
Development of a Real-Time Quantitative PCR Based on a TaqMan-MGB Probe for the Rapid Detection of Theileria haneyi

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
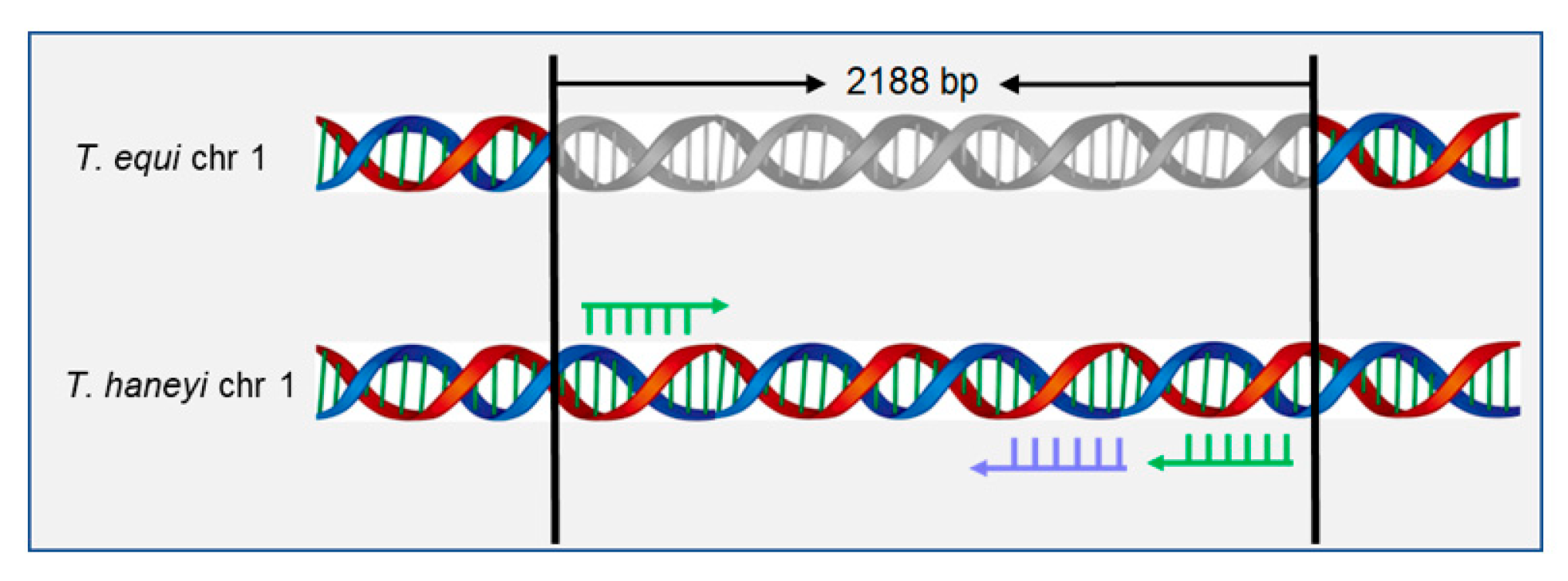
2.2. Selection for Specific Primers and Probes
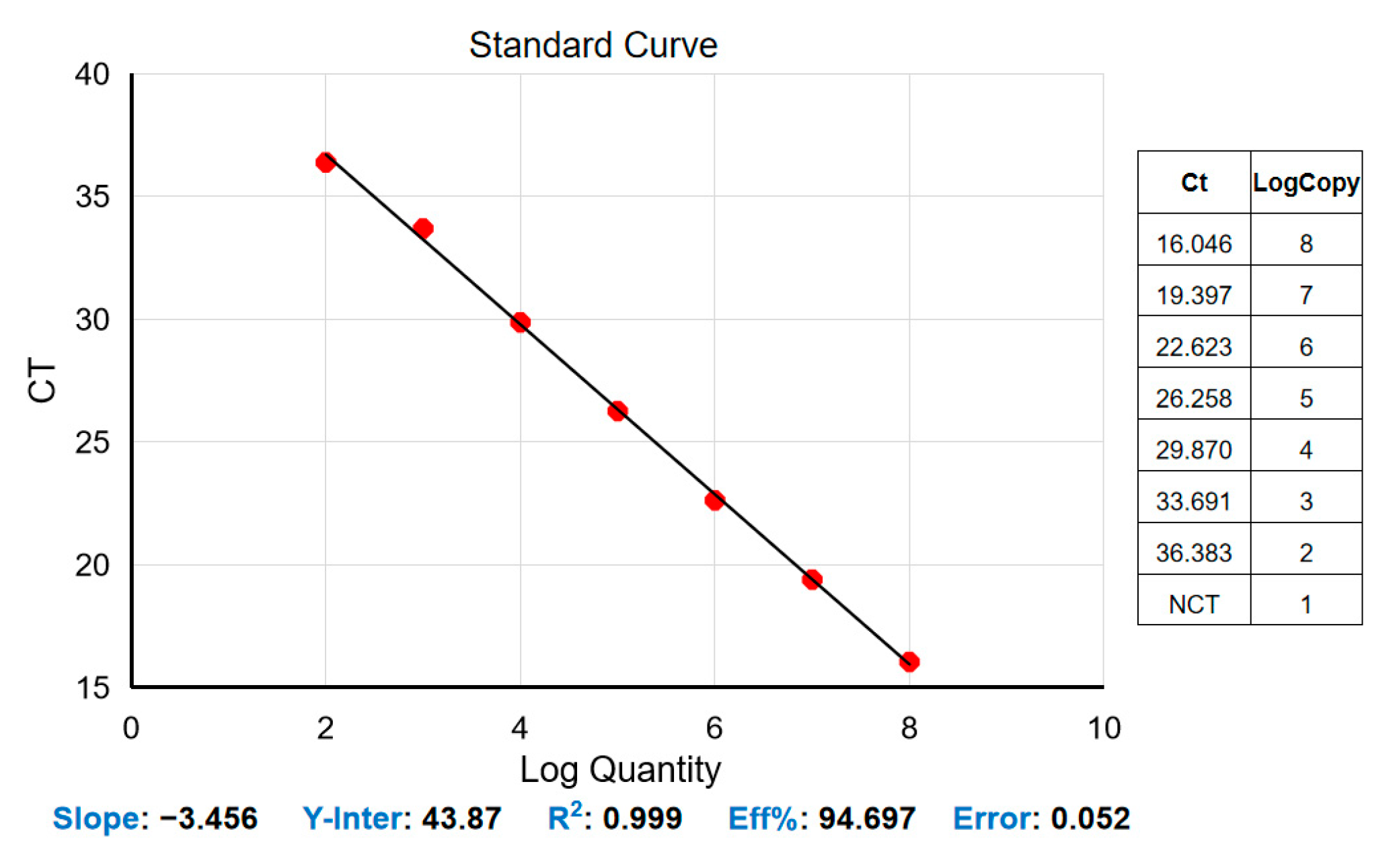
2.3. Plasmid Construction and Standard Curve Generation
2.4. Real-Time Quantitative PCR
2.5. Nested PCR Amplification of the Chr1sco of T. haneyi
2.6. Sensitivity and Specificity
3. Results
3.1. Standard Curve for the Real-Time Quantitative PCR Assay
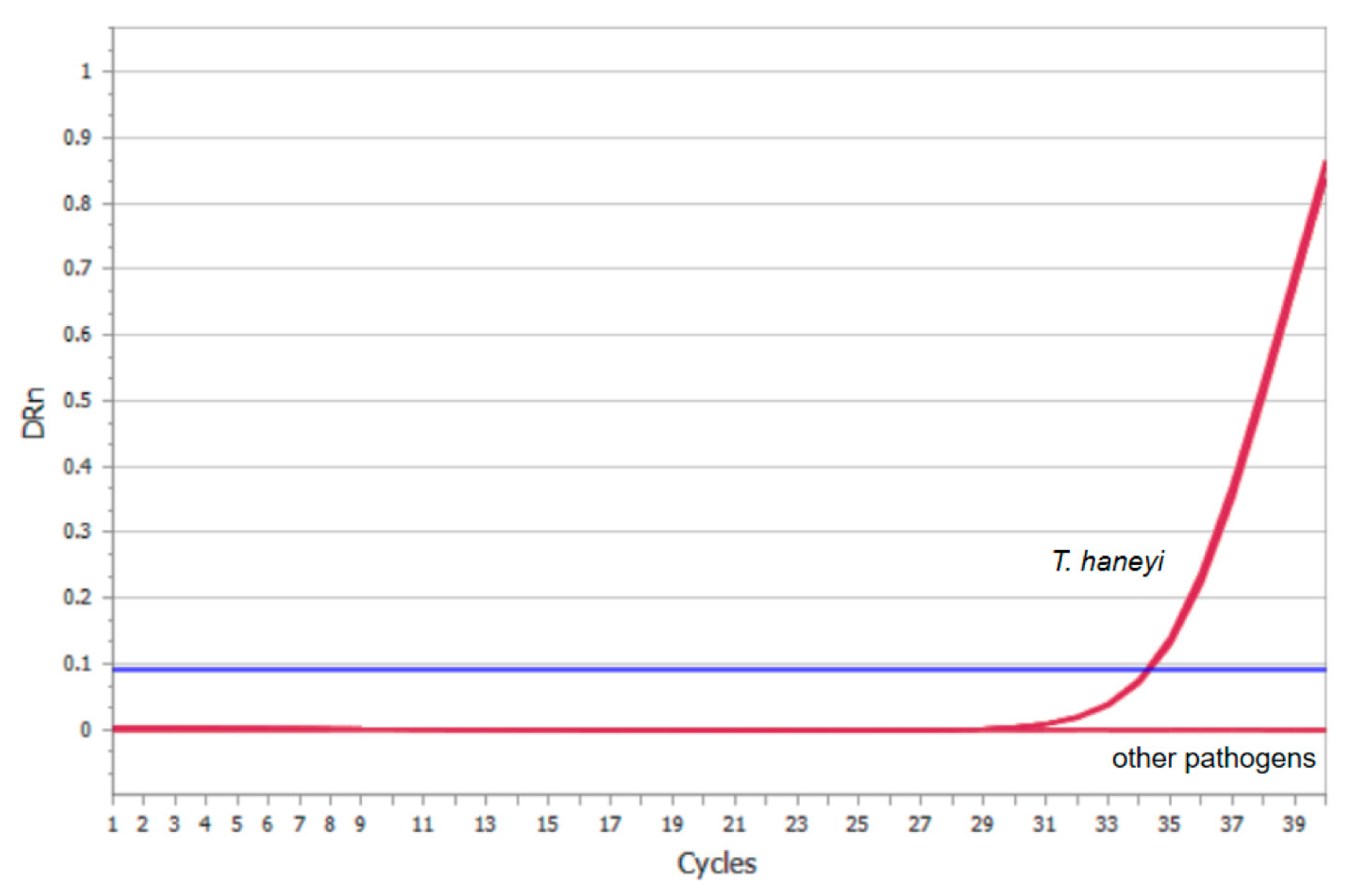
3.2. Analytical Sensitivity of the Real-Time Quantitative PCR Assay
3.3. Specificity of the Real-Time Quantitative PCR Assay
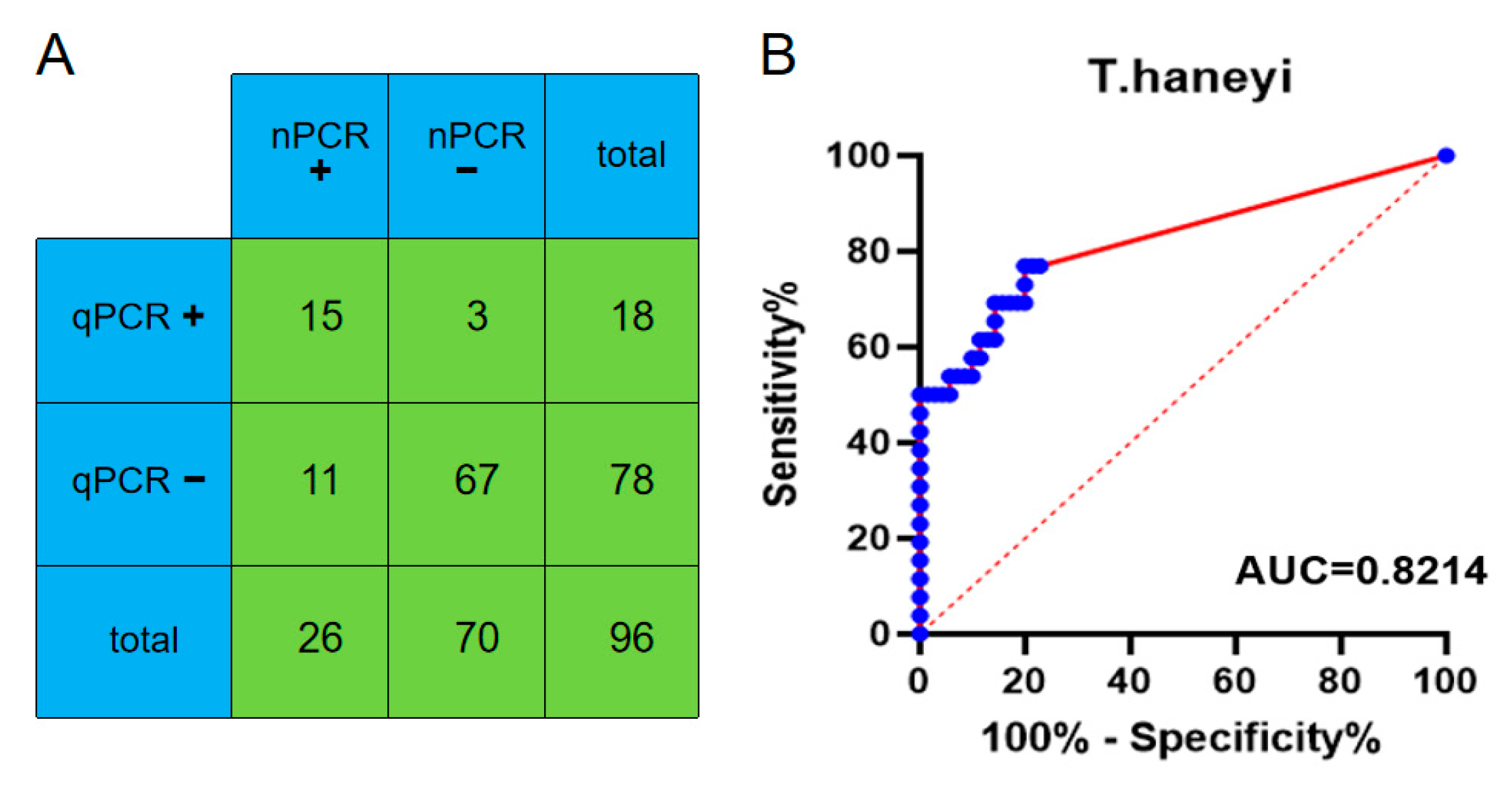
3.4. Clinical Performance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wise, L.N.; Kappmeyer, L.S.; Mealey, R.H.; Knowles, D.P. Review of equine piroplasmosis. J. Vet. Intern. Med. 2013, 27, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.N.; Pelzel-McCluskey, A.M.; Mealey, R.H.; Knowles, D.P. Equine piroplasmosis. Vet. Clin. N. Am. Equine Pract. 2014, 30, 677–693. [Google Scholar] [CrossRef] [PubMed]
- Scoles, G.A.; Ueti, M.W. Vector ecology of equine piroplasmosis. Annu. Rev. Entomol. 2015, 60, 561–580. [Google Scholar] [CrossRef]
