
In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of Target Sequences
2.2. Analysis of Physicochemical Properties of the Proteins
2.3. Signal Peptide Prediction and Subcellular Localization Identification
2.4. Modeling of 3D Protein Structures and its Evaluation
2.5. Functional and Structural Annotations of HBFII Proteins
2.6. Sequence Alignment and Evolutionary Analysis
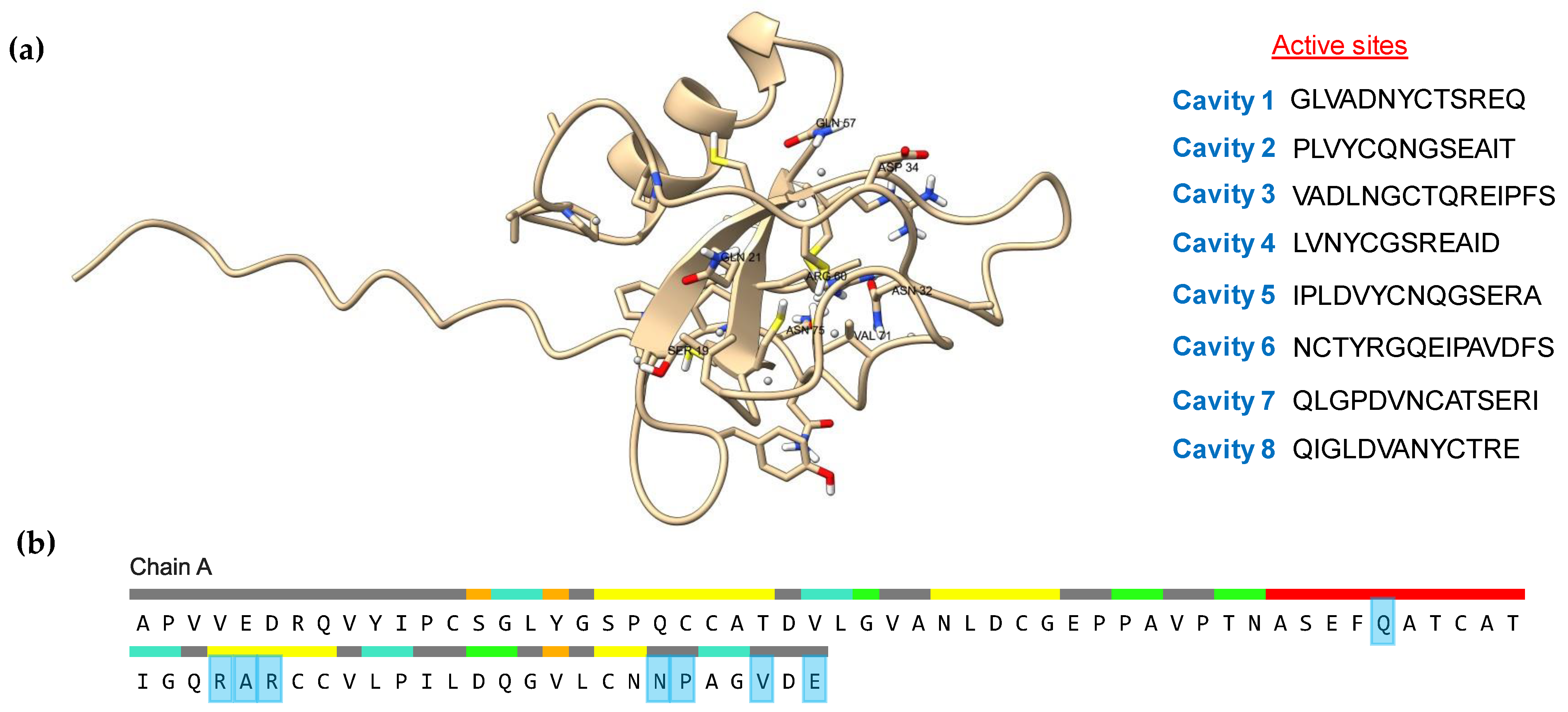
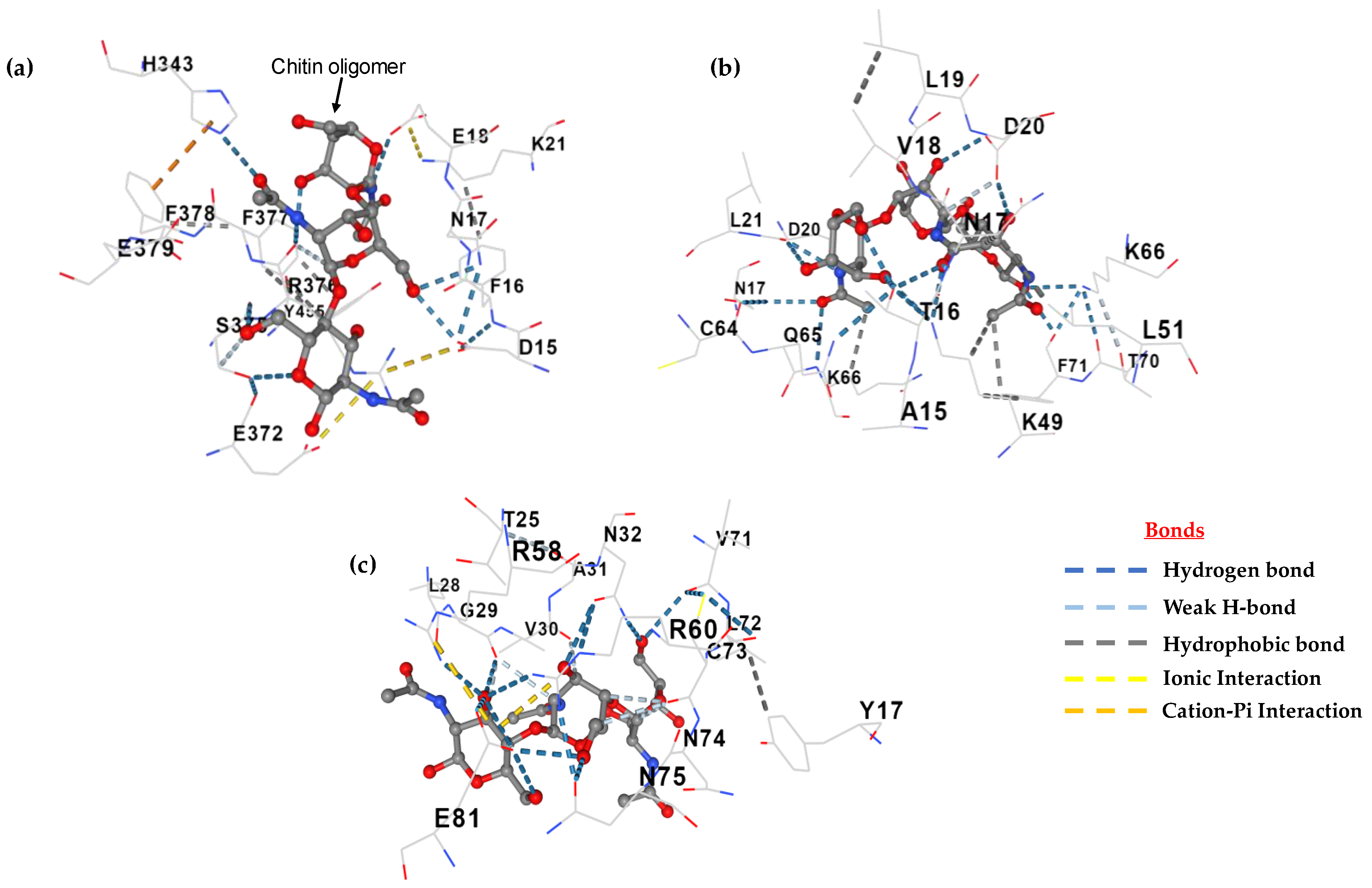
2.7. Active Site and Protein Docking Analysis
3. Results and Discussion
3.1. Detection of Physicochemical Characters of HFBII Proteins
3.2. Signal Peptide Prediction and Subcellular Localization Identification
3.3. Modeling of 3D Protein Structures and Model Evaluation
3.4. Functional and Structural Annotations of HBFII Proteins
3.5. Sequence Alignment and Evolutionary Analysis
3.6. Active Site and Protein Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Wang, F.; Xu, Y.; Liu, G.; Dong, C. Cysteine-rich hydrophobin gene family: Genome wide analysis, phylogeny and transcript Profiling in Cordyceps militaris. Int. J. Mol. Sci. 2021, 22, 643. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Aimanianda, V.; Guijarro, J.I.; Sunde, M.; Latge´, J.P. Hydrophobins—Unique fungal proteins. PLoS Pathog. 2012, 8, e1002700. [Google Scholar] [CrossRef] [PubMed]
- Quarantin, A.; Hadeler, B.; Kröger, C.; Schäfer, W.; Favaron, F.; Sella, L.; Martínez-Rocha, A.L. Different hydrophobins of Fusarium graminearum are involved in hyphal growth, attachment, water-Air interface penetration and plant infection. Front. Microbiol. 2019, 10, 751. [Google Scholar] [CrossRef]
- Vergunst, K.L.; Kenward, C.; Langelaan, D.N. Characterization of the structure and self-assembly of two distinct class IB hydrophobins. Appl. Microbiol. Biotechnol. 2022, 106, 7831–7843. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Terauchi, Y.; Yoshimi, A.; Abe, K. Aspergillus hydrophobins: Physicochemical properties, biochemical properties, and functions in solid polymer degradation. Microorganisms 2022, 10, 1498. [Google Scholar] [CrossRef]
- Ren, Q.; Kwan, A.H.; Sunde, M. Two forms and two faces, multiple states and multiple uses: Properties and applications of the self-assembling fungal hydrophobins. Biopolymers 2013, 100, 601–612. [Google Scholar] [CrossRef]
- Lovett, B.; Kasson, M.T.; Gandier, J. Ecology drives the observed spectrum of hydrophobin protein diversity across Kingdom Fungi. bioRxiv 2022. [Google Scholar] [CrossRef]
- Berger, B.W.; Sallada, N.D. Hydrophobins: Multifunctional biosurfactants for interface engineering. J. Biol. Eng. 2019, 13, 10. [Google Scholar] [CrossRef]
