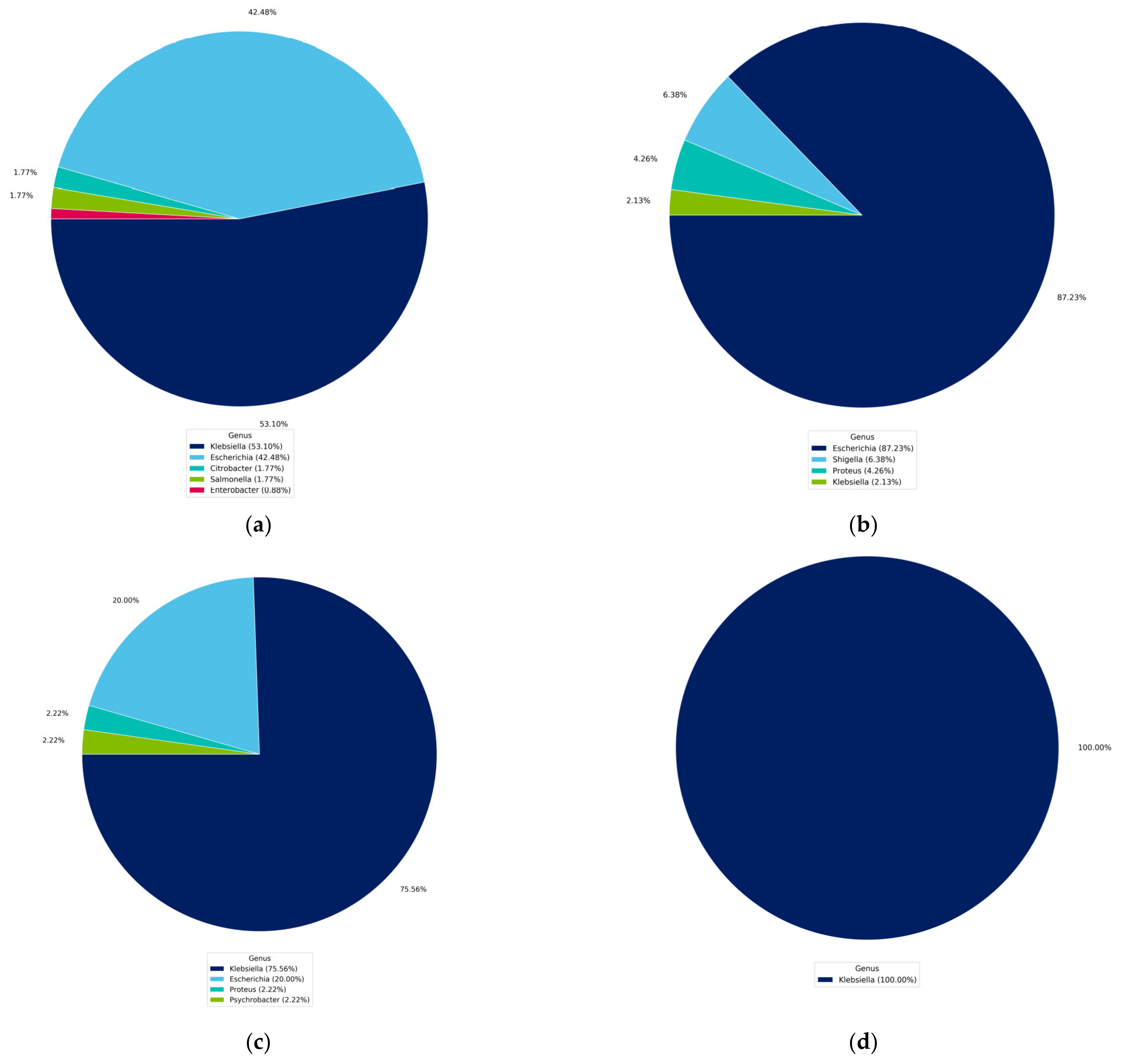
4.1. Identification of Isolated Bacteria
4.2. Identification Based on 16S rRNA Gene and Whole Genome Sequencing
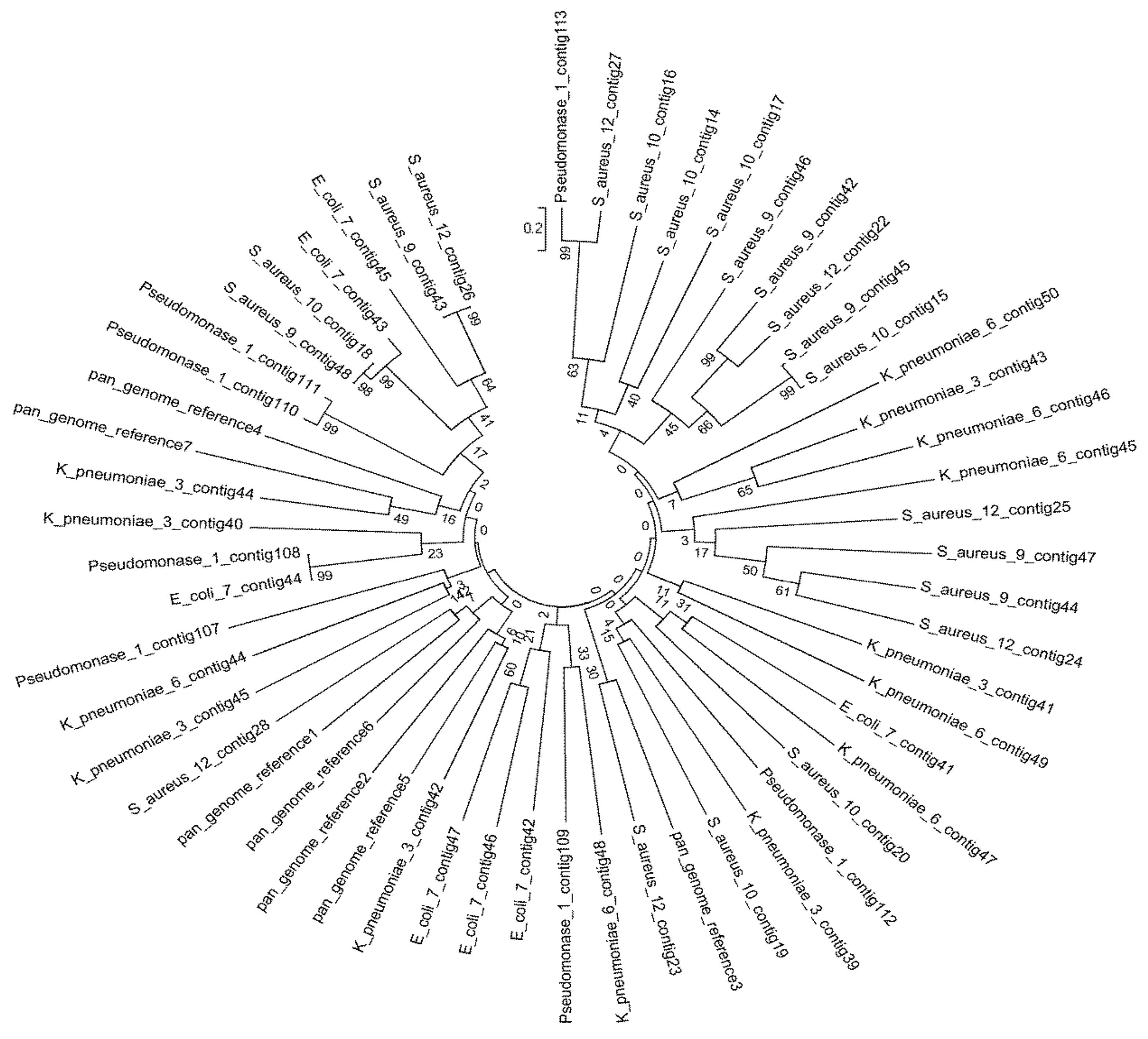
4.3. Phylogenetic Tree of the Seven Selected MDR Bacteria
4.4. Detection of MDR Genes
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bentivegna, E.; Luciani, M.; Arcari, L.; Santino, I.; Simmaco, M.; Martelletti, P. Reduction of multidrug-resistant (MDR) bacterial infections during the COVID-19 pandemic: A retrospective study. Int. J. Environ. Res. Public Health 2021, 18, 1003. [Google Scholar] [CrossRef]
- Terreni, M.; Taccani, M.; Pregnolato, M. New antibiotics for multidrug-resistant bacterial strains: Latest research developments and future perspectives. Molecules 2021, 26, 2671. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Antimicrobial Resistance. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 November 2021).
