
Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship
 , and
, and 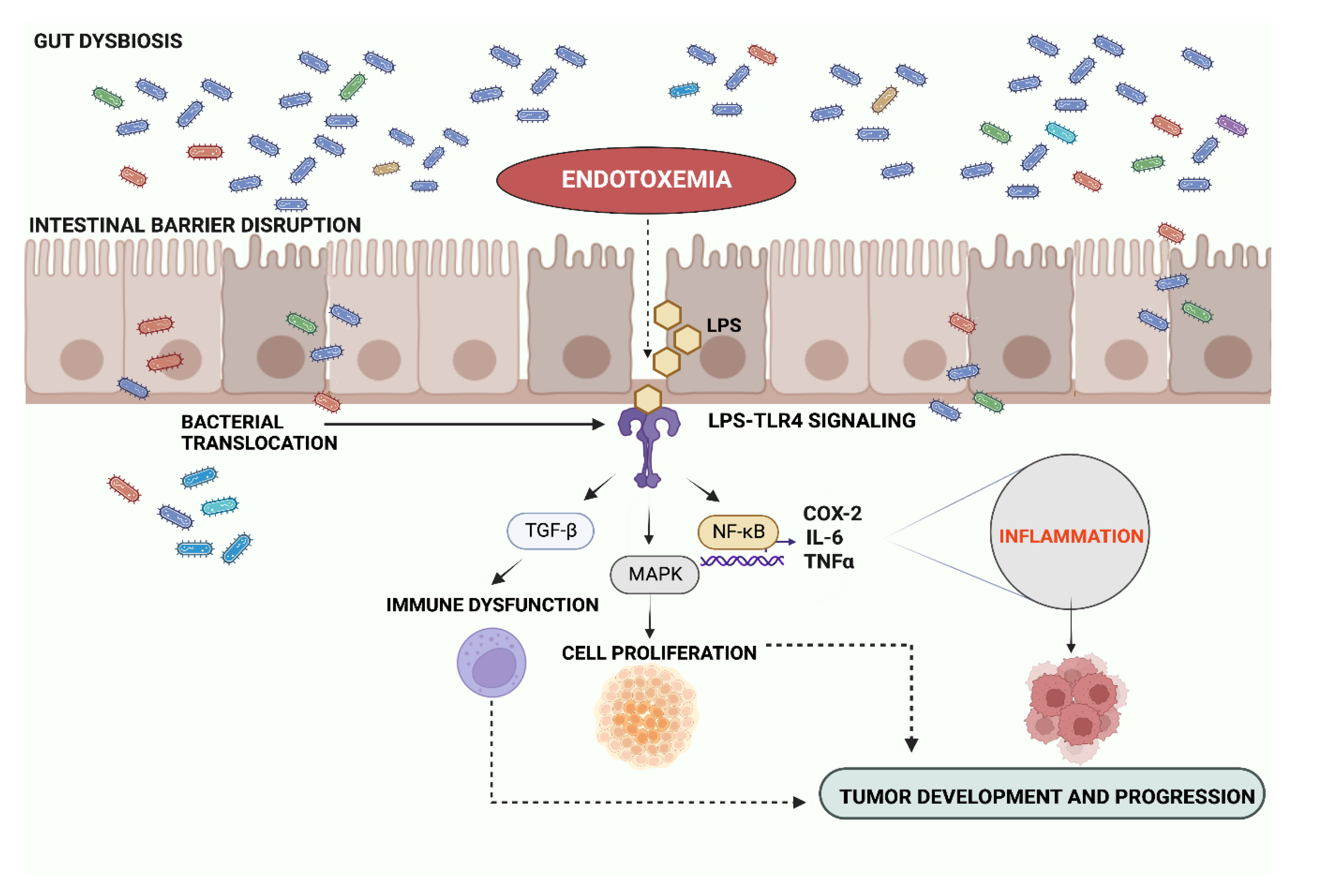{kind=link}
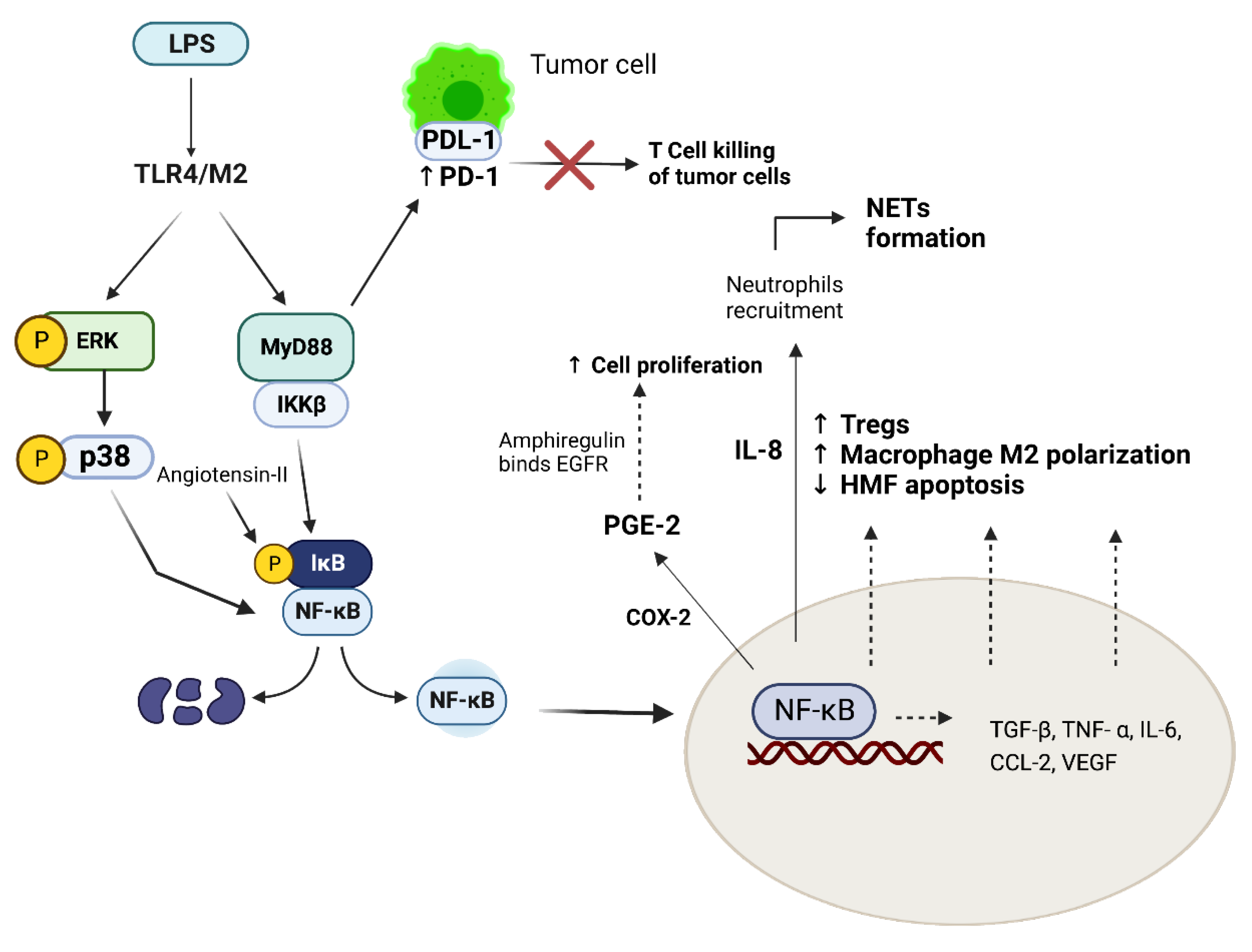{kind=link}
Abstract
:1. Introduction
2. Endotoxemia and Low-Grade Inflammation
3. Endotoxemia and Gastrointestinal Cancers
3.1. LPS and Esophageal Cancer
3.2. LPS and Gastric Cancer
3.3. LPS and Colorectal Cancer
3.4. LPS and Hepatocellular Carcinoma
3.5. LPS and Pancreatic Cancer
4. Endotoxin and its Role in Cancer Progression and Development of Metastasis
5. Blocking LPS/TLR-4/M2 Cascade: Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sampath, V. Bacterial endotoxin-lipopolysaccharide; structure, function and its role in immunity in vertebrates and invertebrates. Agric. Nat. Resour. 2018, 52, 115–120. [Google Scholar] [CrossRef]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Lerouge, I.; Vanderleyden, J. O-antigen structural variation: Mechanisms and possible roles in animal/plant–microbe interactions. FEMS Microbiol. Rev. 2002, 26, 17–47. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emaeshima, N.; Fernandez, R.C. Recognition of lipid A variants by the TLR-4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR-4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR-4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Akashi, S.; Nagafuku, M.; Ogata, M.; Iwakura, Y.; Akira, S.; Kitamura, T.; Kosugi, A.; Kimoto, M.; Miyake, K. Essential role of MD-2 in LPS responsiveness and TLR-4 distribution. Nat. Immunol. 2002, 3, 667–672. [Google Scholar] [CrossRef]
- Ryu, J.-K.; Kim, S.J.; Rah, S.-H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.-O.; Park, B.S.; Yoon, T.-Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR-4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-I.; Lee, C.J.; Jin, M.S.; Lee, C.-H.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal Structure of CD14 and Its Implications for Lipopolysaccharide Signaling. J. Biol. Chem. 2005, 280, 11347–11351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Medzhitov, R. Toll-Like Receptor Signaling Pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.; Durso, D.F.; Bryant, C.E.; Kurt-Jones, E.A.; Silverman, N.; Golenbock, D.T.; Gazzinelli, R.T. The IRAK4 scaffold integrates TLR-4-driven TRIF and MYD88 signaling pathways. Cell Rep. 2022, 40, 111225. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR-4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 Receptor Domain-Containing Adaptor Inducing IFN-β (TRIF) Associates with TNF Receptor-Associated Factor 6 and TANK-Binding Kinase 1, and Activates Two Distinct Transcription Factors, NF-κB and IFN-Regulatory Factor-3, in the Toll-Like Receptor Signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [CrossRef] [Green Version]
- Mohr, A.E.; Crawford, M.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the gut microbiota: Considering structural variation. FEBS Lett. 2022, 596, 849–875. [Google Scholar] [CrossRef]
- Opal, S.M. Endotoxins and Other Sepsis Triggers. Contrib. Nephrol. 2010, 167, 14–24. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Zhou, Y.; Zhang, H.; Zhou, H.; Zhang, X. Slimy partners: The mucus barrier and gut microbiome in ulcerative colitis. Exp. Mol. Med. 2021, 53, 772–787. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qamar, N.; Castano, D.; Patt, C.; Chu, T.; Cottrell, J.; Chang, S.L. Meta-analysis of alcohol induced gut dysbiosis and the resulting behavioral impact. Behav. Brain Res. 2019, 376, 112196. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.B.; Leone, V.; Chang, E.B. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microbes 2017, 8, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Proffitt, C.; Bidkhori, G.; Moyes, D.; Shoaie, S. Disease, Drugs and Dysbiosis: Understanding Microbial Signatures in Metabolic Disease and Medical Interventions. Microorganisms 2020, 8, 1381. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Breton, J.; Galmiche, M.; Déchelotte, P. Dysbiotic Gut Bacteria in Obesity: An Overview of the Metabolic Mechanisms and Therapeutic Perspectives of Next-Generation Probiotics. Microorganisms 2022, 10, 452. [Google Scholar] [CrossRef]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef]