- Gandier, J.A.; Langelaan, D.N.; Won, A.; O’Donnell, K.; Grondin, J.L.; Spencer, H.L.; Wong, P.; Tillier, E.; Yip, C.; Smith, S.P.; et al. Characterization of a Basidiomycota hydrophobin reveals the structural basis for a high-similarity Class I subdivision. Sci. Rep. 2017, 7, 45863. [Google Scholar] [CrossRef]
- He, R.; Li, C.; Feng, J.; Zhang, D. A class II hydrophobin gene, Trhfb3, participates in fungal asexual development of Trichoderma reesei. FEMS Microbiol. Lett. 2017, 364, fnw297. [Google Scholar] [CrossRef]
- Linder, M.B. Hydrophobins: Proteins that self assemble at interfaces. Curr. Opin. Colloid Interface Sci. 2009, 14, 356–363. [Google Scholar] [CrossRef]
- Jensen, B.G.; Andersen, M.R.; Pedersen, M.H.; Frisvad, J.C.; Søndergaard, I. Hydrophobins from Aspergillus species cannot be clearly divided into two classes. BMC Res. Notes 2010, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Seidl-Seiboth, V.; Gruber, S.; Sezerman, U.; Schwecke, T.; Albayrak, A.; Neuhof, T.; von Döhren, H.; Baker, S.E.; Kubicek, C.P. Novel hydrophobins from Trichoderma define a new hydrophobin subclass: Protein properties, evolution, regulation and processing. J. Mol. Evol. 2011, 72, 339–351. [Google Scholar] [CrossRef]
- Bowden, C.G.; Hintz, W.E.; Jeng, R.; Hubbes, M.; Horgen, P.A. Isolation and characterization of the cerato-ulmin toxin gene of the Dutch elm disease pathogen, Ophiostoma ulmi. Curr. Genet. 1994, 25, 323–329. [Google Scholar] [CrossRef] [PubMed]
- McCabe, P.M.; Van Alfen, N.K. Secretion of cryparin, a fungal hydrophobin. Appl. Environ. Microbiol. 1999, 65, 5431–5435. [Google Scholar] [CrossRef]
- De Vries, O.M.; Moore, S.; Arntz, C.; Wessels, J.G.; Tudzynski, P. Identification and characterization of a tri-partite hydrophobin from Claviceps fusiformis. A novel type of class II hydrophobin. Eur. J. Biochem. 1999, 262, 377–385. [Google Scholar] [CrossRef]
- Takai, S. Pathogenicity and cerato-ulmin production in Ceratocystis ulmi. Nature 1974, 252, 124–126. [Google Scholar] [CrossRef]
- Temple, B.; Horgen, P.A.; Bernier, L.; Hintz, W.E. Cerato-ulmin, a hydrophobin secreted by the causal agents of Dutch elm disease, is a parasitic fitness factor. Fungal Genet. Biol. 1997, 22, 39–53. [Google Scholar] [CrossRef]
- Gallo, M.; Luti, S.; Baroni, F.; Baccelli, I.; Cilli, E.M.; Cicchi, C.; Leri, M.; Spisni, A.; Pertinhez, T.A.; Pazzagli, L. Plant defense elicitation by the hydrophobin cerato-ulmin and correlation with its structural features. Int. J. Mol. Sci. 2023, 24, 2251. [Google Scholar] [CrossRef]