- Falagas, M.E.; Tansarli, G.S.; Karageorgopoulos, D.E.; Vardakas, K.Z. Deaths attributable to carbapenem-resistant Enterobacteriaceae infections. Emerg. Infect. Dis. 2014, 20, 1170–1175. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin. Infect. Dis. 2019, 69 (Suppl. 7), S538–S543. [Google Scholar] [CrossRef]
- Pandey, N.; Cascella, M. Beta Lactam Antibiotics. StatPearls. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545311/ (accessed on 26 September 2022).
- Formosa-Dague, C.; Speziale, P.; Foster, T.J.; Geoghegan, J.A.; Dufrêne, Y.F. Zinc-dependent mechanical properties of Staphylococcus aureus biofilm-forming surface protein SasG. Proc. Natl. Acad. Sci. USA 2016, 113, 410–415. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Hackel, M.A.; Tsuji, M.; Yamano, Y.; Echols, R.; Sahm, D.F. In vitro activity of cefiderocol, a siderophore cephalosporin, against Gram-negative bacilli isolated by clinical laboratories in North America and Europe in 2015–2016: SIDERO-WT-2015. Int. J. Antimicrob. Agents 2019, 53, 456–466. [Google Scholar] [CrossRef]
- Bukhari, S.A.R.; Irfan, M.; Ahmad, I.; Chen, L. Comparative genomics and pan-genome driven prediction of a reduced genome of Akkermansia muciniphila. Microorganisms 2022, 10, 1350. [Google Scholar] [CrossRef] [PubMed]
- Hassoun-Kheir, N.; Stabholz, Y.; Kreft, J.U.; de la Cruz, R.; Romalde, J.L.; Nesme, J.; Sørensen, S.J.; Smets, B.F.; Graham, D.; Paul, M. Comparison of antibiotic-resistant bacteria and antibiotic resistance genes abundance in hospital and community wastewater: A systematic review. Sci. Total Environ. 2020, 743, 140804. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, E.; Baricz, A.; Chiriac, C.M.; Farkas, A.; Opris, O.; Soran, M.L.; Andrei, A.S.; Rudi, K.; Balcázar, J.L.; Dragos, N.; et al. Abundance of antibiotics, antibiotic resistance genes and bacterial community composition in wastewater effluents from different Romanian hospitals. Environ. Pollut. 2017, 225, 304–315. [Google Scholar] [PubMed]
- The Pew Charitable Trust Tracking the Global Pipeline of Antibiotics in Development. March 2021. Available online: https://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2021/03/tracking-the-global-pipeline-of-antibiotics-in[1]development (accessed on 1 May 2021).
- World Health Organization. Meeting Report of the World Health Organization Expert Consultation on the Definition of Extensively Drug-Resistant Tuberculosis, 27–29 October 2020; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Zowawi, H.M. Antimicrobial resistance in Saudi Arabia: An urgent call for an immediate action. Saudi Med. J. 2016, 37, 935–940. [Google Scholar] [CrossRef]
- Enani, M.A.; Alzahrani, R.M.; Alzubaidy, K.A.; Ajjaj, R.G.; Saeed, A.M. The prevalence and characters of hospital acquired infections in three private hospital, Jeddah, Saudi Arabia. Int. J. Adv. Res. 2019, 7, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Halwani, M.A.; Tashkandy, N.A.J.; Aly, M.M.; Al Masoudi, S.B.; Dhafar, O.O. Incidence of antibiotic resistance bacteria in Jeddah’s Ministry of Health Hospitals, Saudi Arabia. Adv. Microbiol. 2015, 5, 780–786. [Google Scholar] [CrossRef]
- Helmi, N.R.; Zaman, R.M.; Aly, M.M. Prevalence of Gram-positive bacteria in Jeddah, Kingdom of Saudi Arabia: Study of antimicrobial resistance patterns and molecular typing. Int. J. Pharm. Biol. Sci. 2013, 4, 1231. [Google Scholar]
- Qiagen. Dneasy Tissue Handbook; Qiagen: Hilden, Germany, 2006. [Google Scholar]