- Nagpal, R.; Newman, T.M.; Wang, S.; Jain, S.; Lovato, J.F.; Yadav, H. Obesity-Linked Gut Microbiome Dysbiosis Associated with Derangements in Gut Permeability and Intestinal Cellular Homeostasis Independent of Diet. J. Diabetes Res. 2018, 2018, 3462092. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suriano, F.; Nyström, E.E.L.; Sergi, D.; Gustafsson, J.K. Diet, microbiota, and the mucus layer: The guardians of our health. Front. Immunol. 2022, 13, 953196. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, K.; Kanmura, S.; Morinaga, Y.; Tanaka, A.; Makino, T.; Fujita, T.; Arima, S.; Sasaki, F.; Nasu, Y.; Tanoue, S.; et al. A high-fructose diet induces epithelial barrier dysfunction and exacerbates the severity of dextran sulfate sodium-induced colitis. Int. J. Mol. Med. 2019, 43, 1487–1496. [Google Scholar] [CrossRef]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef] [Green Version]
- Violi, F.; Cammisotto, V.; Bartimoccia, S.; Pignatelli, P.; Carnevale, R.; Nocella, C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat. Rev. Cardiol. 2022, 20, 1–14. [Google Scholar] [CrossRef]
- Donohoe, C.L.; Doyle, S.L.; Reynolds, J.V. Visceral adiposity, insulin resistance and cancer risk. Diabetol. Metab. Syndr. 2011, 3, 12. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Sang, H.; Zhou, Y.; Shang, C.; Wang, Y.; Zhu, H. Effects of hyperglycemia on the progression of tumor diseases. J. Exp. Clin. Cancer Res. 2019, 38, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.-H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.; Sung, J.J.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e2. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.D.; Atay, C.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C.; et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Gao, H.; Qi, Q.; Liu, X.; Li, J.; Gao, J.; Li, P.; Wang, Y.; Du, L.; Wang, C. High fat diet, gut microbiome and gastrointestinal cancer. Theranostics 2021, 11, 5889–5910. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Núñez-Sánchez, M.A.; Ávila-Gálvez, M.A.; Monedero-Saiz, T.; Rodríguez-Gil, F.J.; Martínez-Díaz, F.; Selma, M.V.; Espín, J.C. Consumption of pomegranate decreases plasma lipopolysaccharide-binding protein levels, a marker of metabolic endotoxemia, in patients with newly diagnosed colorectal cancer: A randomized controlled clinical trial. Food Funct. 2018, 9, 2617–2622. [Google Scholar] [CrossRef]
- Zeng, H.; Lazarova, D.L.; Bordonaro, M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J. Gastrointest. Oncol. 2014, 6, 41–51. [Google Scholar] [CrossRef]
- Abreu, A.A.Y.; Milke-García, M.; Argüello-Arévalo, G.; la Barca, A.C.-D.; Carmona-Sánchez, R.; Consuelo-Sánchez, A.; Coss-Adame, E.; García-Cedillo, M.; Hernández-Rosiles, V.; Icaza-Chávez, M.; et al. Dietary fiber and the microbiota: A narrative review by a group of experts from the Asociación Mexicana de Gastroenterología. Rev. de Gastroenterol. de Mex. (Engl. Ed. ) 2021, 86, 287–304. [Google Scholar] [CrossRef]
- Hagland, H.R.; Søreide, K. Cellular metabolism in colorectal carcinogenesis: Influence of lifestyle, gut microbiome and metabolic pathways. Cancer Lett. 2015, 356 Pt A, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef]
- Rivas-Domínguez, A.; Pastor, N.; Martínez-López, L.; Colón-Pérez, J.; Bermúdez, B.; Orta, M. The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer. Cells 2021, 10, 1934. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; Van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal Inflammation and Cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Klooster, C.C.V.; Ridker, P.M.; Hjortnaes, J.; van der Graaf, Y.; Asselbergs, F.W.; Westerink, J.; Aerts, J.G.J.V.; Visseren, F.L.J. The relation between systemic inflammation and incident cancer in patients with stable cardiovascular disease: A cohort study. Eur. Heart J. 2019, 40, 3901–3909. [Google Scholar] [CrossRef] [Green Version]