- Kazmierczak, P.; Kim, D.H.; Turina, M.; Van Alfen, N.K. A Hydrophobin of the chestnut blight fungus, Cryphonectria parasitica, is required for stromal pustule eruption. Eukaryot. Cell 2005, 4, 931–936. [Google Scholar] [CrossRef]
- Littlejohn, K.A.; Hooley, P.; Coxa, P.W. Bioinformatics predicts diverse Aspergillus hydrophobins with novel properties. Food Hydrocoll. 2012, 27, 503–516. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.; Ghosh, S.; Sahoo, D.; Jha, G. Fungal effectors, the double edge sword of phytopathogens. Curr. Genet. 2021, 67, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Guzmán, P.; Alemán-Duarte, M.I.; Delaye, L.; Herrera-Estrella, A.; Olmedo-Monfil, V. Identification of effector-like proteins in Trichoderma spp. and role of a hydrophobin in the plant-fungus interaction and mycoparasitism. BMC Genet. 2017, 18, 16. [Google Scholar] [CrossRef]
- Bouqellah, N.A.; Elkady, N.A.; Farag, P.F. Secretome analysis for a new strain of the blackleg fungus Plenodomus lingam reveals candidate proteins for effectors and virulence factors. J. Fungi 2023, 9, 740. [Google Scholar] [CrossRef]
- Dubey, M.K.; Jensen, D.F.; Karlsson, M. Hydrophobins are required for conidial hydrophobicity and plant root colonization in the fungal biocontrol agent Clonostachys rosea. BMC Microbiol. 2014, 14, 18. [Google Scholar] [CrossRef]
- Cai, F.; Zhao, Z.; Gao, R.; Chen, P.; Ding, M.; Jiang, S.; Fu, Z.; Xu, P.; Chenthamara, K.; Shen, Q.; et al. The pleiotropic functions of intracellular hydrophobins in aerial hyphae and fungal spores. PLoS Genet. 2021, 17, e1009924. [Google Scholar] [CrossRef]
- Santhoshkumar, R.; Yusuf, A. In silico structural modeling and analysis of physicochemical properties of curcumin synthase (CURS1, CURS2, and CURS3) proteins of Curcuma longa. J. Genet. Eng. Biotechnol. 2020, 18, 24. [Google Scholar] [CrossRef]
- Ren, Q.; Kwan, A.H.; Sunde, M. Solution structure and interface-driven self-assembly of NC2, a new member of the Class II hydrophobin proteins. Proteins 2014, 82, 990–1003. [Google Scholar] [CrossRef]
- Hakanpää, J.; Linder, M.; Popov, A.; Schmidt, A.; Rouvinen, J. Hydrophobin HFBII in detail: Ultrahigh-resolution structure at 0.75 A. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 356–367. [Google Scholar] [CrossRef]