- Sakdapetsiri, C.; Ngaemthao, W.; Suriyachadkun, C.; Pinyakong, O. Paeniglutamicibacter quisquiliarum sp. nov., isolated from midden soil waste. Int. J. Sys. Evo. Micr. 2022, 72, 005651. [Google Scholar] [CrossRef]
- Guillen, I.A.; Camacho, H.; Tuero, A.D.; Bacardí, D.; Palenzuela, D.O.; Aguilera, A.; Silva, J.A.; Estrada, R.; Gell, O.; Suárez, J.; et al. PCR conditions for 16S primers for analysis of microbes in the colon of rats. J. Biomol. Technol. 2016, 27, 105–112. [Google Scholar] [CrossRef]
- Ekblom, R.; Wolf, J.B. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 2014, 7, 1026–1042. [Google Scholar] [CrossRef]
- Peker, N.; Garcia-Croes, S.; Dijkhuizen, B.; Wiersma, H.H.; Van Zanten, E.; Wisselink, G.; Friedrich, A.W.; Kooistra-Smid, M.; Sinha, B.; Rossen, J.W.A.; et al. A comparison of three different bioinformatics analyses of the 16S–23S rRNA encoding region for bacterial identification. Front. Microbiol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. In pheatmap: Pretty Heatmaps version 1.0.12 from CRAN; For. Rev. Version 1.0.10; 2018; Available online: Rdrr.io (accessed on 12 May 2022).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010; Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 22 May 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Jaén-Luchoro, D.; Kahnamouei, A.; Yazdanshenas, S.; Lindblom, A.; Samuelsson, E.; Åhrén, C.; Karami, N. Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing, E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections. Microorganisms 2023, 11, 1622. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Elankumaran, P.; Kudinha, T.; Kidsley, A.K.; Trott, D.J.; Jarocki, V.M.; Djordjevic, S.P. Dominance of Escherichia coli sequence types ST73, ST95, ST127 and ST131 in Australian urine isolates: A genomic analysis of antimicrobial resistance and virulence linked to F plasmids. Microb. Genom. 2023, 9, 001068. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Polo, J.A.; Hernández-García, M.; Morosini, M.I.; Pérez-Viso, B.; Soriano, C.; De Pablo, R.; Cantón, R.; Ruiz-Garbajosa, P. Outbreak by KPC-62-producing ST307 Klebsiella pneumoniae isolates resistant to ceftazidime/avibactam and cefiderocol in a university hospital in Madrid, Spain. J. Antimicrob. Chemother. 2023, 78, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.H.; Yeo, C.C.; Che Hamzah, A.M.; Al-Trad, E.A.; Jones, S.U.; Chua, K.H.; Puah, S.M. Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus Associated with Hospitalized Newborn Infants. Diagnostics 2023, 13, 1050. [Google Scholar] [CrossRef]
- Krüger, H.; Ji, X.; Hanke, D.; Schink, A.K.; Fiedler, S.; Kaspar, H.; Wang, Y.; Schwarz, S.; Wu, C.; Feßler, A.T. Novel macrolide-lincosamide-streptogramin B resistance gene erm (54). Int. J. Antimicrob. Chemother. 2022, 77, 2296–2298. [Google Scholar] [CrossRef]
- Szymanek-Majchrzak, K.; Młynarczyk, G. Genomic Insights of first erm B-positive ST338-SCC mec VT/CC59 Taiwan clone of community-associated methicillin-resistant Staphylococcus aureus in Poland. Int. J. Mol. Sci. 2022, 23, 8755. [Google Scholar] [CrossRef]
- Mahesh, S.; Kalyan, R.; Gupta, P.; Veram, S.; Venkatesh, V.; Trİpathİ, P. MecA and ermA gene discrepancy from their phenotypic profile in Staphylococcus aureus isolates. J. Microbiol. Infect. Dis. 2022, 12, 6–11. [Google Scholar] [CrossRef]
- Suma, T.A.; Alam, N.; Raihan, S.Z.; Zahid, M.A.; Mandal, S.C.; Suchana, F.J.; Kundu, R.; Hossain, A.; Muhit, M.A. Association of antibacterial susceptibility profile with the prevalence of genes encoding efflux proteins in the Bangladeshi clinical isolates of Staphylococcus aureus. Antibiotics 2023, 12, 305. [Google Scholar] [CrossRef]