- Giubega, S.; Ilie, M.S.; Luca, I.; Florea, T.; Dreghiciu, C.; Oprescu, I.; Morariu, S.; Dărăbuș, G. Seroprevalence of Anti-Theileria equi Antibodies in Horses from Three Geographically Distinct Areas of Romania. Pathogens 2022, 11, 669. [Google Scholar] [CrossRef]
- Camino, E.; de la Cruz, M.L.; Dominguez, L.; Carvajal, K.A.; Fores, P.; de Juan, L.; Cruz-Lopez, F. Epidemiological Situation of the Exposure to Agents Causing Equine Piroplasmosis in Spanish Purebred Horses in Spain: Seroprevalence and Associated Risk Factors. J. Equine Vet. Sci. 2018, 67, 81–86. [Google Scholar] [CrossRef]
- Onyiche, T.E.; Taioe, M.O.; Molefe, N.I.; Biu, A.A.; Luka, J.; Omeh, I.J.; Yokoyama, N.; Thekisoe, O. Equine piroplasmosis: An insight into global exposure of equids from 1990 to 2019 by systematic review and meta-analysis. Parasitology 2020, 147, 1411–1424. [Google Scholar] [CrossRef]
- Zobba, R.; Ardu, M.; Niccolini, S.; Chessa, B.; Manna, L.; Cocco, R.; Pinna Parpaglia, M.L. Clinical and Laboratory Findings in Equine Piroplasmosis. J. Equine Vet. Sci. 2008, 28, 301–308. [Google Scholar] [CrossRef]
- Almazán, C.; Scimeca, R.C.; Reichard, M.V.; Mosqueda, J. Babesiosis and Theileriosis in North America. Pathogens 2022, 11, 168. [Google Scholar] [CrossRef]
- Allsopp, M.T.; Cavalier-Smith, T.; De Waal, D.T.; Allsopp, B.A. Phylogeny and evolution of the piroplasms. Parasitology 1994, 108 Pt 2, 147–152. [Google Scholar] [CrossRef]