- Courtney, J.M.; Ye, Q.; Nesbitt, A.E.; Tang, M.; Tuttle, M.D.; Watt, E.D.; Nuzzio, K.M.; Sperling, L.J.; Comellas, G.; Peterson, J.R.; et al. Experimental protein structure verification by scoring with a single, unassigned NMR spectrum. Structure 2015, 23, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Naresh, K.; Khan, K.A.; Umer, R.; Cantwell, W.J. The use of X-ray computed tomography for design and process modeling of aerospace composites: A review. Mater. Des. 2020, 190, 108553. [Google Scholar] [CrossRef]
- Deng, H.; Jia, Y.; Zhang, Y. Protein structure prediction. Int. J. Mod. Phys. B 2018, 32, 1840009. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022, arXiv:08.487609. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Edmunds, N.S.; Genc, A.G.; Alharbi, S.M.A.; Salehe, B.R.; Adiyaman, R. Prediction of protein structures, functions and interactions using the IntFOLD7, MultiFOLD and ModFOLDdock servers. Nucleic Acids Res. 2023, 51, W274–W280. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.; Krasileva, K.V. Prediction of effector protein structures from fungal phytopathogens enables evolutionary analyses. Nat. Microbiol. 2023, 8, 174–187. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Aldowsari, F.; Alharbi, S.; Adiyaman, R. ModFOLD8: Accurate global and local quality estimates for 3D protein models. Nucleic Acids Res. 2021, 49, W425–W430. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-align: Universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Lavezzo, E.; Falda, M.; Fontana, P.; Bianco, L.; Toppo, S. Enhancing protein function prediction with taxonomic constraints—The Argot2.5 web server. Methods 2016, 93, 15–23. [Google Scholar] [CrossRef]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Lotun, D.P.; Cochard, C.; Vieira, F.R.J.; Bernardes, J.S. 2dSS: A web server for protein secondary structure visualization. bioRxiv 2019. [Google Scholar] [CrossRef]
- Dass, R.; Mulder, F.A.A.; Nielsen, J.T. ODiNPred: Comprehensive prediction of protein order and disorder. Sci. Rep. 2020, 10, 14780. [Google Scholar] [CrossRef]
- Ayoub, R.; Lee, Y. RUPEE: A fast and accurate purely geometric protein structure search. PLoS ONE 2019, 14, e0213712. [Google Scholar] [CrossRef]
- Ayoub, R.; Lee, Y. Protein structure search to support the development of protein structure prediction method. Proteins Struct. Funct. 2021, 89, 648–658. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Doron-Faigenboim, A.; Stern, A.; Mayrose, I.; Bacharach, E.; Pupko, T. Selecton: A server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 2005, 21, 2101–2103. [Google Scholar] [CrossRef]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Chorin, A.B.; Martz, E. Using evolutionary data to make sense of macromolecules with a "face-lifted" ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Kavya, N.; Prasannakumar, M.K.; Venkateshbabu, G.; Niranjan, V.; Uttarkar, A.; Buela Parivallal, P.; Banakar, S.N.; Mahesh, H.B.; Devanna, P.; Manasa, K.G.; et al. Insights on novel effectors and characterization of metacaspase (RS107_6) as a potential cell death-inducing protein in Rhizoctonia solani. Microorganisms 2023, 11, 920. [Google Scholar] [CrossRef] [PubMed]
- Enany, S. Structural and functional analysis of hypothetical and conserved proteins of Clostridium tetani. J. Infect. Public Health 2014, 7, 296–307. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Kaur, A.; Pati, P.K.; Pati, A.M.; Nagpal, A.K. Physico-chemical characterization and topological analysis of pathogenesis-related proteins from Arabidopsis thaliana and Oryza sativa using in-silico approaches. PLoS ONE 2020, 15, e0239836. [Google Scholar] [CrossRef] [PubMed]
- Nene, T.; Yadav, M.; Yadav, H.S. Plant catalase in silico characterization and phylogenetic analysis with structural modeling. J. Genet. Eng. Biotechnol. 2022, 20, 125. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Keerio, A.A.; Ma, A. Identification of hydrophobin genes and their physiological functions related to growth and development in Pleurotus ostreatus. Microbiol. Res. 2021, 247, 126723. [Google Scholar] [CrossRef]
- Huang, Y.; Mijiti, G.; Wang, Z.; Yu, W.; Fan, H.; Zhang, R.; Liu, Z. Functional analysis of the class II hydrophobin gene HFB2-6 from the biocontrol agent Trichoderma asperellum ACCC30536. Microbiol. Res. 2015, 171, 8–20. [Google Scholar] [CrossRef]
- Neuhof, T.; Dieckmann, R.; Druzhinina, I.S.; Kubicek, C.P.; Nakari-Setälä, T.; Penttilä, M.; von Döhren, H. Direct identification of hydrophobins and their processing in Trichoderma using intact-cell MALDI-TOF MS. FEBS J. 2007, 274, 841–852. [Google Scholar] [CrossRef]
- Mei, J.; Ning, N.; Wu, H.; Chen, X.; Li, Z.; Liu, W. Glycosylphosphatidylinositol anchor biosynthesis pathway-related protein GPI7 is required for the vegetative Growth and pathogenicity of Colletotrichum graminicola. Int. J. Mol. Sci. 2022, 23, 2985. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Ko, Y.H.; So, K.K.; Cho, S.H.; Kim, D.H. A fungal GPI-anchored protein gene functions as a virulence and antiviral factor. Cell Rep. 2022, 41, 111481. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, B.; De Las Peñas, A.; Castaño, I.; Van Dijck, P. Adhesins in Candida Glabrata. J. Fungi 2018, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.; Krasileva, K.V. Computational structural genomics unravels common folds and novel families in the secretome of fungal phytopathogen Magnaporthe oryzae. Mol. Plant Microbe Interact. 