- Yesudhas, D.; Gosu, V.; Anwar, M.A.; Choi, S. Multiple Roles of Toll-Like Receptor 4 in Colorectal Cancer. Front. Immunol. 2014, 5, 334. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yin, J.; Shen, W.; Gao, R.; Liu, Y.; Chen, Y.; Li, X.; Liu, C.; Xiang, R.; Luo, N. TLR-4 Promotes Breast Cancer Metastasis via Akt/GSK3β/β-Catenin Pathway upon LPS Stimulation. Anat. Rec. 2017, 300, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, R.Y.; Chan, C.H.; Spicer, J.D.; Rousseau, M.C.; Giannias, B.; Rousseau, S.; Ferri, L.E. LPS-Induced TLR-4 Signaling in Human Colorectal Cancer Cells Increases β1 Integrin-Mediated Cell Adhesion and Liver Metastasis. Cancer Res. 2011, 71, 1989–1998. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Dash, P.; Minz, A.P.; Satpathi, S.; Samal, A.G.; Behera, P.K.; Satpathi, P.S.; Senapati, S. Lipopolysaccharide (LPS) enhances prostate cancer metastasis potentially through NF-κB activation and recurrent dexamethasone administration fails to suppress it in vivo. Prostate 2019, 79, 168–182. [Google Scholar] [CrossRef]
- Rajamanickam, V.; Yan, T.; Xu, S.; Hui, J.; Xu, X.; Ren, L.; Liu, Z.; Liang, G.; Wang, O.; Wang, Y. Selective targeting of the TLR-4 co-receptor, MD2, prevents colon cancer growth and lung metastasis. Int. J. Biol. Sci. 2020, 16, 1288–1302. [Google Scholar] [CrossRef]
- Gonçalves, M.; Cappellari, .R.; Junior, A.A.D.S.; De Marchi, F.O.; Macchi, F.S.; Antunes, K.H.; De Souza, A.P.D.; Morrone, F.B.; BioMedicina, B.I.D.P.; Machi, F.S. Effect of LPS on the Viability and Proliferation of Human Oral and Esophageal Cancer Cell Lines. Braz. Arch. Biol. Technol. 2016, 59, e16150485, ISSN 1678-4324. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.B.; Coburn, S.B.; Lam, J.R.; Taylor, P.R.; Schneider, J.L.; A Corley, D. Cancer incidence and mortality risks in a large US Barrett’s oesophagus cohort. Gut 2018, 67, 418–529. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Guo, L.; Liu, J.-J.; Zhao, H.-P.; Zhang, J.; Wang, J.-H. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2019, 25, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.E.; Siersema, P.D.; Kate, F.J.T.; Fluiter, K.; Souza, R.F.; Vleggaar, F.P.; Bus, P.; van Baal, J.W.P.M. Toll-like receptor 4 activation in Barrett’s esophagus results in a strong increase in COX-2 expression. J. Gastroenterol. 2014, 49, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Ping, W.; Deng, T.; Zhang, N.; Fu, X.; Sun, W. Lipopolysaccharide-induced toll-like receptor 4 signaling in esophageal squamous cell carcinoma promotes tumor proliferation and regulates inflammatory cytokines expression. Dis. Esophagus 2017, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.C.; Hsu, R.Y.; Spicer, J.D.; McDonald, B.; Chan, C.H.; Perera, R.M.; Giannias, B.; Chow, S.C.; Rousseau, S.; Law, S.; et al. Lipopolysaccharide-induced toll-like receptor 4 signaling enhances the migratory ability of human esophageal cancer cells in a selectin-dependent manner. Surgery 2013, 154, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-P.; Chakder, S.; Gao, F.; Rattan, S. Inducible and neuronal nitric oxide synthase involvement in lipopolysaccharide-induced sphincteric dysfunction. Am. J. Physiol. Liver Physiol. 2001, 280, G32–G42. [Google Scholar] [CrossRef] [Green Version]
- Calatayud, S.; García-Zaragozá, E.; Hernández, C.; Quintana, E.; Felipo, V.; Esplugues, J.V.; Barrachina, M.D. Downregulation of nNOS and synthesis of PGs associated with endotoxin-induced delay in gastric emptying. Am. J. Physiol. Liver Physiol. 2002, 283, G1360–G1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Francois, F.; Pei, Z. Molecular Pathways: Pathogenesis and Clinical Implications of Microbiome Alteration in Esophagitis and Barrett Esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar] [CrossRef] [Green Version]
- Kakelar, H.M.; Barzegari, A.; Dehghani, J.; Hanifian, S.; Saeedi, N.; Barar, J.; Omidi, Y. Pathogenicity of Helicobacter pylori in cancer development and impacts of vaccination. Gastric Cancer 2019, 22, 23–36. [Google Scholar] [CrossRef]