- Medis, S.; Dissanayake, T.; Weerasekera, M.; Namali, D.; Gunasekara, S.; Kottahachchi, J. Distribution of mecA and qacA/B genes among coagulase negative staphylococci isolated from central venous catheters of intensive care unit patients. Indian J. Med. Microbiol. 2022, 40, 505–509. [Google Scholar] [CrossRef]
- Mohamad Farook, N.A.; Argimón, S.; Abdul Samat, M.N.; Salleh, S.A.; Sulaiman, S.; Tan, T.L.; Periyasamy, P.; Lau, C.L.; Ismail, Z.; Muhammad Azami, N.A.; et al. Diversity and dissemination of methicillin-resistant Staphylococcus aureus (MRSA) genotypes in Southeast Asia. Trop. Med. Infect. Dis. 2022, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kashikar, A.; Bush, K. In vitro activity of plazomicin against β-lactamase-producing carbapenem-resistant Enterobacteriaceae (CRE). J. Antimicrob. Chemother. 2017, 72, 2792–2795. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In vitro antibacterial properties of cefiderocol, a novel siderophore cephalosporin, against Gram-negative bacteria. Antimicrob. Agents Chemother. 2018, 62, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Behroozynia, M. Knowledge, attitude and practice of intensive care unit nurses about prevention and control of nosocomial infections. Sci. J. Iranshahr Univ. Med. Sci. 2022, 1, 11–15. [Google Scholar]
- Burnham, J.P.; Feldman, M.F.; Calix, J.J. Seasonal changes in the prevalence of antibiotic-susceptible Acinetobacter calcoaceticus-baumannii complex isolates result in increased multidrug resistance rates during winter months. Open Forum Infect. Dis. 2019, 6, ofz245. [Google Scholar] [CrossRef]
- Schwab, F.; Gastmeier, P.; Hoffmann, P.; Meyer, E. Summer, sun and sepsis—The influence of outside temperature on nosocomial bloodstream infections: A cohort study and review of the literature. PLoS ONE 2020, 15, e0234656. [Google Scholar] [CrossRef]
- Yamaguto, G.E.; Zhen, F.; Moreira, M.M.; Montesanti, B.M.; Raboni, S.M. Community respiratory viruses and healthcare-associated infections: Epidemiological and clinical aspects. J. Hosp. Infect. 2022, 122, 187–193. [Google Scholar] [CrossRef]
- Zhou, Z.C.; Liu, Y.; Lin, Z.J.; Shuai, X.Y.; Zhu, L.; Xu, L.; Meng, L.X.; Sun, Y.J.; Chen, H. Spread of antibiotic resistance genes and microbiota in airborne particulate matter, dust, and human airways in the urban hospital. Environ. Int. 2021, 153, 106501. [Google Scholar] [CrossRef]
- Adalbert, J.R.; Varshney, K.; Tobin, R.; Pajaro, R. Clinical outcomes in patients co-infected with COVID-19 and Staphylococcus aureus: A scoping review. BMC Infect. Dis. 2021, 21, 985. [Google Scholar] [CrossRef]
- Hsu, B.M.; Chen, J.S.; Lin, I.C.; Hsu, G.J.; Koner, S.; Hussain, B.; Huang, S.W.; Tsai, H.C. Molecular and antimicrobial Re-sistance (AMR) profiling of methicillin-Resistant Staphylococcus aureus (MRSA) from hospital and long-term care facilities (LTCF) environment. Antibiotics 2021, 10, 748. [Google Scholar] [CrossRef]
- Yu, C.H.; Shen, S.; Huang, K.-Y.A.; Huang, Y.C. The trend of environmental and clinical methicillin-resistant Staphylococcus aureus in a hospital in Taiwan: Impact of USA300. J. Microbiol. Immunol. Infect. 2022, 55, 241. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Khadka, S.; Sapkota, S.; Rana, J.C.; Khanal, S.; Neupane, A.; Sharma, B. Prevalence and antibiograms of uropathogens from the suspected cases of urinary tract infections in Bharatpur Hospital, Nepal. J. Coll. Med. Sci. 2019, 15, 260–266. [Google Scholar] [CrossRef]