2021, 34, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rony, M.N.H.; Ahmed, R. In silico characterization and structural modeling of bacterial metalloprotease of family M4. J. Genet. Eng. Biotechnol. 2021, 19, 25. [Google Scholar] [CrossRef]
- Chikkerur, J.; Samanta, A.K.; Dhali, A.; Kolte, A.P.; Roy, S.; Maria, P. In Silico evaluation and identification of fungi capable of producing endo-inulinase enzyme. PLoS ONE 2018, 13, e0200607. [Google Scholar] [CrossRef]
- Dalkiran, A.; Rifaioglu, A.S.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Dogan, T. ECPred: A tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature. BMC Bioinform. 2018, 19, 334. [Google Scholar] [CrossRef]
- Baldwin, T.K.; Urban, M.; Brown, N.; Hammond-Kosack, K.E.; Lee, Y.; Min, K.; Son, H.; Park, A.R.; Kim, J.-C.; Choi, G.J.; et al. A role for topoisomerase I in Fusarium graminearum and F. culmorum pathogenesis and sporulation. Mol. Plant Microbe Interact. 2010, 23, 566–577. [Google Scholar] [CrossRef]
- Neu, E.; Debener, T. Prediction of the Diplocarpon rosae secretome reveals candidate genes for effectors and virulence factors. Fungal Biol. 2019, 123, 231–239. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; deWit, P.J. Fungal Effector Proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef]
- Winnenburg, R.; Urban, M.; Beacham, A.; Baldwin, T.K.; Holland, S.; Lindeberg, M.; Hansen, H.; Rawlings, C.; Hammond-Kosack, K.E.; Köhler, J. PHI-base update: Additions to the pathogen host interaction database. Nucleic Acids Res. 2007, 36, D572–D576. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, B.V.; El-Ganainy, S.M.; Alrajeh, H.S.; Al-Sheikh, H. In Silico characterization of the secretome of the fungal pathogen Thielaviopsis punctulata, the causal agent of date palm black scorch disease. J. Fungi 2023, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO family GTPases: Mechanisms of regulation and signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef] [PubMed]
- Ates, L.S.; van der Woude, A.D.; Bestebroer, J.; van Stempvoort, G.; Musters, R.J.; Garcia-Vallejo, J.J.; Picavet, D.I.; Weerd, R.V.; Maletta, M.; Kuijl, C.P.; et al. The ESX-5 system of pathogenic Mycobacteria is involved in capsule integrity and virulence through its substrate PPE10. PLoS Pathog. 2016, 12, e1005696. [Google Scholar] [CrossRef] [PubMed]