- Knowles, D.P.; Kappmeyer, L.S.; Haney, D.; Herndon, D.R.; Fry, L.M.; Munro, J.B.; Sears, K.; Ueti, M.W.; Wise, L.N.; Silva, M.; et al. Discovery of a novel species, Theileria haneyi n. sp., infective to equids, highlights exceptional genomic diversity within the genus Theileria: Implications for apicomplexan parasite surveillance. Int. J. Parasitol. 2018, 48, 679–690. [Google Scholar] [CrossRef]
- Cunha, C.W.; Kappmeyer, L.S.; McGuire, T.C.; Dellagostin, O.A.; Knowles, D.P. Conformational dependence and conservation of an immunodominant epitope within the babesia equi erythrocyte-stage surface protein equi merozoite antigen 1. Clin. Diagn. Lab. Immunol. 2002, 9, 1301–1306. [Google Scholar] [CrossRef]
- Ueti, M.W.; Palmer, G.H.; Kappmeyer, L.S.; Scoles, G.A.; Knowles, D.P. Expression of equi merozoite antigen 2 during development of Babesia equi in the midgut and salivary gland of the vector tick Boophilus microplus. J. Clin. Microbiol. 2003, 41, 5803–5809. [Google Scholar] [CrossRef]
- Bastos, R.G.; Sears, K.P.; Dinkel, K.D.; Kappmeyer, L.; Ueti, M.W.; Knowles, D.P.; Fry, L.M. Development of an Indirect ELISA to Detect Equine Antibodies to Theileria haneyi. Pathogens 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Coultous, R.M.; McDonald, M.; Raftery, A.G.; Shiels, B.R.; Sutton, D.G.M.; Weir, W. Analysis of Theileria equi diversity in The Gambia using a novel genotyping method. Transbound. Emerg. Dis. 2020, 67, 1213–1221. [Google Scholar] [CrossRef]