- Pop, R.; Tăbăran, A.-F.; Ungur, A.P.; Negoescu, A.; Cătoi, C. Helicobacter Pylori-Induced Gastric Infections: From Pathogenesis to Novel Therapeutic Approaches Using Silver Nanoparticles. Pharmaceutics 2022, 14, 1463. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Suda, Y.; Kashihara, W.; Hayashi, T.; Shimoyama, T.; Kusumoto, S.; Tamura, T. Immunobiological activities of chemically defined lipid A from Helicobacter pylori LPS in comparison with Porphyromonas gingivalis lipid A and Escherichia coli-type synthetic lipid A (compound 506). Vaccine 1997, 15, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.; Ferris, J.A.; Szponar, B.; Wadstrom, T.; Fox, J.G.; O’Rourke, J.; Larsson, L.; Yaquian, E.; Ljungh, A.; Clyne, M.; et al. Comparative Chemical and Biological Characterization of the Lipopolysaccharides of Gastric and Enterohepatic Helicobacters. Helicobacter 2004, 9, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.-I.; Okabayashi, T.; Rehli, M.; Fujii, N.; Amano, K.-I. Helicobacter pylori Lipopolysaccharides Upregulate Toll-Like Receptor 4 Expression and Proliferation of Gastric Epithelial Cells via the MEK1/2-ERK1/2 Mitogen-Activated Protein Kinase Pathway. Infect. Immun. 2010, 78, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.F., Jr.; Mitchell, A.; Li, G.; Ding, S.; Fitzmaurice, A.M.; Ryan, K.; Crowe, S.; Goldberg, J.B. Toll-like Receptor (TLR) 2 and TLR-5, but Not TLR-4, Are Required for Helicobacter pylori-induced NF-κB Activation and Chemokine Expression by Epithelial Cells. J. Biol. Chem. 2003, 278, 32552–32560. [Google Scholar] [CrossRef] [Green Version]
- Toshchakov, V.; Jones, B.W.; Lentschat, A.; Silva, A.; Perera, P.-Y.; Thomas, K.; Cody, M.J.; Zhang, S.; Williams, B.R.G.; Major, J.; et al. TLR-2 and TLR-4 agonists stimulate unique repertoires of host resistance genes in murine macrophages: Interferon-β-dependent signaling in TLR-4-mediated responses. J. Endotoxin Res. 2003, 9, 169–175. [Google Scholar] [CrossRef]
- Kawahara, T.; Teshima, S.; Oka, A.; Sugiyama, T.; Kishi, K.; Rokutan, K. Type I Helicobacter pylori Lipopolysaccharide Stimulates Toll-Like Receptor 4 and Activates Mitogen Oxidase 1 in Gastric Pit Cells. Infect. Immun. 2001, 69, 4382–4389. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.; Amano, K.; Hayashi, S.; Fujii, N. Low antigenicity of the polysaccharide region of Helicobacter pylori lipopolysaccharides derived from tumors of patients with gastric cancer. Infect. Immun. 1997, 65, 3509–3512. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xu, H.; Ou, Y.; Feng, Z.; Zhang, Q.; Zhu, Q.; Cai, Z. LPS-induced CXCR7 expression promotes gastric Cancer proliferation and migration via the TLR-4/MD-2 pathway. Diagn. Pathol. 2019, 14, 3. [Google Scholar] [CrossRef]
- Ito, N.; Tsujimoto, H.; Ueno, H.; Xie, Q.; Shinomiya, N. Helicobacter pylori-Mediated Immunity and Signaling Transduction in Gastric Cancer. J. Clin. Med. 2020, 9, 3699. [Google Scholar] [CrossRef]
- de Waal, G.M.; de Villiers, W.J.S.; Forgan, T.; Roberts, T.; Pretorius, E. Colorectal cancer is associated with increased circulating lipopolysaccharide, inflammation and hypercoagulability. Sci. Rep. 2020, 10, 8777. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples from Patients with Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.H. Lipopolysaccharide: Basic Biochemistry, Intracellular Signaling, and Physiological Impacts in the Gut. Intest. Res. 2014, 12, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-Like Receptor-4 Promotes the Development of Colitis-Associated Colorectal Tumors. Gastroenterology 2007, 133, 1869–1869.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Mayer, R.J. Chemoprevention of Colorectal Cancer. N. Engl. J. Med. 2000, 342, 1960–1968. [Google Scholar] [CrossRef]
- Hardwick, J.C.; van den Brink, G.R.; Offerhaus, G.J.; van Deventer, S.J.; Peppelenbosch, M.P. NF-kappaB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene 2001, 20, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Moser, A.; Luongo, C.; Gould, K.; McNeley, M.; Shoemaker, A.; Dove, W. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 1995, 31, 1061–1064. [Google Scholar] [CrossRef]