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-resistant bacteria and alternative methods to control them: An overview. Microb. Drug Resist. 2019, 25, 890–908. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, N.; Zhou, C.; Lin, Y.; Zhang, Y.; Wang, L.; Zheng, X.; Zhou, T.; Wang, Z. Molecular mechanisms and epidemiology of carbapenem-resistant Enterobacter cloacae complex isolated from Chinese patients during 2004–2018. Infect. Drug Resist. 2021, 14, 3647–3658. [Google Scholar] [CrossRef]
- Pot, M.; Reynaud, Y.; Couvin, D.; Ducat, C.; Ferdinand, S.; Gravey, F.; Gruel, G.; Guérin, F.; Malpote, E.; Breurec, S.; et al. Wide distribution and specific resistance pattern to third-generation cephalosporins of Enterobacter cloacae complex members in humans and in the environment in Guadeloupe (French West Indies). Front. Microbiol. 2021, 12, 628058. [Google Scholar] [CrossRef]
- Manandhar, S.; Nguyen, Q.; Nguyen Thi Nguyen, T.; Pham, D.T.; Rabaa, M.A.; Dongol, S.; Basnyat, B.; Dixit, S.M.; Baker, S.; Karkey, A. Genomic epidemiology, antimicrobial resistance and virulence factors of Enterobacter cloacae complex causing potential community-onset bloodstream infections in a tertiary care hospital of Nepal. JAC Antimicrob Resist. 2022, 4, dlac050. [Google Scholar] [CrossRef]
- Baiou, A.; Elbuzidi, A.A.; Bakdach, D.; Zaqout, A.; Alarbi, K.M.; Bintaher, A.A.; Ali, M.M.B.; Elarabi, A.M.; Ali, G.A.M.; Daghfal, J.; et al. Clinical characteristics and risk factors for the isolation of multi-drug-resistant Gram-negative bacteria from critically ill patients with COVID-19. J. Hosp. Infect. 2021, 110, 165–171. [Google Scholar] [CrossRef]
- Derakhshan, S.; Navidinia, M.; Haghi, F. Antibiotic susceptibility of human-associated Staphylococcus aureus and its relation to agr typing, virulence genes, and biofilm formation. BMC Infect. Dis. 2021, 21, 627. [Google Scholar] [CrossRef]
- Miklasińska-Majdanik, M. Mechanisms of resistance to macrolide antibiotics among Staphylococcus aureus. Antibiotics 2021, 10, 1406. [Google Scholar] [CrossRef]
- Nji, E.; Kazibwe, J.; Hambridge, T.; Joko, C.A.; Larbi, A.A.; Damptey, L.A.O.; Nkansa-Gyamfi, N.A.; Stålsby Lundborg, C.S.; Lien, T.Q. High prevalence of antibiotic resistance in commensal Escherichia coli from healthy human sources in community settings. Sci. Rep. 2021, 11, 3372. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, Z.; Xia, B.; Zhang, Y.; Liu, X.; Yu, Y.; Tang, N.; Tong, X.; Wang, M.; Ye, X.; et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol. 2022, 40, 921. [Google Scholar] [CrossRef] [PubMed]
- Nasrin, S.; Hegerle, N.; Sen, S.; Nkeze, J.; Sen, S.; Permala-Booth, J.; Choi, M.; Sinclair, J.; Tapia, M.D.; Johnson, J.K.; et al. Distribution of serotypes and antibiotic resistance of invasive Pseudomonas aeruginosa in a multi-country collection. BMC Microbiol. 2022, 22, 13. [Google Scholar] [CrossRef]
- Hwang, W.; Yoon, S.S. Virulence characteristics and an action mode of antibiotic resistance in multidrug-resistant Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.O.E.; Campolina, G.A.; Batista, L.R.; Alves, E.; Caetano, A.R.S.; Brandão, R.M.; Nelson, D.L.; Cardoso, M.D.G. Mechanism of action of various terpenes and phenylpropanoids against Escherichia coli and Staphylococcus aureus. FEMS Microbiol. Lett. 2021, 368, fnab052. [Google Scholar] [CrossRef]