- Mshelia, P.W.; Kappmeyer, L.; Johnson, W.C.; Kudi, C.A.; Oluyinka, O.O.; Balogun, E.O.; Richard, E.E.; Onoja, E.; Sears, K.P.; Ueti, M.W. Molecular detection of Theileria species and Babesia caballi from horses in Nigeria. Parasitol. Res. 2020, 119, 2955–2963. [Google Scholar] [CrossRef]
- Bhoora, R.V.; Collins, N.E.; Schnittger, L.; Troskie, C.; Marumo, R.; Labuschagne, K.; Smith, R.M.; Dalton, D.L.; Mbizeni, S. Molecular genotyping and epidemiology of equine piroplasmids in South Africa. Ticks Tick-Borne Dis. 2020, 11, 101358. [Google Scholar] [CrossRef]
- Navarro, E.; Serrano-Heras, G.; Castaño, M.J.; Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta 2015, 439, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Chen, K.; Hu, Z.; Li, J.; Wang, J.; Liu, D.; Qi, T.; Guo, W.; Du, C.; Wang, X. Prevalence and molecular epidemiology of equine piroplasmosis in China: A neglected tick-borne disease. Sci. China Life Sci. 2021, 65, 445–447. [Google Scholar] [CrossRef]
- Whelan, J.A.; Russell, N.B.; Whelan, M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods 2003, 278, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.P.; Kappmeyer, L.S.; Onzere, C.K.; Odongo, D.O.; Githaka, N.; Sears, K.P.; Knowles, D.P.; Fry, L.M. Equid infective Theileria cluster in distinct 18S rRNA gene clades comprising multiple taxa with unusually broad mammalian host ranges. Parasit. Vectors 