- Zhao, R.; Coker, O.O.; Wu, J.; Zhou, Y.; Zhao, L.; Nakatsu, G.; Bian, X.; Wei, H.; Chan, A.W.; Sung, J.J.; et al. Aspirin Reduces Colorectal Tumor Development in Mice and Gut Microbes Reduce its Bioavailability and Chemopreventive Effects. Gastroenterology 2020, 159, 969–983.e4. [Google Scholar] [CrossRef]
- Liu, W.-T.; Jing, Y.-Y.; Yu, G.-F.; Han, Z.-P.; Yu, D.-D.; Fan, Q.-M.; Ye, F.; Li, R.; Gao, L.; Zhao, Q.-D.; et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015, 358, 136–143. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taurá, P.; Fuster, J.; García-Valdecasas, J.C.; Lacy, A.; Suárez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Angrisano, T.; Pero, R.; Peluso, S.; Keller, S.; Sacchetti, S.; Bruni, C.B.; Chiariotti, L.; Lembo, F. LPS-induced IL-8 activation in human intestinal epithelial cells is accompanied by specific histone H3 acetylation and methylation changes. BMC Microbiol. 2010, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Chand, H.S.; Harris, J.F.; Tesfaigzi, Y. IL-13 in LPS-Induced Inflammation Causes Bcl-2 Expression to Sustain Hyperplastic Mucous cells. Sci. Rep. 2018, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.; Schwabe, R.F. TLR-4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Yu, L.-X.; Yan, H.-X.; Liu, Q.; Yang, W.; Wu, H.-P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.-Q.; Zou, S.-S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Yu, L.-X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.-X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradère, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR-4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, G.L. Lipopolysaccharides in liver injury: Molecular mechanisms of Kupffer cell activation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 283, G256–G265. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Ghosh, S. NF-κB, an Evolutionarily Conserved Mediator of Immune and Inflammatory Responses. Adv. Exp. Med. Biol. 2005, 560, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Hatano, E.; Bennett, B.L.; Manning, A.M.; Qian, T.; Lemasters, J.J.; Brenner, D.A. NF-κB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-α– and Fas-mediated apoptosis. Gastroenterology 2001, 120, 1251–1262. [Google Scholar] [CrossRef]
- Watson, M.R.; Wallace, K.; Gieling, R.G.; Manas, D.M.; Jaffray, E.; Hay, R.T.; Mann, D.A.; Oakley, F. NF-κB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J. Hepatol. 2008, 48, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Karin, M. Regulation and Function of IKK and IKK-Related Kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Israël, A. The IKK Complex, a Central Regulator of NF- B Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, J.R.; Clouston, A.D.; Ando, Y.; Kelemen, L.I.; Horn, M.J.; Adamson, M.D.; Purdie, D.M.; Powell, E.E. Angiotensin-Converting Enzyme Inhibition Attenuates the Progression of Rat Hepatic Fibrosis. Gastroenterology 2001, 121, 148–155. [Google Scholar] [CrossRef]
- Oakley, F.; Teoh, V.; Ching–A–Sue, G.; Bataller, R.; Colmenero, J.; Jonsson, J.R.; Eliopoulos, A.G.; Watson, M.R.; Manas, D.; Mann, D.A. Angiotensin II Activates IκB Kinase Phosphorylation of RelA at Ser536 to Promote Myofibroblast Survival and Liver Fibrosis. Gastroenterology 2009, 136, 2334–2344.e1. [Google Scholar] [CrossRef]
- Karbach, S.H.; Schönfelder, T.; Brandão, I.; Wilms, E.; Hörmann, N.; Jäckel, S.; Schüler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II–Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5, e003698. [Google Scholar] [CrossRef] [Green Version]
- Santisteban, M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef]
- Colmenero, J.; Bataller, R.; Sancho-Bru, P.; Domínguez, M.; Moreno, M.; Forns, X.; Bruguera, M.; Arroyo, V.; Brenner, D.A.; Ginès, P. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 297, G726–G734. [Google Scholar] [CrossRef] [Green Version]
- Identifier: NCT00990639. Available online: ClinicalTrials.gov (accessed on 15 January 2023).
- Identifier: NCT00298714. Available online: ClinicalTrials.gov (accessed on 15 January 2023).