- Mgbeahuruike, A.C.; Kovalchuk, A.; Chen, H.; Ubhayasekera, W.; Asiegbu, F. Evolutionary analysis of hydrophobin gene family in two wood-degrading basidiomycetes, Phlebia brevispora and Heterobasidion annosum s.l. BMC Evol. Biol. 2013, 13, 240. [Google Scholar] [CrossRef]
- Bywater, R.P. Prediction of protein structural features from sequence data based on Shannon entropy and Kolmogorovc complexity. PLoS ONE 2015, 10, e0119306. [Google Scholar] [CrossRef]
- Jin, R.; Wang, J.; Guo, B.; Yang, T.; Hu, J.; Wang, B.; Yu, Q. Identification and expression analysis of the Alfin-like gene family in tomato and the role of SlAL3 in salt and drought stresses. Plants 2023, 12, 2829. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Khan, F.A.; Khan, M.A.; Imran, S.; Akhtar, R.W.; Pandupuspitasari, N.S.; Negara, W.; Chen, J. Molecular evolution of the bactericidal/permeability-increasing protein (BPIFA1) regulating the innate immune responses in mammals. Genes 2023, 14, 15. [Google Scholar] [CrossRef]
- Liao, J.; Wang, Q.; Wu, F.; Huang, Z. In silico methods for identification of potential active sites of therapeutic targets. Molecules 2022, 27, 7103. [Google Scholar] [CrossRef]
- Tanaka, S.; Kahmann, R. Cell wall-associated effectors of plant-colonizing fungi. Mycologia 2021, 113, 247–260. [Google Scholar] [CrossRef]
- Zhang, S.; Li, C.; Si, J.; Han, Z.; Chen, D. Action mechanisms of effectors in plant-pathogen interaction. Int. J. Mol. Sci. 2022, 23, 6758. [Google Scholar] [CrossRef] [PubMed]
- Frischmann, A.; Neudl, S.; Gaderer, R.; Bonazza, K.; Zach, S.; Gruber, S.; Spadiut, O.; Friedbacher, G.; Grothe, H.; Seidl-Seiboth, V. Self-assembly at air/water interfaces and carbohydrate binding properties of the small secreted protein EPL1 from the fungus Trichoderma atroviride. J. Biol. Chem. 2013, 288, 4278–4287. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Luti, S.; Bernardi, R.; Scala, A.; Pazzagli, L. Cerato-platanin shows expansin-like activity on cellulosic materials. Appl. Microbiol. Biotechnol. 2014, 98, 175–184. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouqellah, N.A.; Farag, P.F. In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens. Microorganisms 2023, 11, 2632. https://doi.org/10.3390/microorganisms11112632
Bouqellah NA, Farag PF. In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens. Microorganisms. 2023; 11(11):2632. https://doi.org/10.3390/microorganisms11112632
Chicago/Turabian StyleBouqellah, Nahla A., and Peter F. Farag. 2023. "In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens" Microorganisms 11, no. 11: 2632. https://doi.org/10.3390/microorganisms11112632