2020, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Sears, K.; Knowles, D.; Dinkel, K.; Mshelia, P.W.; Onzere, C.; Silva, M.; Fry, L. Imidocarb Dipropionate Lacks Efficacy against Theileria haneyi and Fails to Consistently Clear Theileria equi in Horses Co-Infected with T. haneyi. Pathogens 2020, 9, 1035. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Criado-Fornelio, A.; Martinez-Marcos, A.; Buling-Sarana, A.; Barba-Carretero, J.C. Molecular studies on Babesia, Theileria and Hepatozoon in Southern Europe. Part I. Epizootiological aspects. Vet. Parasitol. 2003, 113, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.S.; Bashiruddin, J.B.; Alexander, D.J. Deduced amino acid sequences at the fusion protein cleavage site of Newcastle disease viruses showing variation in antigenicity and pathogenicity. Arch. Virol. 1993, 128, 363–370. [Google Scholar] [CrossRef]
- Valera, M.J.; Torija, M.J.; Mas, A.; Mateo, E. Acetobacter malorum and Acetobacter cerevisiae identification and quantification by Real-Time PCR with TaqMan-MGB probes. Food Microbiol. 2013, 36, 30–39. [Google Scholar] [CrossRef]
- Watzinger, F.; Ebner, K.; Lion, T. Detection and monitoring of virus infections by real-time PCR. Mol. Asp. Med. 2006, 27, 254–298. [Google Scholar] [CrossRef]