- Rahman, M.A.; Amirkhani, A.; Chowdhury, D.; Mempin, M.; Molloy, M.P.; Deva, A.K.; Vickery, K.; Hu, H. Proteome of Staphylococcus aureus biofilm changes significantly with aging. Int. J. Mol. Sci. 2022, 23, 6415. [Google Scholar] [CrossRef]
- Tu, I.F.; Lin, T.L.; Yang, F.L.; Lee, I.M.; Tu, W.L.; Liao, J.H.; Ko, T.P.; Wu, W.J.; Jan, J.T.; Ho, M.R.; et al. Structural and biological insights into Klebsiella pneumoniae surface polysaccharide degradation by a bacteriophage K1 lyase: Implications for clinical use. J. Biomed. Sci. 2022, 29, 9. [Google Scholar] [CrossRef]
- Fesseha, H.; Mathewos, M.; Aliye, S.; Mekonnen, E. Isolation and antibiogram of Escherichia coli O157: H7 from diarrhoeic calves in urban and peri-urban dairy farms of Hawassa town. Vet. Med. Sci. 2022, 8, 864–876. [Google Scholar] [CrossRef]
- Gebremedhin, E.Z.; Merga, D.; Sarba, E.J.; Marami, L.M.; Tola, G.K.; Endale, S.S. Prevalence, risk factors and antibiogram of Escherichia coli isolated from dogs in Ambo, Gojo and Bako towns of Oromia region, Ethiopia. Ethiop. Vet. J. 2021, 25, 1–22. [Google Scholar] [CrossRef]
- Mapipa, Q.; Digban, T.O.; Nnolim, N.E.; Nwodo, U.U. Antibiogram profile and virulence signatures of pseudomonas aeruginosa isolates recovered from selected agrestic hospital effluents. Sci. Rep. 2021, 11, 11800. [Google Scholar] [CrossRef]
- KuKanich, K.S.; Bagladi-Swanson, M.; KuKanich, B. Pseudomonas aeruginosa susceptibility, antibiogram and clinical interpretation, and antimicrobial prescribing behaviors for dogs with otitis in the Midwestern United States. J. Vet. Pharmacol. Ther. 2022, 45, 440–449. [Google Scholar] [CrossRef]
- Founou, L.L.; Amoako, D.G.; Founou, R.C.; Essack, S.Y. Antibiotic resistance in food animals in Africa: A systematic review and meta-analysis. Microb. Drug Resist. 2018, 24, 648–665. [Google Scholar] [CrossRef]
- Cazer, C.L.; Westblade, L.F.; Simon, M.S.; Magleby, R.; Castanheira, M.; Booth, J.G.; Jenkins, S.G.; Gröhn, Y.T. Analysis of multidrug resistance in Staphylococcus aureus with a machine learning-generated antibiogram. Antimicrob. Agents Chemother. 2021, 65, e02132-20. [Google Scholar] [CrossRef]
- Roth, B.M.; Laps, A.; Yamba, K.; Heil, E.L.; Johnson, J.K.; Stafford, K.; Hachaambwa, L.M.; Kalumbi, M.; Mulenga, L.; Patel, D.M.; et al. Antibiogram development in the setting of a high frequency of multi-drug resistant organisms at University Teaching Hospital, Lusaka, Zambia. Antibiotics 2021, 10, 782. [Google Scholar] [CrossRef]
- Baron, S.A.; Diene, S.M.; Rolain, J.-M. Human microbiomes and antibiotic resistance. Hum. Microbiome J. 2018, 10, 43–52. [Google Scholar] [CrossRef]
- Alemayehu, T. Prevalence of multidrug-resistant bacteria in Ethiopia: A systematic review and meta-analysis. J. Glob. Antimicrob. Resist. 2021, 26, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- Ito-Horiyama, T.; Ishii, Y.; Ito, A.; Sato, T.; Nakamura, R.; Fukuhara, N.; Tsuji, M.; Yamano, Y.; Yamaguchi, K.; Tateda, K. Stability of novel siderophore cephalosporin S-649266 against clinically relevant carbapenemases. Antimicrob. Agen. chemoth. 2016, 60, 4384–4386. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, D.; Chetri, S.; Das, B.J.; Dhar Chanda, D.D.; Bhattacharjee, A. Distribution of virulence genes and SCCmec types among methicillin-resistant Staphylococcus aureus of clinical and environmental origin: A study from community of Assam, India. BMC Res. Notes 2021, 14, 58. [Google Scholar] [CrossRef]