- Massoumi, R.L.; Teper, Y.; Ako, S.M.; Ye, L.; Wang, E.B.; Hines, O.J.; Eibl, G. Direct Effects of Lipopolysaccharide on Human Pancreatic Cancer Cells. Pancreas 2021, 50, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, D.; Wang, Q.; Zheng, D.; Jiang, X.; Xu, L. LPS Induced miR-181a Promotes Pancreatic Cancer Cell Migration via Targeting PTEN and MAP2K4. Dig. Dis. Sci. 2014, 59, 1452–1460. [Google Scholar] [CrossRef]
- Ping, P.H.; Bo, T.F.; Li, L.; Hui, Y.N.; Hong, Z. IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Chang, H.-H.; Eibl, G. Obesity-Induced Adipose Tissue Inflammation as a Strong Promotional Factor for Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 673. [Google Scholar] [CrossRef] [Green Version]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Lesina, M.; Wörmann, S.M.; Neuhöfer, P.; Song, L.; Algül, H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin. Immunol. 2014, 26, 80–87. [Google Scholar] [CrossRef]
- Zhuang, Z.; Ju, H.-Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-κB Activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.; Wang, J.; Winter, D.; Laug, W.; Redmond, H. Tumor Cell Adhesion to Endothelial Cells Is Increased by Endotoxin via an Upregulation of β-1 Integrin Expression. J. Surg. Res. 2001, 97, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Quondamatteo, F.; Scherf, C.; Miosge, N.; Herken, R. Immunohistochemical localization of laminin, nidogen, and type IV collagen during the early development of human liver. Histochem. Cell Biol. 1999, 111, 39–47. [Google Scholar] [CrossRef]
- Zheng, Q.C.; Yang, J.; Li, M. Emerging role of Toll-like receptor 4 in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2015, 2, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Goto, Y.; Narita, N.; Hoon, D.S. Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment. Cancer Microenviron. 2009, 2 (Suppl. S1), 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-M.; Xiao, X.; Lao, X.-M.; Wei, Y.; Liu, R.-X.; Zeng, Q.-H.; Wang, J.-C.; Ouyang, F.-Z.; Chen, D.-P.; Chan, K.-W.; et al. Polarization of Tissue-Resident TFH-Like Cells in Human Hepatoma Bridges Innate Monocyte Inflammation and M2b Macrophage Polarization. Cancer Discov. 2016, 6, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Yao, Y.; Yang, C.; Zhang, S. Identification of mannose receptor and CD163 as novel biomarkers for colorectal cancer. Cancer Biomark. 2018, 21, 689–700. [Google Scholar] [CrossRef]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR-4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Killeen, S.D.; Wang, J.H.; Andrews, E.J.; Redmond, H.P. Bacterial endotoxin enhances colorectal cancer cell adhesion and invasion through TLR-4 and NF-κB-dependent activation of the urokinase plasminogen activator system. Br. J. Cancer 2009, 100, 1589–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nespoli, A.; Gianotti, L.; Totis, M.; Bovo, G.; Nespoli, L.; Chiodini, P.; Brivio, F. Correlation between Postoperative Infections and Long-term Survival after Colorectal Resection for Cancer. Tumori J. 2004, 90, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Schietroma, M.; Pessia, B.; Carlei, F.; Amicucci, G. Intestinal permeability changes, systemic endotoxemia, inflammatory serum markers and sepsis after Whipple’s operation for carcinoma of the pancreas head. Pancreatology 2017, 17, 839–846. [Google Scholar] [CrossRef]
- Richards, C.H.; Platt, J.J.; Anderson, J.H.; McKee, R.F.; Horgan, P.G.; McMillan, D.C. The Impact of Perioperative Risk, Tumor Pathology and Surgical Complications on Disease Recurrence following Potentially Curative Resection of Colorectal Cancer. Ann. Surg. 2011, 254, 83–89. [Google Scholar] [CrossRef]