- Kutyavin, I.V.; Afonina, I.A.; Mills, A.; Gorn, V.V.; Lukhtanov, E.A.; Belousov, E.S.; Singer, M.J.; Walburger, D.K.; Lokhov, S.G.; Gall, A.A.; et al. 3′-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 2000, 28, 655–661. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, S.; Qin, H.; Meng, S.; Deng, X.; Lin, H.; Xin, X.; Liang, Y.; Chen, B.; Cui, Y.; et al. Establishment of a quantitative RT-PCR detection of SARS-CoV-2 virus. Eur. J. Med. Res. 2021, 26, 147. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequences (5′-3′) | Products |
|---|---|---|
| T. haneyi-F: | AATCCAAAACCAGCT | 97 bp |
| T. haneyi-R: | GTACAAATCTCCCTAGAG | |
| T. haneyi-Probe: | FAM-CCTCCAAGTCGTCGT-MGB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, B.; Yang, G.; Hu, Z.; Chen, K.; Guo, W.; Wang, X.; Du, C. Development of a Real-Time Quantitative PCR Based on a TaqMan-MGB Probe for the Rapid Detection of Theileria haneyi. Microorganisms 2023, 11, 2633. https://doi.org/10.3390/microorganisms11112633
Zhou B, Yang G, Hu Z, Chen K, Guo W, Wang X, Du C. Development of a Real-Time Quantitative PCR Based on a TaqMan-MGB Probe for the Rapid Detection of Theileria haneyi. Microorganisms. 2023; 11(11):2633. https://doi.org/10.3390/microorganisms11112633
Chicago/Turabian StyleZhou, Bingqian, Guangpu Yang, Zhe Hu, Kewei Chen, Wei Guo, Xiaojun Wang, and Cheng Du. 2023. "Development of a Real-Time Quantitative PCR Based on a TaqMan-MGB Probe for the Rapid Detection of Theileria haneyi" Microorganisms 11, no. 11: 2633. https://doi.org/10.3390/microorganisms11112633