- de Souza, G.H.A.; Dos Santos Radai, J.A.; Mattos Vaz, M.S.; Esther da Silva, K.; Fraga, T.L.; Barbosa, L.S.; Simionatto, S. In vitro and in vivo antibacterial activity assays of carvacrol: A candidate for development of innovative treatments against KPC-producing Klebsiella pneumoniae. PLoS ONE 2021, 16, e0246003. [Google Scholar] [CrossRef]
- Meirelles, L.A.; Perry, E.K.; Bergkessel, M.; Newman, D.K. Bacterial defenses against a natural antibiotic promote collateral resilience to clinical antibiotics. PLOS Biol. 2021, 19, e3001093. [Google Scholar] [CrossRef]
- Roemhild, R.; Bollenbach, T.; Andersson, D.I. The physiology and genetics of bacterial responses to antibiotic combinations. Nat. Rev. Microbiol. 2022, 20, 478. [Google Scholar] [CrossRef] [PubMed]
- Bojar, B.; Sheridan, J.; Beattie, R.; Cahak, C.; Liedhegner, E.; Munoz-Price, L.S.; Hristova, K.R.; Skwor, T. Antibiotic resistance patterns of Escherichia coli isolates from the clinic through the wastewater pathway. Int. J. Hyg. Environ. Health 2021, 238, 113863. [Google Scholar] [CrossRef] [PubMed]
- Langendonk, R.F.; Neill, D.R.; Fothergill, J.L. The building blocks of antimicrobial resistance in Pseudomonas aeruginosa: Implications for current resistance-breaking therapies. Front. Cell. Infect. Microbiol. 2021, 11, 665759. [Google Scholar] [CrossRef] [PubMed]
- Sharahi, J.Y.; Hashemi, A.; Ardebili, A.; Davoudabadi, S. Molecular characteristics of antibiotic-resistant Escherichia coli and Klebsiella pneumoniae strains isolated from hospitalized patients in Tehran, Iran. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 32. [Google Scholar] [CrossRef]
- Haeili, M.; Shoghi, Y.; Moghimi, M.; Ghodousi, A.; Omrani, M.; Cirillo, D.M. Genomic features of in vitro selected mutants of Escherichia coli with decreased susceptibility to tigecycline. J. Glob. Antimicrob. Resist. 2022, 31, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Van An, N.; Hoang, L.H.; Le, H.H.; Thai Son, N.; Hong, L.T.; Viet, T.T.; Le, T.D.; Thang, T.B.; Vu, L.H.; Nguyen, V.T.; et al. Distribution and antibiotic resistance characteristics of bacteria isolated from blood culture in a teaching hospital in Vietnam during 2014–2021. Infec. Drug Resist. 2023, 16, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, C.; Sørum, V.; Tokuriki, N.; Johnsen, P.J.; Samuelsen, Ø. Evolution of β-lactamase-mediated cefiderocol resistance. J. Antimicrob. Chemoth. 2022, 77, 2429–2436. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Index 5 | Index 7 |
|---|---|---|
| Pseudomonas_1 | CTAGGCAA | CTCACCAA |
| K_pneumoniae_3 | CTGTATTA | ATATGGAT |
| K_pneumoniae_6 | TCACGCCG | GCGCAAGC |
| E_coli_7 | ACTTACAT | AAGATACT |
| S_aureus_9 | GTCCGTGC | GGAGCGTC |
| S_aureus_10 | AAGGTACC | ATGGCATG |
| S_aureus_12 | GGAACGTT | GCAATGCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghamdi, M.; Al-Judaibi, E.; Al-Rashede, M.; Al-Judaibi, A. Comparative De Novo and Pan-Genome Analysis of MDR Nosocomial Bacteria Isolated from Hospitals in Jeddah, Saudi Arabia. Microorganisms 2023, 11, 2432. https://doi.org/10.3390/microorganisms11102432
Alghamdi M, Al-Judaibi E, Al-Rashede M, Al-Judaibi A. Comparative De Novo and Pan-Genome Analysis of MDR Nosocomial Bacteria Isolated from Hospitals in Jeddah, Saudi Arabia. Microorganisms. 2023; 11(10):2432. https://doi.org/10.3390/microorganisms11102432
Chicago/Turabian StyleAlghamdi, Molook, Effat Al-Judaibi, Mohammed Al-Rashede, and Awatif Al-Judaibi. 2023. "Comparative De Novo and Pan-Genome Analysis of MDR Nosocomial Bacteria Isolated from Hospitals in Jeddah, Saudi Arabia" Microorganisms 11, no. 10: 2432. https://doi.org/10.3390/microorganisms11102432