- Wang, W.-W.; Wu, L.; Lu, W.; Chen, W.; Yan, W.; Qi, C.; Xuan, S.; Shang, A. Lipopolysaccharides increase the risk of colorectal cancer recurrence and metastasis due to the induction of neutrophil extracellular traps after curative resection. J. Cancer Res. Clin. Oncol. 2021, 147, 2609–2619. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. PLOS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Belančić, A. Gut microbiome dysbiosis and endotoxemia—Additional pathophysiological explanation for increased COVID-19 severity in obesity. Obes. Med. 2020, 20, 100302. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- E Naugler, W.; Karin, M. NF-κB and cancer—Identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Vinelli, V.; Biscotti, P.; Martini, D.; Del Bo’, C.; Marino, M.; Meroño, T.; Nikoloudaki, O.; Calabrese, F.M.; Turroni, S.; Taverniti, V.; et al. Effects of Dietary Fibers on Short-Chain Fatty Acids and Gut Microbiota Composition in Healthy Adults: A Systematic Review. Nutrients 2022, 14, 2559. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Tsujikawa, T.; Fujiyama, Y. Role of Dietary Fiber and Short-Chain Fatty Acids in the Colon. Curr. Pharm. Des. 2003, 9, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Reytor, D.; Puebla, C.; Karahanian, E.; García, K. Use of Short-Chain Fatty Acids for the Recovery of the Intestinal Epithelial Barrier Affected by Bacterial Toxins. Front. Physiol. 2021, 12, 650313. [Google Scholar] [CrossRef]
- Russo, E.; Giudici, F.; Fiorindi, C.; Ficari, F.; Scaringi, S.; Amedei, A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 2754. [Google Scholar] [CrossRef] [Green Version]
- Bailie, L.; Loughrey, M.B.; Coleman, H.G. Lifestyle Risk Factors for Serrated Colorectal Polyps: A Systematic Review and Meta-analysis. Gastroenterology 2017, 152, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, T.; Maehara, N.; Kai, T.; Arai, S.; Miyazaki, T. Dietary fructose-induced hepatocellular carcinoma development manifested in mice lacking apoptosis inhibitor of macrophage (AIM). Genes Cells 2016, 21, 1320–1332. [Google Scholar] [CrossRef]
- Kong, C.; Gao, R.; Yan, X.; Huang, L.; Qin, H. Probiotics improve gut microbiota dysbiosis in obese mice fed a high-fat or high-sucrose diet. Nutrition 2019, 60, 175–184. [Google Scholar] [CrossRef]
- Lu, K.; Dong, S.; Wu, X.; Jin, R.; Chen, H. Probiotics in Cancer. Front. Oncol. 2021, 11, 638148. [Google Scholar] [CrossRef]
- Tripathy, A.; Dash, J.; Kancharla, S.; Kolli, P.; Mahajan, D.; Senapati, S.; Jena, M. Probiotics: A Promising Candidate for Management of Colorectal Cancer. Cancers 2021, 13, 3178. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, W.; Wang, J.; Shi, J.; Sun, Y.; Wang, W.; Ning, G.; Liu, R.-X.; Hong, J. Akkermansia muciniphila improves metabolic profiles by reducing inflammation in chow diet-fed mice. J. Mol. Endocrinol. 2017, 58, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yue, Y.; Ma, C.; Dong, L.; Chen, F. Pasteurized Akkermansia muciniphila Ameliorate the LPS-Induced Intestinal Barrier Dysfunction via Modulating AMPK and NF-κB through TLR-2 in Caco-2 Cells. Nutrients 2022, 14, 764. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Choi, Y.; Kim, D.-K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.-S.; Jee, Y.-K.; Gho, Y.S.; et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, G.Y.; Kane, A.; Lee, K.; Xu, Q.; Wu, X.; Roper, J.; Mason, J.B.; Crott, J.W. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and Akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int. J. Cancer 2018, 143, 1797–1805. [Google Scholar] [CrossRef] [Green Version]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-Like Receptor (TLR) 4 Signaling, Binds Selectively to TLR-4 and Interferes with Interactions between TLR-4 and Its Adaptor Molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Pastille, E.; Faßnacht, T.; Adamczyk, A.; Phuong, N.N.T.; Buer, J.; Westendorf, A.M. Inhibition of TLR-4 Signaling Impedes Tumor Growth in Colitis-Associated Colon Cancer. Front. Immunol. 2021, 12, 669747. [Google Scholar] [CrossRef]
- Schneider, K.M.; Mohs, A.; Kilic, K.; Candels, L.S.; Elfers, C.; Bennek, E.; Schneider, L.B.; Heymann, F.; Gassler, N.; Penders, J.; et al. Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. Int. J. Mol. Sci. 2019, 20, 308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, C.; Chai, D.; Li, C.; Guan, Y.; Liu, L.; Kuang, T.; Deng, W.; Wang, W. The association between antibiotic use and outcomes of HCC patients treated with immune checkpoint inhibitors. Front. Immunol. 2022, 13, 956533. [Google Scholar] [CrossRef]
- Pinato, D.J.J.; Li, X.; Mishra-Kalyani, P.S.; D’Alessio, A.; Fulgenzi, C.A.; Wei, G.; Schneider, J.A.; Rivera, D.; Pazdur, R.; Theoret, M.R.; et al. Antibiotic therapy and association with oncological outcomes from targeted and immune-based therapy in hepatocellular carcinoma (HCC). J. Clin. Oncol. 2022, 40, 4089. [Google Scholar] [CrossRef]
- Cheung, K.S.; Lam, L.K.; Seto, W.K.; Leung, W.K. Use of Antibiotics during Immune Checkpoint Inhibitor Treatment Is Associated with Lower Survival in Hepatocellular Carcinoma. Liver Cancer 2021, 10, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Pichika, M.R.; Mai, C.W.; Kang, Y.B. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: Its expression and effects in the ten most common cancers. OncoTargets Ther. 2013, 6, 1573–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Kaji, K.; Nishimura, N.; Ishida, K.; Ogawa, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. The Angiotensin II Receptor Blocker Losartan Sensitizes Human Liver Cancer Cells to Lenvatinib-Mediated Cytostatic and Angiostatic Effects. Cells 2021, 10, 575. [Google Scholar] [CrossRef]
- Fujinaga, Y.; Kawaratani, H.; Kaya, D.; Tsuji, Y.; Ozutsumi, T.; Furukawa, M.; Kitagawa, K.; Sato, S.; Nishimura, N.; Sawada, Y.; et al. Effective Combination Therapy of Angiotensin-II Receptor Blocker and Rifaximin for Hepatic Fibrosis in Rat Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5589. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, X.; Jiang, Y.; Wu, Z.; Zhu, X.; Liu, H.; Wu, X.; Cai, J.; Ding, X.; Gong, W. The synergistic antitumor effect of IL-6 neutralization with NVP-BEZ235 in hepatocellular carcinoma. Cell Death Dis. 2022, 13, 146. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Miyashita, A.; Fukushima, S.; Ikeda, T.; Kubo, Y.; Senju, S.; Ihn, H.; Nishimura, Y.; Oshiumi, H. Combined Blockade of IL6 and PD-1/PD-L1 Signaling Abrogates Mutual Regulation of Their Immunosuppressive Effects in the Tumor Microenvironment. Cancer Res. 2018, 78, 5011–5022. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Pu, N.; Chen, Q.; Zhang, J.; Zhao, G.; Xu, X.; Wang, D.; Kuang, T.; Jin, D.; Lou, W.; et al. Gut-derived lipopolysaccharide remodels tumoral microenvironment and synergizes with PD-L1 checkpoint blockade via TLR-4/MyD88/AKT/NF-κB pathway in pancreatic cancer. Cell Death Dis. 2021, 12, 1033. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zeng, Z.; Shen, X.; Wu, Z.; Dong, Y.; Cheng, J.C.-H. MicroRNA-146a-5p Negatively Regulates Pro-Inflammatory Cytokine Secretion and Cell Activation in Lipopolysaccharide Stimulated Human Hepatic Stellate Cells through Inhibition of Toll-Like Receptor 4 Signaling Pathways. Int. J. Mol. Sci. 2016, 17, 1076. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-W.; Huang, Y.-T. Inhibitory Effect of Tanshinone IIA on Rat Hepatic Stellate Cells. PLoS ONE 2014, 9, e103229. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, H.; Burke, L.J.; Bridle, K.R.; Li, X.; Zhao, C.-X.; Crawford, D.H.G.; Roberts, M.; Liang, X. Therapeutic modulators of hepatic stellate cells for hepatocellular carcinoma. Int. J. Cancer 2020, 147, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, X.; Cheng, X.; Zhang, Y.; Chen, L.; Shi, L.; Liu, Z.; Qian, H.; Wu, M.; Yin, Z. Intratumoral Hepatic Stellate Cells as a Poor Prognostic Marker and a New Treatment Target for Hepatocellular Carcinoma. PLoS ONE 2013, 8, e80212. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manilla, V.; Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship. Microorganisms 2023, 11, 267. https://doi.org/10.3390/microorganisms11020267
Manilla V, Di Tommaso N, Santopaolo F, Gasbarrini A, Ponziani FR. Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship. Microorganisms. 2023; 11(2):267. https://doi.org/10.3390/microorganisms11020267
Chicago/Turabian StyleManilla, Vittoria, Natalia Di Tommaso, Francesco Santopaolo, Antonio Gasbarrini, and Francesca Romana Ponziani. 2023. "Endotoxemia and Gastrointestinal Cancers: Insight into the Mechanisms Underlying a Dangerous Relationship" Microorganisms 11, no. 2: 267. https://doi.org/10.3390/microorganisms11020267